
Abstract
Abstract
Background:
In general, patients with sepsis die from the host response to the infecting pathogen rather than from the infecting pathogen itself. Four patterns of death have been identified in sepsis, namely vasoplegic shock, single-organ respiratory failure (acute respiratory distress syndrome [ARDS]), multi-system organ failure (MSOF), and persistent MSOF with ongoing inflammation and immunosuppression with recurrent infections (persistent inflammation-immunosuppression and catabolism syndrome [PICS]). To improve the outcome of sepsis adjunctive therapies that modulate the immune system have been tested; these therapies that have targeted specific molecules or pathways have universally failed.
Conclusion:
We propose that the combination of hydrocortisone, intravenous ascorbic acid, and thiamine (HAT therapy), which synergistically targets multiple pathways, restores the dysregulated immune system and organ injury, and reduces the risk of death and organ failure following sepsis.
T

The four patterns of sepsis related deaths. SIRS = systemic inflammatory response syndrome; CARS = compensatory anti-inflammatory response syndrome; MSOF = multi-system organ failure; PICS = persistent inflammation-immunosuppression and catabolism syndrome.
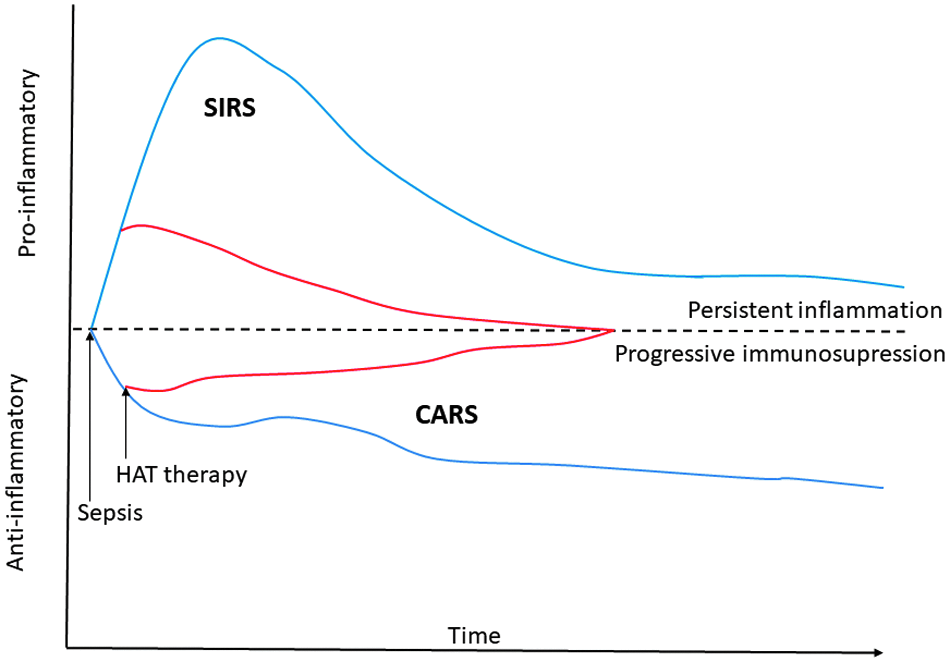
Treatment with hydrocortisone, ascorbic acid, and thiamine (HAT therapy) attenuates both the pro- and anti-inflammatory response in patients with sepsis.
The first pattern is that of refractory vasoplegic septic shock caused by a cytokine storm, leading to death within 96 hours [10,11]. The second pattern is that of death caused by single-organ respiratory failure (acute respiratory distress syndrome [ARDS]) resulting from the combined effects of an exuberant proinflammatory response combined with volume overload; death usually occurs between three and seven days of onset [9,12,13]. The third pattern of death is from multi-system organ failure (MSOF) caused by cellular bioenergetic failure with cell hibernation, with death typically occurring between four days and two weeks [9,14]. The final pattern is one of prolonged and persistent MSOF with ongoing inflammation and immunosuppression with recurrent and unresolved infection (persistent inflammation-immunosuppression and catabolism syndrome [PICS]) [15,16]. It is likely that the characteristics of the pathogen (type of pathogen and microbial load) together with host factors (site of infection, age, immune status, comorbidities, and genetic makeup) as well as treatment factors (delay in initiating treatment, inappropriate antibiotic agents, fluid overload, adjunctive treatment modalities) interact to determine the outcome and pattern of death.
