
Abstract
Abstract
Background:
Hyperglycemia associated with insulin resistance is common among critically ill patients. Interleukin (IL)-18 has been linked with hyperglycemia and insulin resistance in chronic disease, but the relation between IL-18 and insulin resistance during critical illness was unexplored. This study investigated whether IL-18 modulates hyperglycemia and insulin resistance during acute inflammation.
Methods:
We injected lipopolysaccharide (LPS) 40 mg/kg into wild-type (WT) and IL-18 knockout (KO) mice to induce endotoxemia and examined insulin resistance and insulin-dependent signaling pathways during the acute phase.
Results:
During the first hour after LPS treatment, IL-18 KO mice showed higher blood glucose and insulin and less insulin receptor substrate-1 and less phosphorylated Akt in the liver compared with WT mice. Interleukin-18 KO mice exhibited better survival after LPS treatment.
Conclusions:
The findings suggest that endogenous IL-18 may attenuate hyperglycemia and modulate insulin signaling in liver. Accordingly, IL-18 may modulate glucose tolerance during acute inflammation.
H
Pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, are elevated in acute inflammation and promote hyperglycemia by inhibiting insulin signaling pathways [9,10]. Interleukin-18 is a pro-inflammatory cytokine and is also elevated dramatically in acute inflammation [11–13]. In addition, some studies suggest that circulating IL-18 concentrations are elevated during chronic insulin resistance, such that seen as a result of obesity and diabetes [14,15]. However, an association between IL-18 and acute insulin resistance had not been reported. In this study, we used IL-18 knockout (KO) mice to investigate the role of IL-18 during acute lethal endotoxemia.
Materials and Methods
Animals
Twenty-week-old male C57BL/6J (wild-type, WT) mice (Clea Japan, Tokyo, Japan) and IL-18 KO mice (B6.129P2-IL18tm1Aki/J; The Jackson Laboratory, Bar Harbor, ME) were studied. All mice were kept at 22°C under a 12-h light/dark cycle and allowed access to a standard diet and water ad libitum. Mice were anesthetized with isoflurane and then injected intraperitoneally with lipopolysaccharide (LPS) 40 mg/kg (Escherichia coli O111: B4, Sigma-Aldrich, St Louis, MO) or phosphate buffered saline (PBS). Either 1 h or 12 h after injection, the mice were killed by cardiac puncture under general anesthesia. Samples of blood, liver, and lung were harvested immediately and homogenized in lysis buffer A (100 mM HEPES [pH 7.4], 1% Triton, 10% glycerol, 150 mM NaCl, 1 mM sodium pyrophosphate, 100 mM sodium fluoride, 2 mM ethylenediaminetetraacetic acid [EDTA], 5 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride [PMSF], 5 mcg/mL aprotinin, and 5 mcg/mL leupeptin) for protein isolation or TRIzol Reagent (Invitrogen, Carlsbad, CA) for total RNA isolation, and stored at −80°C until analysis. All experiments involving mice were conducted in accordance with the approval of the Animal Welfare Committee of Kobe University Graduate School of Health Sciences.
Glucose, hormones, and cytokines
Venous blood was obtained from the tail vein before LPS injection and 15, 30, and 60 min and 12 h after LPS injection. Blood glucose concentrations were measured using a portable glucose meter (Glutest Sensor; Sanwa Kagaku Kenkyusho, Nagoya, Japan). Plasma IL-18 concentrations were assessed using an enzyme-linked immunosorbent assay (ELISA) kit (MBL, Nagoya, Japan) specific for the 18 kDa bioactive form of IL-18 according to the manufacturer's instructions. Plasma glucagon (WAKO, Osaka, Japan) and insulin (Shibayagi, Gunma, Japan) concentrations were also measured using ELISA according to the manufacturer's instructions. Plasma TNF-α and IL-6 concentrations were assayed using Cytometric Bead Array (CBA) flex sets (BD Pharmingen Corp., San Diego, CA). Flow cytometric analysis was performed using a FACS Array flow cytometer (BD Immunocytometry Systems, Franklin Lakes, NJ). Data were acquired and analyzed by BD FACS Array software and FCAP Array software, version 1.0 (BD Immunocytometry Systems). The limit of detection for TNF-α was 17.1 pg/mL and the limit for detection of IL-6 was 6.5 pg/mL.
