
Abstract
Abstract
The sepsis syndrome is a systemic host inflammatory response accompanied by organ dysfunction in response to invading microbial pathogens. The host recognizes both danger and pathogens through its pattern recognition receptors on immune cells. These receptors bind to pathogen- (PAMP) and danger- (DAMP) associated molecular patterns derived from microbes and host tissues, respectively. These processes set in motion a cascade of events in host cells and tissue, which activate multiple cytokines that serve as activators of the host inflammatory response as well as eventually lead to resolution of the response if the host recovers. The following article describes some of these DAMPs and PAMPs, and how they activate pathways that activate the host cytokine immune response to injury and infection.
S
SIRS is likely initiated by a complex neuroendocrine response initiated by tissue injury, be it mechanical or infectious [2]. Because the immune system likely evolved as a response to local injury or infection, SIRS is essentially a mal-adaptive response by the host to a major challenge. Such challenges were lethal until intravenous resuscitation, development of antibiotic agents, organ support systems, and the intensive care unit (ICU) of the 20th century. At the beginning of that century, it was noted that “Except on few occasions, the patient appears to die from the body's response to infection rather than from it.” This remark, made by William Osler (1849–1919), is still widely cited [3], and thus the host response to serious infectious challenge was recognized as a major detriment that accounted for death [4]. Paul Erhlich, Nobel laureate, warned of a “horror autotoxicus” if host mechanisms were not in place to recognize self-antigens and therefore contain the host defense response [5].
An explosion of research into the genomics and proteomics of SIRS and sepsis has produced extensive insight into the arcane endocrine and paracrine mechanisms that drive the early innate immune response including cell-cell interactions, sub-cellular communication, and generation of products that amplify (necrosis) or turn off (apoptosis) the systemic inflammatory response [2]. There are multiple redundant and complex means for the host to recognize danger [6] in the form of injury or infection, which involve multiple different molecules both released from host cells, collectively named danger-associated molecular patterns (DAMPs) and microbes (pathogen-associated molecular pattern or PAMPs). These complex processes lead to well-defined sub-cellar activation pathways that eventually lead to development of an innate immune response.
Pathogenesis of the Host Septic Response
Sepsis is a life-threatening physiologic state accompanied by organ dysfunction as a result of a dysregulated host response to infection [7]. The progression from infection to sepsis has been a focus of investigation, and this progression is initiated by the detection of DAMPs or PAMPs [3]. Host tissues responsible for this continuous screening possess pattern recognition receptors (PRRs) both on the cell surface and in the cytosol. There are between 20 and 40 of these, and most are within the Toll-like receptor (TLR), C-type lectin receptor (CLR), NOD-like receptor (NLR), and retinoic acid-inducible gene (RIG)-I-like receptor (RLR) families [8]. These include Dectin, Mincel, receptors for advanced glycation end products (RAGE), RIG1, melanoma differentiation-associated gene 5 (MDA5), NALP3, and mannose-binding lectin [9]. The bound complexes of such DAMPs and PAMPs with their respective PRRs are collectively referred to as the inflammasome pathway, which eventually stimulates the production of pro-inflammatory cytokines and amplifies the local inflammatory response by host cell pyroptosis (Fig. 1).