A limited number of studies have evaluated the causes of death in patients with sepsis. Vincent et al. [7] analyzed the cause of death in 4,459 patients with severe sepsis. In this study, 27% of patients died during the 28-day study period. Twenty-two percent of patients died of refractory septic shock, 13% from respiratory failure, and 43% died from MSOF. Frencken et al. [17] studied the patterns of death among 708 patients with severe sepsis and septic shock admitted to two Dutch intensive care units (ICUs). The one-year mortality was 53.8% with 12% of patients dying within four days of admission, 20% between days four and 28, and 22% between day 29 and one year. Weiss et al. [18] evaluated the patterns of death in pediatric sepsis. The median time to death was eight days. The most common cause of death was refractory shock (34%), followed by MSOF (27%) and single-organ respiratory failure (9%). Early deaths (≤3 d) were mostly caused by refractory septic shock whereas MSOF predominated after three days.
Synergistic Anti-Inflammatory Properties of HAT Therapy
Sepsis is fundamentally an inflammatory disease mediated by the activation of the innate immune system by both pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Calvano et al. [19] demonstrated that exposure of blood leukocytes to endotoxin altered the expression of 3,714 genes. These include genes for pro- and anti-inflammatory cytokines, chemokines, adhesion molecules, transcription factors, enzymes, clotting factors, stress proteins, and anti-apoptotic molecules [20]. The excess production of proinflammatory mediators plays an important role in four pathogenetic mechanism leading to death in patients with sepsis. We believe that HAT therapy has potent anti-inflammatory properties that underlies the benefit of this treatment in patients with sepsis.
Vitamin C suppresses activation of nuclear factor kappa-B (NF-κB) by inhibiting tumor necrosis factor-α (TNF-α)–induced phosphorylation of inhibitory kappa-B kinase (IκB kinase) [21]. The primary anti-inflammatory action of glucocorticoids is to repress the transcriptional activity of NF-κB and activator protein-1 (AP-1). Nuclear factor kappa-B and AP-1 increase the transcription of multiple proinflammatory mediators that contribute to the generation of reactive oxygen species (ROS), cellular injury, coagulation activation, and the endothelial and microvascular dysfunction characteristic of sepsis. Ascorbic acid decreases high mobility group box 1 (HMGB1) secretion [22]; HMGB1 is an important late proinflammatory cytokine. Histamine has been shown to play an important role in sepsis [23]. Vitamin C has been demonstrated to decrease the synthesis and to inactivate histamine [24].
Sepsis is characterized by the excessive production of ROS by the induction of enzymes such as nicotinamide adenine dinucleotide phosphate-oxidase (NOX) and the uncoupling of mitochondrial oxidative phosphorylation and iNOS [25]. In addition, ROS are produced by xanthine oxidase, lipoxygenase, and cyclo-oxygenase. When the antioxidant defenses are overwhelmed, ROS can induce injury to lipids, proteins, and nucleic acids thereby resulting in widespread endothelial dysfunction, mitochondrial dysfunction, cellular injury, and multiple organ dysfunction. Unopposed ROS oxidizes tetrahydrobiopterin (BH4), the cofactor of endothelial nitric oxide synthase (eNOS), and thereby reduces eNOS activity, the enzyme producing endothelial nitric oxide [25]. Endothelial nitric oxide (eNO) maintains microcirculatory flow and inhibits platelet aggregation and adhesion of activated platelets and leukocytes. In the absence of BH4, eNOS becomes uncoupled, producing superoxide (O2-) rather than nitric oxide (NO). Superoxide (O2-) and NO yield peroxynitrite, a more damaging ROS. Vitamin C is a key cellular antioxidant, detoxifying exogenous oxidant radical species that have entered cells or that have arisen within cells because of excess O2-generation by mitochondrial metabolism, by NOX, xanthine oxidase, or by uncoupled nitric oxide synthase (NOS) [25,26]. In addition, vitamin C recycles other antioxidants including α-tocopherol (vitamin E) and BH4. Tetrahydrobiopterin plays a critical role in the function of eNOS. Vitamin C deficiency results in the incomplete regeneration of BH4 resulting in the uncoupling of eNOS and the generation of superoxide and peroxynitrite.[25] Thiamine may act together with vitamin C to decrease oxidative injury [27,28].