Western blotting
Anti-insulin receptor substrate (IRS)-1, anti-phosphorylated IRS-1 (Tyr895), anti-Akt and anti-phosphorylated Akt (Ser473) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-β-actin antibody was purchased from Sigma-Aldrich. Western blotting was performed as described previously (16). Membranes were incubated overnight at 4°C with primary antibodies diluted in Can Get Signal immunoreaction enhancer solution 1 (Toyobo, Osaka, Japan), then with horseradish peroxidase-conjugated anti-rabbit secondary antibodies diluted in Can Get Signal solution 2 for 1 h at room temperature. Antibody binding was detected using enhanced chemiluminescence plus reagents (GE Healthcare, Buckinghamshire, United Kingdom) and exposed on Hyperfilm (GE Healthcare, Fairfield, CT). The band intensity was quantified by Image J software version 1.47 (National Institutes of Health, Bethesda, MD).
Hematoxylin and eosin staining
To evaluate liver and lung injury 12 h after LPS or PBS injection, we performed hematoxylin and eosin (H&E) staining. The liver and lung samples were immersed in fixative solution (4% paraformaldehyde in PBS) for 24 h, dehydrated through graded alcohol dilutions, and embedded in paraffin. Tissue was sectioned and stained with H&E using standard protocols.
Statistical analysis
The data were expressed as mean±standard error (SEM). The blood glucose and plasma IL-18 concentrations were examined using two-factor analysis of variance (ANOVA). For other experiments, ANOVA with the Tukey-Kramer post-hoc test was performed. Survival was examined by Kaplan-Meier analysis and the log-rank test. A probability level of p<0.05 was considered statistically significant.
Results
Blood glucose and plasma insulin
To assess the effects of IL-18 alteration on glucose metabolism, we measured blood glucose, plasma insulin, and glucagon concentrations in WT and IL-18 KO mice after LPS administration to induce endotoxemia. High-dose LPS induced transient hyperglycemia within 1 h of administration, followed by hypoglycemia 12 h after administration in both WT and IL-18 KO mice (Fig. 1A). Blood glucose concentrations in the IL-18 KO mice were significantly increased compared with WT during the first hour after LPS injection (Fig. 1A). Plasma insulin concentrations were increased in the IL-18 KO mice 1 h after LPS administration then returned to baseline concentrations after 12 h (Fig. 1B). In contrast, plasma insulin concentrations in WT were not altered by LPS injection. Plasma glucagon concentrations increased during the first hour after LPS injection in both the WT and IL-18 KO mice and were beneath the assay detection threshold in all animals 12 h after LPS administration (Fig. 1C).