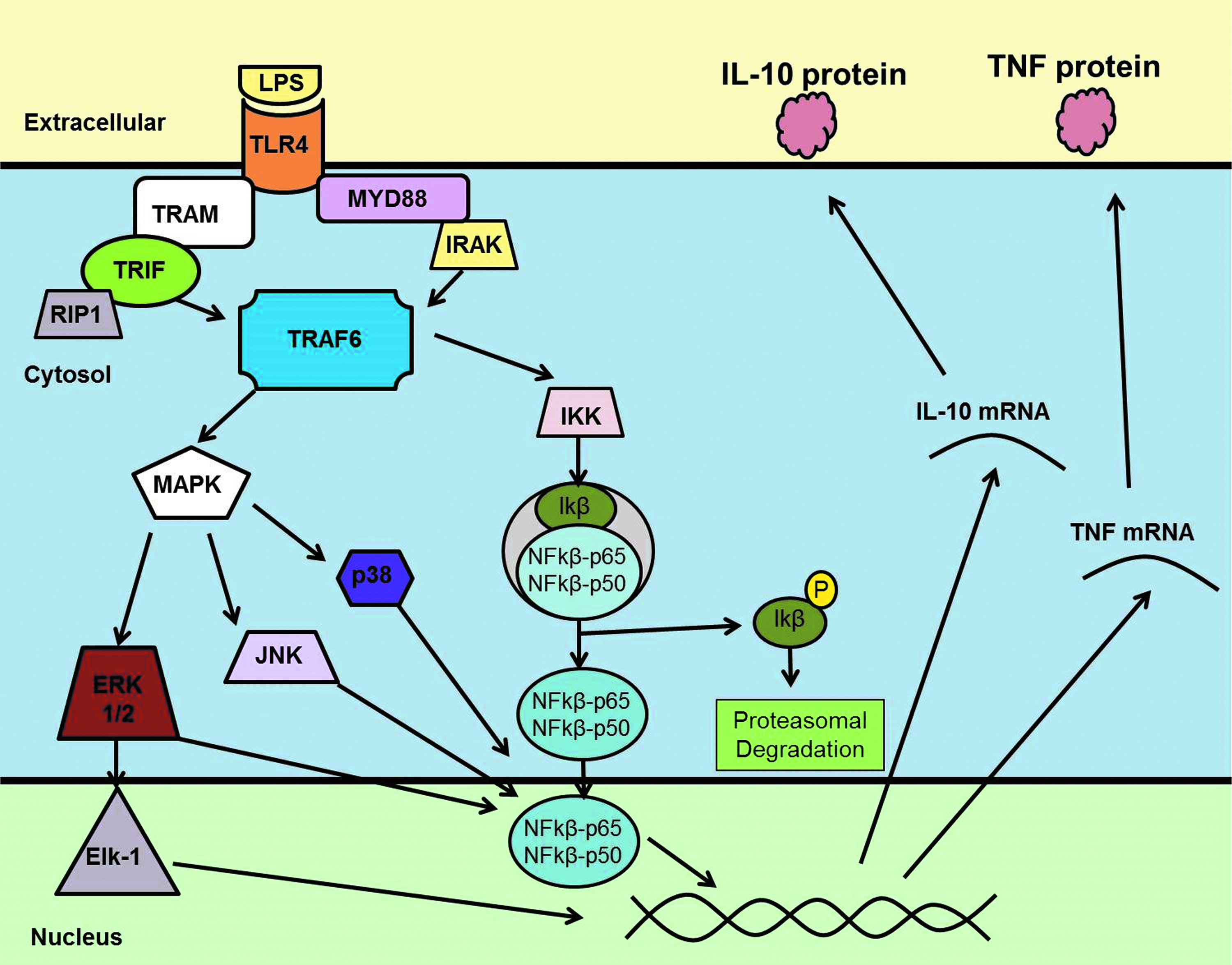
Toll-like receptor-4 signaling. Example of pathogen-associated molecular pattern- oligerimized sensor protein (PAMP-PRR) cell signaling. Toll-Like Receptor 4 (TLR-4) signaling through MyD88 dependent and independent pathways. Lipopolysaccharide (LPS), TRIF-related adapter molecule (TRAM), TIR-domain-containing adaptor-inducing interferon β (TRIF), myeloid differentiation primary response gene 88 (MyD88), IL-1 receptor associated kinase (IRAK), TNF-receptor associated factor 6 (TRAF6), mitogen activated protein kinases pathway (MAPK), extracellular signal related kinase (ERK), c-Jun N-terminal kinase (JNK), inhibitory kappa kinase (IKK), inhibitory kappa beta (Iκβ), nuclear factor kappa beta (NF-κB), interleukin-10 (IL-10) and tumor necrosis factor-α.
The inflammasome assemblies consist of oligerimized sensor proteins (PRRs) associated caspase 1 and can persist in the local environment to provide a sustained inflammatory response. The sensing of altered host cellular homeostasis by intracellular inflammasomes has been termed “homeostasis altering molecular processes (HAMPs)” and reflects sensing of danger by altered homeostasis rather than molecular patterns [8]. While this interaction is meant to work in favor of the host by activation of protective immune and inflammatory processes, it often triggers pathologic tissue and organ injury associated with severe sepsis and trauma [2]. In either case, a compensatory anti-inflammatory response syndrome is initiated along with the pro-inflammatory response [10], just as Ehrlich had predicted more than 100 years ago. The exaggerated immune response seen in sepsis is mediated by a complex interplay of cytokines, chemokines, complement and coagulation factors, as well as inflammatory and immune regulatory cells including the neuroendocrine response of the host as well. There is growing evidence that sepsis includes progression from the initial hyper-inflammatory state to an immunosuppressive state [11].