We propose that HAT therapy may have synergistic anti-inflammatory and anti-oxidative properties thereby modulating the proinflammatory response of sepsis. This postulate is supported by experimental studies. Barabutis et al. [29]. have demonstrated that hydrocortisone together with vitamin C protects the vascular endothelium from damage by endotoxin while neither agent alone had this effect. Azari et al. [30] compared the protective effects of vitamin C, vitamin E, and hydrocortisone alone and in combination, in a renal and intestinal ischemia-reperfusion model [31]. In these studies both vitamin C and hydrocortisone reduced the ischemia-reperfusion injury with the combination having synergistic protective effects. Many interventions in critically ill patients are time sensitive; the use of antibiotic agents and source control in patients with sepsis are examples in which a delay in the initiation of therapy may adversely affect outcomes [32,33]. Similarly, it is likely the benefits of HAT therapy may diminish with delays in administration. It is important to emphasize that early initiation of HAT therapy is critical to a good outcome as the pathologic processes leading to organ failure become less reversible with time. Furthermore, it is likely that fluid overload with an aggressive approach to fluid resuscitation attenuates the benefit of HAT treatment [34–36].
Refractory Vasoplegic Shock and the Cytokine Storm
The early phase of sepsis is generally believed to result from the uncontrolled production of proinflammatory mediators, the so-called cytokine storm, which may lead to refractory vasoplegic shock [37,38]. The early proinflammatory response to bacteremic shock can be reproduced by administration of proinflammatory cytokines [39–41]. Using advanced informatics techniques from sepsis transcriptomic datasets, Sweeney et al. [8] identified three functional transcriptomic subtypes that they termed “inflammopathic, adaptive, and coagulopathic.” The inflammopathic subtype was characterized by overactivation of the innate immune system with increased expression of proinflammatory signaling pathways, pattern recognition receptor activity, and complement activation. Clinical features of this subtype included a high disease severity with shock, high bandemia, and high mortality. Although the early systemic inflammatory response has been considered the hallmark of sepsis, immunosuppression occurs both early and late in the host sepsis response.
Refractory vasodilatory shock develops from uncontrolled vasodilation and vascular hyporesponsiveness to endogenous vasoconstrictors, causing failure of physiologic vasoregulatory mechanisms [11]. Vasoplegic shock results from failure of the vascular smooth muscle to constrict with profound arterial and venodilatation [10]. Vasoplegic shock is believed to be caused by the massive production of proinflammatory cytokines with increased expression of inducible nitric oxide synthetase with increased production of nitric oxide (NO), activation of KATP channels resulting in hyperpolarization of the muscle cell membrane, increased production of natriuretic peptides (which act synergistically with NO), and a relative vasopressin deficiency [10]. These factors lead to profound vasodilation with hypotension and vasopressor resistance. This hemodynamic pattern is complicated by decreased contractility with decreased myocardial performance and diastolic dysfunction [42].
Prevention of vasoplegic shock with HAT therapy
Vitamin C and hydrocortisone act via multiple mechanism to reverse vasoplegic shock and restore organ perfusion. Both molecules suppress activation of NF-κB; NF-κB increase the transcription of multiple proinflammatory mediators responsible for the cytokine storm [21]. Vitamin C is an essential cofactor for the synthesis of norepinephrine, epinephrine, and vasopressin; in addition, vitamin C increases adrenergic transmission [43]. In a murine cecal ligation and puncture (CLP) model, prophylactic bolus injection of vitamin C attenuated the loss of arteriolar and blood pressure responsiveness to the vasoconstrictors norepinephrine and angiotensin [44–46]. Glucocorticoids are potent anti-inflammatory agents that suppress inflammation by multiple mechanisms that involve both the innate and adaptive immune responses. In addition to attenuating the cytokine storm, low-dose glucocorticoids have immune-stimulating effects that may limit the anti-inflammatory immunosuppressive state. In addition, glucocorticoids have additional beneficial effects including increasing adrenergic responsiveness [47] and preserving the endothelial glycocalyx [48]. Based on the overlapping beneficial effects of vitamin C and glucocorticoids in experimental models of sepsis [49,50], we have postulated that these two agents may act synergistically to reverse vasoplegic shock [51,52]. In a retrospective before and after study we have demonstrated that HAT therapy rapidly reserves vasoplegic shock with a reduction in the dose of vasopressors within two hours of the initiation of treatment with vasopressor independence within 24 hours [52].
Acute Respiratory Distress Syndrome
According to the Berlin Definition, ARDS is defined as an “acute diffuse, inflammatory lung injury, leading to increased pulmonary vascular permeability, increased lung weight, and loss of aerated lung tissue with hypoxemia and bilateral radiographic opacities” [53]. Sepsis is the most common cause of ARDS [54] and results from intra-alveolar macrophage activation with elaboration of a diverse array of bioactive mediators including proteases, ROS, cytokines, and chemokines [13]. Because ARDS is caused by the excess production of proinflammatory mediators, HAT therapy rapidly reverses the pathogenic mechanism of ARDS [52,55].