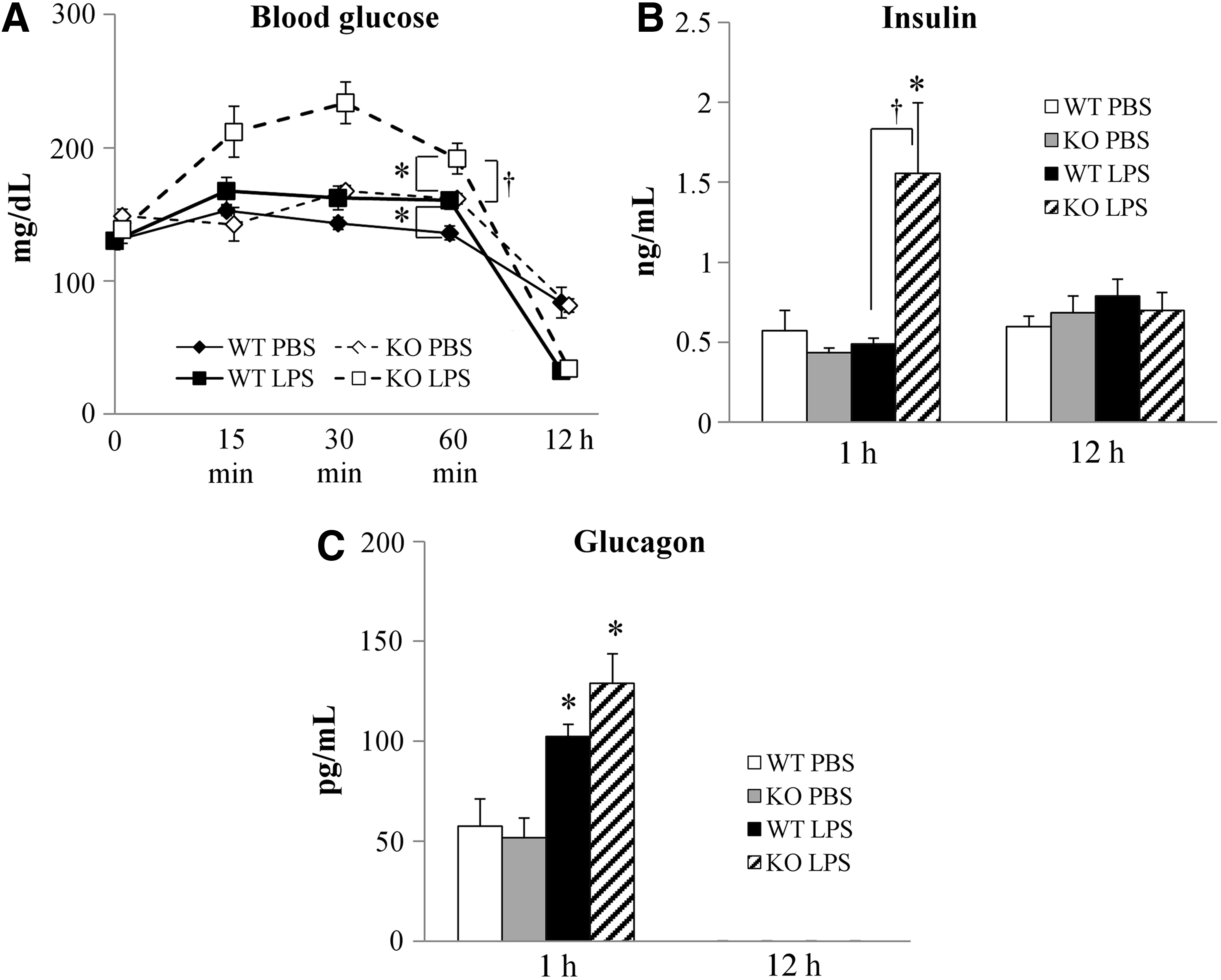
Blood glucose and plasma hormones concentrations after lipopolysaccharide (LPS) injection. (
Cytokine levels in plasma
To assess cytokine expression during hyperglycemia induced by endotoxemia, we measured plasma concentrations of IL-18, IL-6, and TNF-α by ELISA or CBA. In WT mice, IL-18 concentrations were not elevated 1 h after LPS administration (100 pg/mL in both PBS- and LPS-treated mice) but increased dramatically to ∼7,000 pg/mL 12 h after LPS administration (Fig. 2A). IL-6 and TNF-α were elevated significantly after LPS administration in both WT and IL-18 KO (Fig. 2B and 2C). Concentrations of IL-6 were much higher 12 h after LPS injection than 1 h after injection, but 1 h after injection the IL-18 KO mice expressed more IL-6 than the WT mice (Fig. 2B). In contrast, TNF-α concentrations were much higher 1 h after LPS injection than 12 h after LPS injection and did not differ between WT and IL-18 KO mice (Fig. 2C).