Host Immune Response to Injury and Microbial Organisms
The immune response occurs in three phases: (1) An immediate innate response, which occurs over seconds to a few hours and is characterized by recognition of the invading pathogen or tissue injury by pre-formed, non-specific, or broadly specific effectors; (2) an early innate response that occurs over hours to a few days, in which host recognition of both DAMPS and PAMPS occurs and leads to recruitment and activation of effector cells (Fig. 2), which further enhances the host inflammatory response; and (3) the adaptive response that occurs over the ensuing days to weeks, which consists of recognition by B and T cells, their clonal expansion and subsequent differentiation into effector cells against specific antigens [12,13].

Neutrophil (polymorphonuclear [PMN]) migration from the intravascular space into the interstitium in response to microbial invasion. The PMNs bind to endothelium via adhesion molecular interaction and migrate across cell gaps along a chemokine gradient where they release granules toxic to both host tissue and microbes. The PMNs then undergo necrosis, which amplifies the inflammatory response, or apoptosis, which leads to its resolution.
The innate immune system has evolved as the first line of response to pathogens and injury. It consists of the mechanical barriers to infection, such as skin epithelium and mucosal linings of the gastrointestinal tract, pulmonary tree, and genitourinary system. The activation of complement and coagulation cascades impairs pathogen function, as well as activating the cellular component of the innate immune system [13]. The PAMPs and DAMPs are released in response to pathogen invasion and tissue injury and are recognized by PRRs on the surface of immune cells [14].
The major cell types of the innate immune system are the macrophage and neutrophil, both of which play critical roles in the phagocytosis of microbes, but the macrophage is the key orchestrator of the local reaction [13,15]. Macrophages reside in the tissues and release mediators such as cytokines and chemokines to induce inflammation as well as directly attacking microbes via release of free radicals, proteolytic enzymes, and phagocytose invading pathogens [16,17]. This release stimulates the influx of neutrophils and subsequently monocytes and macrophages from the peripheral circulation to assist in fighting the infection and ultimately facilitating tissue repair and wound healing. The third main function of macrophages is antigen processing after phagocytosis and presentation to other immune cells, which initiates the adaptive immune response with eventual specific antibody production [13].
The pro-inflammatory response is followed by a regulated anti-inflammatory response that dampens down any excessive inflammation to reduce the damage to surrounding normal tissue [13]. Often there is a dysfunctional early innate immune response that does not respond appropriately to injury or infection, which can lead to an imbalance in the pro- and anti-inflammatory responses, persistent organ dysfunction, and host morbidity and death [18].
DAMPs (Alarmins)
Alarmins are constitutively available endogenous molecules that are released on tissue damage and take part in immune system activation. The PAMPs, during early sepsis and via PRRs stimulation, orchestrate the innate immune response. They activate a signaling cascade that results in the release of early pro-inflammatory cytokines, including DAMPs, also known as alarmins [19,20]. The following are DAMPs that have been most studied (Table 1).
DAMPS = danger-associated molecular patterns; PRR = oligerimized sensor proteins ; HMGB1 = high mobility group box protein1; TLR = Toll-like receptor; RAGE = receptor for advanced glycation end products; mtDNA = mitochondrial deoxyribonucleic acid; HSP = heat shock protein; IL = interleukin; ST2 = serum stimulation-2.
High mobility group box protein 1 (HMGB1)
The HMGB1 is a highly conserved nuclear protein. It is expressed on almost all human cells except those without nuclei. The HMGB1 was known for its function as a transcription factor. In 1999, however, Wang et al [21] demonstrated its role as a pro-inflammatory cytokine in the immune response to tissue damage. The HMGB1 exhibits diverse functions based on its location in the cell. After cell damage, it is released and then recognized by RAGE. It also interacts directly with plasminogen and tissue type plasminogen activator (tPA), which produces plasmin at the cell surface and causes extracellular proteolysis during cell invasion and tissue injury [22,23]. The HMGB1 is also released after tissue necrosis, thereby leading to an enhanced local inflammatory response [24].