Multi-System Organ Failure
Multi-system organ failure is the most common cause of death in patients with sepsis. These patients usually develop progressive and irreversible organ failure requiring the increasing use of advanced life support measures. Because of the high burden of ongoing treatment with the extremely grave prognosis families will often elected to limit care allowing for a more peaceful death.
What is the mechanism of MSOF in sepsis? The prevailing theory suggests that MSOF results from impaired oxygen delivery at the cellular level because of altered microcirculatory blood flow [56,57]. Although global oxygen delivery is typically increased in sepsis, proponents of this theory suggest that many tissue capillary beds do not receive adequate oxygen supplies because of microvascular endothelial dysfunction. However, there are few data that tissue hypoxia occurs in patients with sepsis. Multiple experimental models have failed to demonstrate cellular hypoxia in sepsis [58–61]. Increasing oxygen delivery in patients with sepsis does not increase oxygen consumption [62–64]. A number of randomized controlled trials have failed to demonstrate an improvement in outcome in patients with sepsis who received therapy that aimed to increase oxygen delivery compared with those who received conventional therapy [65–67]. These studies suggest that microcirculatory impairment with shunting and maldistribution of blood flow are unlikely to be central to the pathophysiology of sepsis-induced organ failure.
Metabolic failure as a cause of sepsis-related MOSF
In patients with sepsis who die of MSOF histopathologic examination of the heart, liver, and kidneys have failed to demonstrate necrotic or apoptotic cell death [68,69]. Takasu et al. [69] performed rapid postmortem cardiac and renal harvest in 44 patients with sepsis and compared the immunohistochemistry features with those of control organs. Immunohistochemistry demonstrated low levels of apoptotic cardiomyocytes in septic and control subjects, however, electron microscopy showed hydropic mitochondria in the septic specimens. It is noteworthy that per milligram of tissue, the heart and kidney consume the most oxygen and possess the greatest abundance of mitochondria [70]. The authors of these studies have concluded that the degree of cell injury and death does not account for severity of sepsis-induced organ dysfunction. Similarly, animal models have failed to reveal widespread cell death in sepsis despite progressive organ failure [71]. To explain these findings Hotchkiss et al. [72] have suggested that these features are consistent with cell stunning or hibernation and that these changes may represent a short-term adaptive response.
Multiple defects in mitochondrial function have been described in animal models and patients with sepsis. These include abnormal autophagy, decreased biogenesis (mitochondrial numbers), decreased activity of the components of the electron transport chain (particularly complex I), and uncoupling of oxidation phosphorylation [73–80]. Alterations in fatty acid metabolism with decreased β-oxidation and abnormalities of the citric acid cycle appear to be a characteristic feature of the mitochondrial dysfunction in sepsis [81,82]. These changes lead to decreased adenosine triphosphate (ATP) production. Furthermore, damaged mitochondria undergo mitophagy, i.e., autophagy directed at the removal of damaged mitochondria [83,84]. Matkovich et al. [85] measured messenger RNA (mRNA) alterations in hearts from patients who died from sepsis and compared these with mRNA expression in non-failing and failing human hearts. In this study, mRNA expression levels for 198 mitochondrially localized energy production components, including Krebs cycle and electron transport genes, were reduced on average by 43%. Except for mitochondrial isocitrate dehydrogenase, all of the other 19 genes involved in the tricarboxylic acid (TCA) cycle were decreased. In a sepsis model, Arulkumaran et al. [61] demonstrated that sepsis-induced renal dysfunction was associated with reduced nicotinamide adenine dinucleotide (NADH) and mitochondrial membrane potential (MMP) with increased renal ROS production. In this study, 4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl, a free radical scavenger was protective against renal tubule injury. It is likely that the unbalanced production of mitochondrial reactive oxygen species (mtROS) impairs mitochondrial structure, enzymatic function and biogenesis and play a role in the mitochondrial dysfunction of sepsis. These data suggest that the bioenergetic failure in sepsis leads to organ failure particularly in those organs with a high metabolic demand and high mitochondrial concentration [79–84,86]. The bioenergetic failure does not lead to widespread cell necrosis or apoptosis but rather a state of cellular hibernation [72]. Furthermore, the mitochondrial oxidative injury releases mitochondrial DNA (mtDNA), which amplifies the inflammatory injury leading to further immune paresis and PICS syndrome [87].