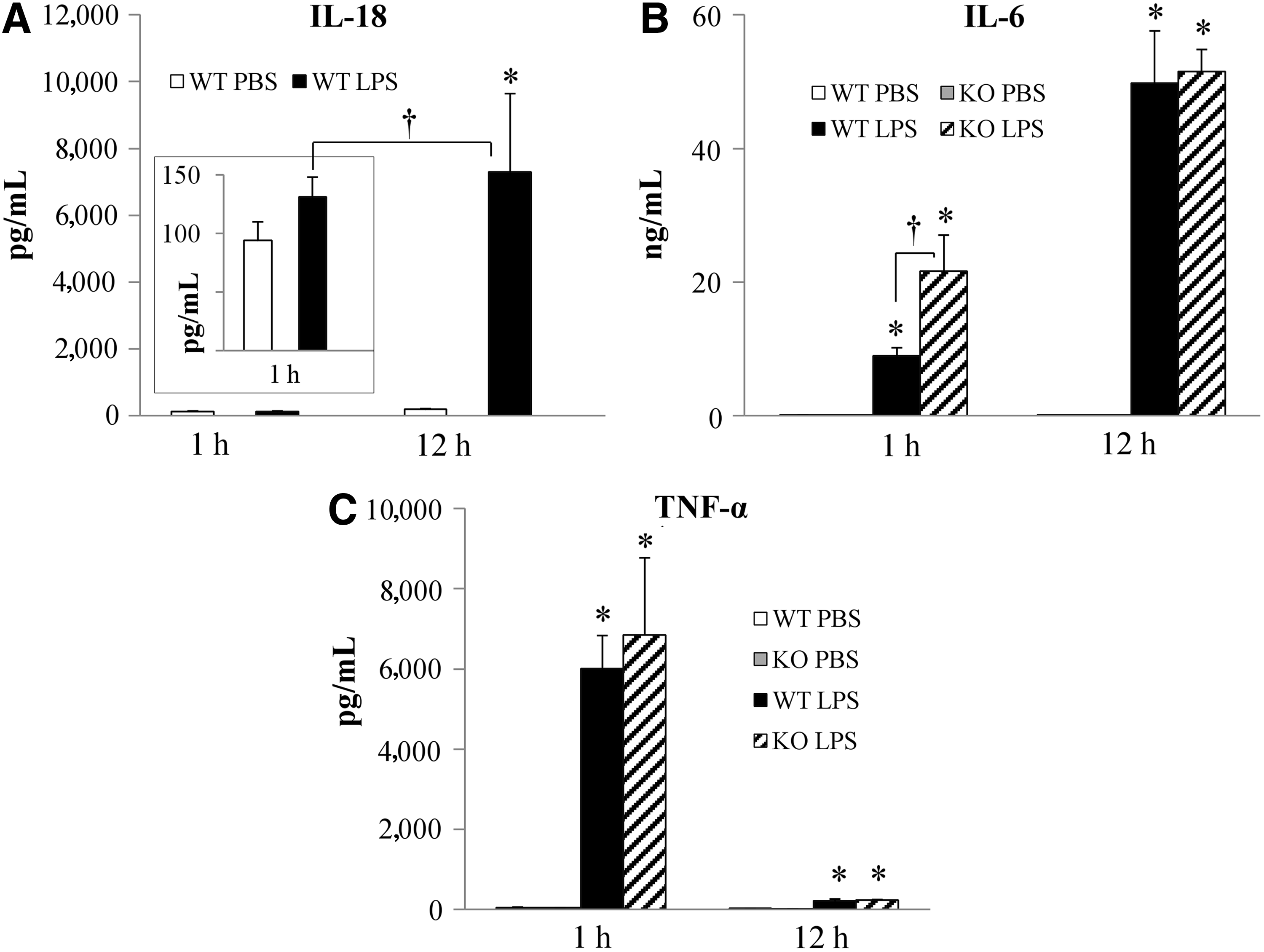
Plasma cytokine concentrations after lipopolysaccharide (LPS) injection. (
Activation of IRS-1 and Akt in the liver
To assess the effects of IL-18 deletion on insulin signaling in response to acute endotoxemia, we analyzed the activation of IRS-1 and Akt. Because the blood glucose and plasma insulin and IL-6 concentrations differed between WT and IL-18 KO 1 h after LPS administration and did not differ 12 h after LPS injection, we chose to analyze the effect of IL-18 deletion on insulin receptor signaling only at the 1-h time point. In PBS-treated mice, total IRS-1 expression was higher in IL-18 KO mice compared with WT mice (Fig. 3A and 3B), and high concentrations of IRS-1 phosphorylation were observed, indicating higher baselines concentrations of activated IRS-1 in the absence of IL-18. After LPS treatment, total IRS-1 and pIRS-1 tended to increase in WT mice but decreased significantly in IL-18 KO mice compared with PBS-treated WT or IL-18 KO mice (Fig. 3A and 3B). The differences likely reflected changes in total IRS-1 protein concentrations, as the pIRS-1/IRS-1 ratio did not differ significantly between WT and KO or in response to LPS administration (Fig. 3A and Fig. 3B, right panel). Total akt expression was not altered significantly by either IL-18 deletion or LPS injection (Fig. 3A and Fig. 3C, left panel). Akt phosphorylation (pAkt) was increased significantly by LPS in WT mice, but did not increase significantly in the IL-18KO mice after LPS treatment. (Fig. 3A and Fig 3C, center and right panel)

Activation of the insulin signaling pathway. (
Liver and lung morphology
Because clinical sepsis can lead to dysfunction of multiple organs, we examined lung and liver morphology after LPS administration in presence and absence of IL-18. Lipopolysaccharide-treated IL-18 KO mice experienced less severe lung injury, such as alveolar septal thickening and neutrophil accumulation, after LPS administration than WT mice (Fig. 4A). The lungs of PBS-treated WT and IL-18 KO mice were similar morphology.