In sepsis, macrophages, monocytes, and neutrophils also release HMGB1, which amplifies both the local and systemic inflammatory response and may result in multi-organ damage [25]. Immune cells, after acetylating HMGB1 in the nucleus, translocate it to the cytoplasm and secrete it via nuclear factor (NF-κB) activation, probably through non-transcriptional mechanisms [24]. Extracellular HMGB1 is recognized by RAGE, TLR2, and TLR4. The HMGB1-induced signaling has pleiotropic effects on immune cells, promoting inflammation and the potentially harmful disruption of epithelial barriers [26]. In addition to the activation of PRRs, HMGB1 increases the pro-inflammatory activity of cytokines (such as interleukin [IL]-1ß) through binding to these mediators. This supports the idea that HMGB1 might not act solely as a pro-inflammatory mediator, but also as a carrier for other DAMPs [27]. Activation of the cholinergic anti-inflammatory pathway, however, suppresses HMGB1 secretion by macrophages in sepsis and improves survival [28].
In summary, clinical and pre-clinical studies have implicated HMGB1 as a mediator of sepsis pathogenesis and death. Because its blockade has shown promising reduction in mortality rates, HMGB1 represents a novel candidate for passive immune targeting in sepsis management [29].
Mitochondrial deoxyribonucleic acid (mtDNA)
The mtDNA encodes essential protein sub-units of the oxidative phosphorylation system. The mtDNA fragments are released from injured or dying cells into the extracellular space, resulting in activation of the immune response cascade and inflammation. Host tissue injury, whether infective or sterile, correlates with increased release of mtDNA [30,31]. Mitochondrial DNA, similar to bacterial and prime neutrophil DNA, contains unmethylated CpG, which can initiate potent immune responses through TLR9 inflammasome and interferon (INF)-1. Inflammasomes activate caspase-1 resulting in the release of pro-inflammatory cytokines IL-1ß and IL-18 [32]. When in the cytosol, mtDNA is recognized by TLR9, MYD88, and NF-κB, stimulating circulating leukocytes to increase production of pro-inflammatory cytokines such as tumor necrosis factor (TNF), IL-6, and adhesion molecules as well [33]. The elicited inflammatory response has the potential to propagate injury to sites distant from the initiating event; however, mtDNA depletion in macrophages impairs inflammasome activation, suggesting a critical role for mtDNA in the immune response [34,35]. The mtDNA and bDNA polymerase chain reactions can quantify tissue injury incurred by septic or sterile mechanisms and have the potential to be used in identification of the source of SIRS of unknown origin [31].
Heat shock proteins (HSPs)
The HSPs are a superfamily of molecular chaperones involved in various steps of protein maturation. A HSP is one of the most evolutionarily conserved proteins from archaebacterials to eukaryotes [36]. Although most of the HSPs are expressed under normal conditions and play vital roles in normal cell function, their expression in T and B lymphocytes can be induced by cellular stress and injury in conditions such as fever, heat-shock, cold-shock, ethanol, oxidative stress, and sepsis [37]. The HSP27, HSP60, HSP70, and HSP90 protein expression is up-regulated in adults with sepsis; the same pattern is seen with HSP60 and HSP72 protein expression in children with sepsis, and their increased levels have been found to correlate directly with prognosis [38,39]. The HSP70 binds to the monocyte cell surface via the TLR4/ TLR2 complex, leading to MyD88-dependent signaling, NF-κB activation, and up-regulation of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α [40].
Studies have shown significantly increased survival in lipopolysaccharide (LPS)-induced shock in murine models pre-treated with HSP90 inhibitors (radicicol and 17-allylaminodemethoxygel-danamycin). The HSP90 inhibitor pre-treatment prevented the rise in monocyte chemo-attractant protein-1 (MCP-1) and TNF-α plasma level, decreasing lung injury and attenuating vascular leak [41]. Although the death rate was higher in HSP70−/− mice after cecal ligation and puncture (CLP) induced sepsis [42], increased serum HSP70 induced by oxidative stress during sepsis is associated with a higher death rate [43]. McConnell et al. [44] argue that higher HSP70 levels mediate survival and decrease the systemic inflammatory response in aged septic mice as TNF-α, IL-6, IL-10, and IL-1β plasma levels increase in aged HSP70 knockout mice after CLP. Therefore, although these results suggest that extracellular HSPs may play a role as DAMPs in sepsis, other studies supports the anti-inflammatory role of HSP70 in response to sepsis and suggest a therapeutic potential in patients with sepsis.