Prevention of MSOF with HAT treatment
Critically ill patients with sepsis typically have low or undetectable serum levels of vitamin C resulting in an acute scorbutic condition [52,88–90]. Recently Carr et al. [91] demonstrated that 100% of patients with sepsis had low vitamin C levels. Low plasma concentrations are associated with the severity of organ failure and mortality [89]. Furthermore, low vitamin C levels have been demonstrated to be predictive of the development of MSOF [90].
The overlapping anti-inflammatory properties of glucocorticoids and vitamin C reduce the production of proinflammatory mediators and ROS, which are linked to the development of MSOF. In experimental models, parenteral vitamin C attenuates organ injury and improves survival in mice with sepsis [92–94]. Dehydroascorbic acid is transported via the GLUT1 transporter into mitochondria, where it converted to ascorbic acid and acts as a potent antioxidant limiting mitochondrial oxidant injury [95,96]. In an experimental model, Dhar-Mascareno et al. [97] demonstrated that oxidant-induced mitochondrial damage and apoptosis in human endothelial cells were inhibited by vitamin C. Studies using N-acetylcysteine have proven to be ineffective and potentially harmful in patients with sepsis possibly because of the limited ability of this drug to enter into the mitochondria and its inability to regenerate BH4 [25,98,99]. Ascorbic acid is required for the synthesis of carnitine, which is required for the transport of fatty acids into the mitochondrial matrix and for β-oxidation [81,95].
Thiamine is the precursor of thiamine pyrophosphate, the essential coenzyme of several decarboxylases required for glucose metabolism, the Krebs cycle, the generation of ATP, the pentose phosphate pathway, and the production of NADPH. Thiamine pyrophosphate (TPP) is a critical coenzyme for the pyruvate dehydrogenase complex, the rate-limiting step in the citric acid cycle. Thiamine deficiency is common among patients with sepsis, with a range in prevalence between 20% and 70%, depending on measurement techniques and inclusion criteria [100–105]. A deficiency in thiamine leads to decreased activity of thiamine-dependent enzymes that triggers a sequence of metabolic events leading to energy compromise and decreased ATP production. It is likely that thiamine deficiency compounds the mitochondrial injury and bioenergetic failure caused by vitamin C depletion. In a pilot randomized controlled trial, Donnino et al. [105] randomly assigned 88 patients with septic shock to receive 200 mg thiamine twice daily for seven days. In the pre-defined subgroup of patients with thiamine deficiency, those in the thiamine treatment group had statistically significantly lower lactate levels at 24 hours and a lower mortality at 30 days. Furthermore, in a secondary analysis of this study the need for renal replacement therapy and the serum creatinine were greater in the placebo group [106]. The observation that thiamine was reno-protective is an important observation considering the high metabolic demand of the kidney and the postulated role of thiamine in improving energy metabolism. This finding is all the more important because correction of mean arterial pressure and macrovascular blood flow is not restorative in septic acute kidney injury (AKI), and supports of the concept that mitochondrial dysfunction and injury may be a critical feature of septic AKI [84].
Several studies suggest that vitamin C alone and when combined with glucocorticoids and thiamine reduce the risk of MSOF. Nathens et al. [107] randomly assigned 595 surgical intensive care (ICU) patients to receive intravenous vitamin C and vitamin E for up to 28 days. The vitamin combination was associated with a significant reduction in the incidence of MSOF (p = 0.04). The Sepsis-Related Organ Failure Assessment (SOFA) score has been used to quantify the severity of organ dysfunction in patients with sepsis [14]. In a pilot study of 24 patients with severe sepsis and septic shock, Fowler et al. [88] demonstrated a significant decrease in the SOFA score in patients randomly assigned to receive intravenous vitamin C. Similarly in our observational study in which we compared treatment with HAT therapy versus standard treatment, we demonstrated a rapid decrease in the SOFA score in patients who received HAT therapy (Fig. 3) [52].

Time course of the Sepsis-Related Organ Failure Assessment (SOFA) score over the four-day treatment period in the treatment group and in the control group survivors and non-survivors. *p < 0.001 for comparison of treatment group vs control group. (Reproduced with permission from Marik PE, Khangoora V, Rivera R, et al. Hydrocortisone, vitamin C and thiamine for the treatment of severe sepsis and septic shock: A retrospective before-after study. Chest 2017;151:1229–1238).