Morphologic changes in lung and liver and mouse survival after lipopolysaccharide (LPS) adminstration. (
In the livers of WT mice, many hemorrhagic changes and large necrosis were observed (Fig. 4B). In contrast, the livers of IL-18 KO mice had few hemorrhagic changes and small necrosis in response to LPS administration. Hemorrhagic changes and necrosis were rarely observed in the livers of WT or IL-18 mice injected with PBS only.
The mortality of lethal endotoxemia
Endotoxemia induced by LPS injection is frequently lethal. More than one-half of the WT mice (60%) died within 2 d of injection. The IL-18 KO mice exhibited better survival with 70% surviving more than 2 d (Fig. 4C).
Discussion
This study showed that a deficiency of IL-18 promoted hyperglycemia and hyperinsulinemia during the hyperacute phase (first hour) of endotoxemia. In addition, IL-18 deficiency inhibited insulin signaling pathway activation in the liver during acute endotoxemia. These data suggest that IL-18 modulates glucose tolerance during endotoxin-induced acute inflammation. Surprisingly, IL-18 deficiency limited lung and liver damage and improved survival after LPS-induced endotoxemia.
Our observations of LPS-induced hyperglycemia 1 h after LPS injection and hypoglycemia 12 h after injection in this study correlate well with previous reports [16]. During the first hour after LPS injection, plasma glucagon, IL-6, and TNF-α concentrations increased compared with mice given a sham injection of PBS only. Because glucagon is an inducer of hyperglycemia and the cytokines are upregulated in response to hyperglycemia, we believe that these compounds may contribute to the transient hyperglycemia after LPS administration.