IL-1
IL-1, previously known as endogenous pyrogen, causes fever by interaction with the hypothalamus [45, 46]. A recent meta-analysis has provided evidence in support of the role of IL-1A-889, IL-1B + 3954, and IL-1RN variable number tandem repeat (VNTR) in sepsis susceptibility [47]. Whether IL-1 and IL-33 are DAMPs or cytokines is a matter of controversy. The IL-1 superfamily contains 11 sub-families (IL-1F1 to IL-1F11 families), and initially cytokines IL-1α and IL-1β were described. The IL-18 and IL-1 receptor antagonist (IL-1ra) were later added to the family and finally IL-33 [48]. Besides structural homology, these sub-families are synthesized primarily as pro-forms (pIL-1), and their maturation process consists of enzymatic cleavage by enzymes such as caspase-1. Precursor forms of IL-1α and IL-33 bind to their respective receptors and trigger signal transduction [49].
Unlike IL-1β, IL-33 and IL-1α are dual function cytokines that possess both nuclear and extracellular functions. Their extracellular form is now recognized to act as DAMP after inflammation induced by necrosis or tissue damage after ischemia. Although IL-1 and IL-33 are produced constantly in normal conditions, their production is increased after cell damage [50], and their excessive production is linked to development of septic shock [51].
The IL-1 family is known as a primary immune system mediator, with both anti- and pro-inflammatory effects [52]. Both IL-1α and IL-1β are produced mainly by macrophages and monocytes after infective stimuli. They are also produced by endothelial and epithelial cells that are the major source for IL-33. After ligand binding to the receptor, these pro-inflammatory cytokines activate NFκB signaling and the JNK and p38 mitogen-activated protein (MAP) kinase pathways to develop an inflammatory response against PAMPs and other DAMPs [53,54].
The IL-1B rs16944 AA genotype in the context of major surgical procedures seems to be related to septic shock and death [55]. Sepsis results in the release of IL-1β from activated microglia [56] and plays a major role in the acute brain inflammatory response (ABIR), inducing a transient synaptic deficit associated with cognitive impairments [57]. During the acute phase of sepsis, persistence of ABIR is associated with long-term hippocampus levels of BDNF and cognitive impairment [58]. During shock, IL-1β down-regulates vascular reactivity via both nitric oxide-dependent and less known nitric oxide-independent mechanisms. The IL-1β down-regulates α1AR expression through activation of the JAK2-STAT3 pathway [59]. It also down-regulates vascular calcium sensitivity through down-regulation of the activities of PKC and Rho kinase [60]. In both cases, IL-1β participates in the regulation of vascular hypo-reactivity during endotoxemia.
IL-33
Extracellular IL-33 inhibits expression of G protein-coupled receptor kinase-2 (GRK2), blocking the TLR4-dependent down-regulation of the chemokine receptor CXCR2 in neutrophils leading to enhanced neutrophil recruitment [61]. The Il-33 has been shown to decrease expression of Fas and increase expression of Bcl-2, and also inhibit apoptosis of CD4(+) and CD8(+) T lymphocytes and CD19(+) B cells in the spleen. Further, IL-33 expression has been associated with reduced levels of IL-6, IL-10, IFN-γ, and TNF-α, and elevated levels of IL-17. Moreover, higher numbers of CD3(+) T cells and elevated levels of active caspase-3, caspase-8, and caspase-9 have been linked to expression of IL-33 [62]. While IL-33 does not alter the local release of pro-inflammatory cytokines, systemic treatment with IL-33 has been associated with reduced cytokine expression in blood and lung, preventing sepsis-induced systemic organ damage [61].
IL-33, as well as Upar and ST2, may have the same clinical application as an acute phase reactant such as C-reactive protein, TNF-α, and IL-6 in the diagnosis of childhood sepsis [63]. Also, patients with sepsis have shown elevated concentration of IL-33 within three hours of ICU admission, correlating with pro-calcitonin levels. Moreover, IL-33 trending is a valuable tool in distinguishing sepsis from SIRS and overall prognosis assessment [64].
PAMPs
The first step in triggering a host immune response is pathogen recognition. PAMPs are the most well-known alerting molecules to the immune system of eukaryotic organisms. Several are among the most studied and include lipopolysaccharides, lipoproteins, peptidoglycans (PGN), lipoteichoic acid, and nucleic acids, which will be discussed below [2] (Table 2).