Our results demonstrating changes in insulin signaling in the liver in response to LPS-induced acute endotoxemia also support the findings of previous studies. Acute insulin resistance with hyperglycemia and hyperinsulinemia has been observed in hepatocytes in response to trauma and hemorrhage with corresponding downregulation of the insulin signaling pathway [17,18]. Additionally, inflammatory cytokines, such as TNF-α and IL-6, are elevated during acute and chronic inflammation, can inhibit insulin signaling pathways, and are associated with insulin resistance [9,10,19]. Although we hypothesized that IL-18 deletion would decrease acute insulin resistance and hyperglycemia, in fact, blood glucose, insulin and IL-6 concentrations were significantly higher in IL-18 KO mice than in WT mice. There are a few experimental studies suggesting that IL-18 is necessary for glucose tolerance in chronic inflammation [20,21]. The present study does not contradict these reports. In our study, the insulin signaling pathway was dampened in the liver of IL-18 KO mice, but not in that of WT mice. Insulin receptor substrate-mediated insulin signaling was altered in the IL-18 KO compared with the WT because the total cellular pool of IRS-1 differed between the IL-18 KO mice and the WT mice both before and after LPS administration. These data suggest that endogenous IL-18 plays an important role in modulating IRS-1 expression. Endogenous IL-18 might suppress hyperinsulinemia by maintaining insulin signaling pathway activity during the hyperacute phase (first hour) of endotoxemia, and IL-18 deletion might accelerate the inhibition of insulin signals. High plasma IL-6 in IL-18 KO mice may have promoted hyperglycemia, because IL-6 induces gluconeogenesis and glycogenolysis in hepatocytes and inhibits the phosphorylation of IRS-1 [10,22]. Maintenance of the insulin signaling pathway by IL-18 may be through the inhibition of IL-6, however, it is unclear how IL-18 might suppress IL-6 production. Negative feedback signaling could play a role because IL-18 can activate signal transducer and activator of transcription (STAT)-3, which is downstream of IL-6 [22,23].
Some reports have shown that increased circulating IL-18 or genetic variation of IL-18 is linked with insulin resistance in patients with type 2 diabetes mellitus, obesity, or polycystic ovary syndrome [14,15,24,25]. By contrast, our study identified insulin resistance during acute endotoxemia only in the absence of IL-18. This contradiction between our results and these reports may derive from differences in IL-18 sensitivity during acute and chronic inflammation. Zilverschoon et al. [26] reported that leukocytes from patients with obesity or type 2 diabetes mellitus had low responses after stimulation with IL-18, despite having high serum IL-18 concentrations. This suggests that IL-18 sensitivity is decreased in chronic inflammation. High IL-18 concentrations in patients with chronic insulin resistance may be compensatory reaction to reverse insulin resistance. Alternatively, the role of IL-18 in insulin resistance in response to chronic or acute inflammation may be understood insufficiently. This is a complex pathway with many redundancies to ensure adequate regulation. Studies of single elements of the pathway should be interpreted conservatively, and care must be taken not to imply causality when in fact only correlations are proven.
Our previous study suggested that endogenous IL-18 promoted lung edema and contributed to poor outcomes during endotoxemia [27]. The present study showed that in the absence of IL-18 (IL-18 KO) lung and liver injury were less severe and a survival was better after LPS injection compared with WT mice. We speculate that dramatically elevated IL-18 may promote organ dysfunction and contribute to a poor prognosis. Additionally, it is possible that the high blood glucose concentrations in the IL-18 KO mice preserved intravascular volume and perfusion and led to better survival.
There were some limitations of this study. First, the LPS-induced sepsis model does not model all aspects of clinical sepsis. We used a lethal endotoxemia model, because we believe it more closely mimics clinical septic shock than non-lethal models. However, we could only reproduce hyperglycemia for a short time. Second, KO mice are known to develop compensatory mechanisms that allow proper development and organ function in the absence of the targeted gene product. We cannot exclude the possibility of compensatory reactions in this study. Thus, the current findings may not be applicable directly to clinical sepsis, and further study is warranted.
In summary, this report revealed new aspects of the relation between IL-18 and glucose tolerance during acute inflammation. In this study, we discovered that IL-18 KO mice exhibited hyperglycemia and hyperinsulinemia after LPS injection. Because overinduction of IL-18 may contribute to high mortality, the possibility that an IL-18 blocking agent could be developed to treat patients with acute inflammation is intriguing. However, this report should also raise an alarm that perfect block of IL-18 may be undesirable and may induce acute insulin resistance and hyperglycemia.
Footnotes
Acknowledgments
The authors thank Dr. Shannon L Wyszomierski for editing the manuscript.
This research was supported by JSPS KAKENHI Grant Number 23659852.
Author Disclosure Statement
No competing financial interests exist.