LPS = lipopolysaccharide; TLR = Toll-like receptor; NOD = nucleotide oligomerization domain; NALP = cryopyrin; RIG = retinoic acid-inducible gene; MDA = melanoma differentiation-associated gene.
LPS
LPS or endotoxin, which is the major component of the outer membrane of gram-negative bacteria, is one of the most important PAMPs. Although it has been recognized as one of the key elements in pathogenesis of sepsis by gram-negative bacteria over a century ago, its role in septic shock is still not fully understood [65]. The mechanism of action of LPS-induced immune stimulation ends in activation of the exracellular-signal-regulated kinase (ERK) signaling pathway and subsequent gene activation. The inflammatory host response to LPS is regulated primarily through TLR4/MD2 complex signaling via either MyD88-dependent or MyD88-independent pathways [66].
The LPS binding protein (LBP) and CD14 catalyze LPS transfer to the TLR4/MD2 complex through a mechanism that is yet to be fully understood [67]. A recent study has shown longitudinal binding of LBP to LPS and multiple CD14s to single LBP/LPS complexes before transfer to the TLR4/MD2 assembly [68]. These molecules are able to differentiate LPS from other structurally similar host molecules and therefore play an important role in autoimmune disorders [65]. The LPS provokes release of multiple pro-inflammatory cytokines such as TNF-α, Type I INF genes, and several interleukins resulting in an inflammatory response that may progress to septic shock. Recently, several different clinical trials are under way to test therapeutic options to control this pathway [69]. In one study, LPS pre-treatment over 24 hours was associated with an augmented increase in pro-inflammatory response in peritonitis-induced sepsis [70].
PGN
The main cell wall component in gram-positive bacteria, PGN is a heteropolymer consisting of a glycan backbone of alternating units of N-acetyl-glucosamine and N-acetyl-muramic acid, with short peptides linked to the lactyl groups of the muramic acid moieties [71]. The TLR2, CD14, nucleotide oligomerization domain (NOD)-containing protein-1 and −2, a family of peptidoglycan recognition proteins (PGRPs) and PGN-lytic enzymes (lysozyme and amidase) recognize PGN and trigger the innate immune system. Signaling via the NOD receptors bypasses MyD88 and induces an inflammatory response primarily through RIP2 (RICK) signaling of NF-κB and MAP kinases. In addition, PGN induces inflammasome assembly and caspase-1 activity [72], which results in pro-inflammatory cytokine production.
Lipoteichoic acid
Lipoteichoic acid (LTA) is another major cell wall component in most gram-positive bacteria and is one of the major immunostimulating PAMPs in gram-positive infections. Diacylglycerol binds LTA to the cell membrane and projects through the PGN layer of most gram-positive bacteria. Lysozyme and leukocytic cationic peptides induce bacteriolysis and LTA release, which then activates NF-κB, MAP kinase, and AP-1 and phosphoinositide 3-kinase through TLR2-CD14 and TLR2 homodimer complexes [73]. Studies suggest that the immunostimulating potential of gram-positive bacteria may explain the different potencies of TLR2 stimulation by various LTAs [74].
Lipoproteins
A component of all bacteria, lipoproteins ligate to TLR2 in multiple ways, resulting in a similar inflammatory response. The importance of this molecule as a member of the PAMP family is highlighted by the fact that lipoprotein-lacking Staphylococcus aureus variants can bypass typical immune recognition and cause life-threatening infections [71]. Lipoprotein can ligate to TLR2 homodimers and TLR1-TLR2, TLR6-TLR2, and CD14-TLR2 heterodimers, which facilitate diversity in TLR recognition of lipoproteins. The resulting inflammatory response is mediated via MyD88 [75].
Nucleic acids
Tissue-residing macrophages and dendritic cells (DCs) detect nucleic acids via PRRs during bacterial phagocytosis, viral replication, or reverse transcription. Activation of PRRs such as TLRs and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) by microbial and host nucleic acids, respectively, can determine whether an infectious state or autoimmunity develops. Unmethylated CpG dinucleotides patterns are unique to bacteria, which distinguish them from host DNA. A potent T-helper-1-like inflammatory response via TLR9 is triggered followed by the activation of NF-κB. The TLR9-/- mice have shown resistance to the lethal effect of CpG DNA without any elevation of serum pro-inflammatory cytokine levels, which further documents the triggering pathway of nucleic acids. Viral DNA is also recognized by TLR9, which induces type I IFNs and the inflammasome. Both TLR7 and TLR8 recognize viral ssRNAs, while TLR3 and RIG-I-like receptors recognize viral dsRNA [76]. A recent study has shown that DNA sensing (at least by the PYHIN proteins) relies on a dsDNA backbone compared with sequence-dependent recognition, and although the innate immune system recognizes pathogenic RNA as “stranger,” DNA is rather recognized as “danger” [77].
Despite similarity in pro-coagulance and platelet-stimulating potential, cell-free DNA (CFDNA) from nuclear, mitochondrial, and bacterial origin has varying pro-inflammatory effects. The role of CFDNA in the development of sepsis and its correlation with death may vary with the source of DNA [78]. Cytoplasmic RNA activates the RLR-MAVS pathway while cytosolic DNA is believed to activate the cGAS-cGAMP STING pathway, potently stimulating the innate immune response in human cells [79].
Flagellin
Flagellin is the main monomeric sub-unit of the flagella that provide motility to both pathogenic and non-pathogenic bacteria. During infection, macrophages and DCs determine the virulence of flagellated bacteria by sensing whether flagellin remains outside the cell or if it gains access into the cytosol [80]. Extracellular flagellin is detected by TLR5, which induces expression of pro-inflammatory cytokines. Delivery of flagellin to the macrophage cytosol, on the other hand, induces the NAIP5/NLRC4 (also known as IPAF) inflammasome, which in turn activates caspase-1–dependent cell death and secretion of IL-1β. The TLR5 responds more generally to flagellated bacteria, while IPAF responds to bacteria that express both flagellin and virulence factors [81].
Cytokines
Cytokines are proteins that are involved in immune cell development, regulation of the overall immune and inflammatory response, and cell to cell communication, usually in a paracrine fashion. Chemokines and growth factors are also cytokines with specific functions such as chemotaxis and augmentation of immune cell differentiation. Most cytokines interact with immune cells via specific receptors, although they shed receptors and may also bind cytokines in the intra-vascular space. There are now many interleukins (IL-1 to IL-37) that have been well studied and regulate the immune response. Excess production of such pro-inflammatory cytokines contributes to the systemic inflammatory response, and mechanisms to inhibit production are important for its resolution. The NLRP3 inflammasome stimulation of IL-1 production can be inhibited at the mitochondrial level through a process termed mitophagy. Such mitochondrial degradation negatively regulates the effects of NLRP3 on caspase-1 to control production of IL-1 [82]. Mast cells and natural killer (NK, and NKT) cells are also a major source of cytokine production in areas such as the gut and respiratory mucosa.
Cytokines can also be produced by mononuclear cells in response to specific antigenic stimulation—e.g., TNF-α is produced by macrophages in response to antigenic stimuli, most notably endotoxin. Its production occurs rapidly, and binding to soluble receptors has probably accounted for the inconsistency in its detection from the blood of patients in gram-negative septic shock. The INFs are a family of proteins that were initially described in the supernatant of virally infected cell cultures, which “interfered” with such viral super-infection. The IFN-γ is produced primarily by mononuclear cells and has been shown to increase class II histocompatibility antigens (HLA-DR, Ia) on the surface of monocyte/macrophages, which may amplify antigen recognition.
T-helper cells have been divided into Th-1 and Th-2 sub-populations, both of which have distinct patterns of cytokine secretion that augment or inhibit various components of the immune system. The Th-1 cells secrete IL-2, IFN-γ, and TNF-α, which augment cell-mediated immunity by activating monocyte/macrophages, T cells, and NK cells. The Th-2 cells secrete IL-4, -10, -13, -35, and -37, which activate B cells and aid in their maturation to plasma cells and antibody production. These cytokines are also anti-inflammatory and inhibit the actions of the Th-1 cytokines. The IL-1 is discussed above, and the following are the most well studied cytokines related to the inflammatory response (Table 3).
IL = interleukin; MIF = macrophage inhibitory factor; MAP = mitogen-activated protein; JAK-STAT = Janus kinase-signal transducer and Activator of Transcription; IFN = interferon; IFNGR = interferon gamma receptor; TGF = transforming growth factor.
TNF-α
Tumor necrosis factor–α is an important pro-inflammatory cytokine with a plethora of effects when injected into animals, including humans, such as fever, hypotension, metabolic acidosis, and capillary leak. The TNF-α plays a regulatory role in promoting local and systemic bacterial clearance, and commercial use of the anti-TNF antibody has been effective in certain chronic diseases. Blockade of TNF was not found to be beneficial in sepsis after similar results with anti-endotoxin antibodies; however, anti-TNF antibodies have found clinical use in various chronic inflammatory states.
IL-4
Interleukin-4 is a potent anti-inflammatory cytokine produced primarily by Th-2 lymphocytes. The IL-4 acts through its specific receptor to down-regulate the host inflammatory response during sepsis, although it also has been implicated in the pathogenesis of asthma and dermatitis in conjunction with immunoglobulin E. The monoclonal antibody dupilomab, which binds to the IL-4 receptor and inhibits IL-4 and -13 signaling, has been approved for the treatment of patients with severe asthma.
IL-6
Interleukin-6 is released from macrophages in response to pathogens. It acts both locally to activate lymphocytes and increase antibody production, as well as systemically to induce fever, the production of acute phase proteins from the liver, and neutrophil mobilization and migration to the focus of infection. The IL-6 correlates with death, and IL-6 levels six hours from injury in a CLP model predicts death [83] and has been shown to be the best biomarker of death in clinical sepsis [84]. The IL-6 is induced by NFκB and augmented by PDCD4 [85].
IL-8
Interleukin -8 is the most potent host chemokine discovered and functions mainly in the role of attracting host neutrophils and macrophages to sights of tissue damage and infection. Neutrophils possess specific high affinity receptors (CXCR1 and 2) that bind to IL-8 and promote such chemotaxis into sights of inflammation. Blockade of IL-8 has been shown to reduce tissue and organ damage in the setting of systemic inflammation that is maladaptive.
IL-10
Interleukin-10, previously named cytokine synthesis inhibitory factor (CSIF), suppresses both macrophage and DC function, including the ability to present antigens and the production of pro-inflammatory cytokines (IL-1β, TNF-α, and IL-12) [86]. The IL-10, as an important part of an anti-inflammatory response, limits the immune and inflammatory responses to pathogens, which might otherwise result in collateral damage to the host organism [87, 88]. The IL-10 is constitutively transcribed in many cells, although post-transcription regulations make the protein levels tightly regulated [89]. Loh et al. [90] show that PDCD4 inhibits translation by blocking the binding of translation initiation factor eIF4Gc to eIF4A in an inactive conformation. This is thought to be part of the mechanism by which IL-10 is suppressed by PDCD4, because PDCD4 inhibits the cap-dependent translation of mRNAs with complex 5' untranslated regions (UTRs), which include IL-10.
IL-12
Cytokines of the IL-12 family include IL-12, -23, -27, and -35 and are important in T helper cell regulation via the receptor-specific JAK-STAT signaling pathway. These have mostly pro-inflammatory functions similar to those of IL-6, but can also be anti-inflammatory [91]. Neuroendocrine modulation of IL-27 production has been shown in LPS-stimulated murine macrophages because catecholamines reduced IL-27 production in a dose dependent fashion [92].
Conclusions
The host responds to both danger and microbial invasion by immune cell pattern receptor mediated recognition of specific molecular patterns associated with host tissue injury (DAMPs) and microbial invasion (PAMPs). These trigger sub-cellular signaling pathways, which result in production of a wide variety of proteins that amplify as well as resolve the host inflammatory response. Host inflammation pathways have evolved to respond to local tissue injury and infection, and when the response is over-exuberant, a systemic inflammatory response develops that is essentially maladaptive and leads to organ dysfunction.
The sepsis syndrome is such a response that is initiated by microbial invasion, and modulation of this response to squelch unnecessary local inflammation away from the focus of infection is a major therapeutic goal. There are numerous targets along the complex and redundant immune response pathways that should lend themselves to novel therapeutic strategies. Selective targeting to organs not involved in the primary infective process, however, has remained elusive because organ-specific targeting in the patient with sepsis has not been developed. Immune and micro-vascular sensing in various organ vascular beds has also not been achieved in humans yet and likely will need to precede organ-specific therapy to limit the maladaptive inflammatory response associated with organ dysfunction in patients with sepsis.
Footnotes
Author Disclosure Statement
No competing financial interests exist.