Persistent Inflammation-Immunosuppression and Catabolism Syndrome
Patients with MSOF who survive the first two weeks of sepsis often have a protracted clinical trajectory and exhibit both chronic immune suppression and inflammation. This syndrome has been termed the persistent inflammation/immunosuppression and catabolism syndrome (PICS) [15,108]. This persistent inflammation is characterized by increased concentration of plasma interleukin (IL)-6, a persistent acute phase response, neutrophilia, with increased immature granulocyte count, anemia, lymphopenia, and, often, tachycardia [108]. Although these patients are profoundly immunosuppressed, inflammation is ongoing. While the exact pathophysiology of PICS syndrome is unclear, PICS is probably driven by DAMPs and alarmins that are produced by injured organs and tissues, such as mtDNA, nucleosides, histones, HMGB1, protein S100A, ATP, adenosine and/or hyaluronan products. The paradoxical immunosuppression and infectious complications in patients with sepsis compound as sepsis progresses, with a shift to infection by opportunistic organisms. Torgersen et al. [16] studied both clinical and postmortem records of patients (n = 235) who died from sepsis or septic shock in their institution over a period of 10 years. Multiple organ dysfunction syndrome (MODS) was considered the main cause of death in 52% of patients. An unresolved infectious focus was present at autopsy in 77% of patients. Moreover, 90% of the patients with prolonged intensive care exceeding seven days had persistent infectious foci.
Prevention of PICS with HAT therapy
The use of HAT therapy reduces the proinflammatory response and the development of MSOF and as such limits progression to PICS syndrome. Moreover, HAT therapy specifically decreases the immunosuppression associated with sepsis. It has been known for more than 60 years that vitamin C has immune-enhancing properties. In 1949. Fred Klenner from Reidsville, North Carolina, reported on the use of intravenous vitamin C in the treatment of polio and other viral illnesses [109]. It was initially assumed that vitamin C was directly viricidal (in vivo) and this mistaken belief underlies the recommendations of Linus Pauling who promoted the use of large doses of oral vitamin C (up to 18 g/d) for the prevention and treatment of the common cold [110]. However, a number of randomized controlled trials have reported that vitamin C supplementation had no effect on the incidence of the common cold [111]. Whereas high-dose vitamin C has in vitro viricidal properties [112,113], there are no data or physiologic rationale to suggest that this occurs in vivo. Rather, the anti-viral effects of vitamin C are likely because vitamin C has specific immune-enhancing effects. Vitamin C is concentrated in leucocytes, lymphocytes, and macrophages, reaching high concentrations in these cells [114]. Vitamin C improves chemotaxis, enhances neutrophil phagocytic capacity and oxidative killing, stimulates interferon production, and supports lymphocyte proliferation [115–117]. In addition, vitamin C has been demonstrated to attenuate the formation of neutrophil-extracellular-traps (NETs) [94,118]. Vitamin C stimulates dendritic cells to secrete IL-12 and thereby drive naïve CD4+ T cells to differentiate into TH1 cells [118]. Vitamin C improves the generation of NK-cell progenitors from T/NK-cell progenitors [119]. Vitamin C promotes T-cell maturation, possibly by epigenetic mechanisms [120]. In a CLP sepsis model, Gao et al. [121] demonstrated that parenteral vitamin C improved the outcome of sepsis and sepsis-induced MODS in wild-type as well as Gulo−/− mice. In this study negative immunoregulation by CD4+CD25+ regulatory T cells (Tregs) and the secretion of inhibitory cytokines (TGF-β and IL-10) were inhibited and T-cell–mediated immunosuppression was improved in those mice treated with vitamin C.
Many physicians are reluctant to prescribe a short course of low-dose glucocorticoid on the basis that these drugs increase the risk of infections. The prolonged (>10 d) use of moderate- to high-dose glucocorticoids (>400 mg hydrocortisone per day) is well known to increase the risk of opportunistic infections. In a landmark study, Keh et al. [122] performed a randomized, cross-over study that evaluated the clinical and immunologic effects of low-dose glucocorticoids in patients with septic shock. These investigators demonstrated that hydrocortisone simultaneously decreased circulating levels of both pro- and anti-inflammatory cytokines. Hydrocortisone downregulated the proinflammatory immune response but did not inhibit the TH1-related response and concentrations of IL-12 and interferon-γ were not suppressed. Paradoxically, by enhancing macrophage function and the innate immune system and blunting the anti-inflammatory response short-term glucocorticoids may reduce the risk of infection and the development of PICS syndrome (Fig. 2). Roquilly et al. [123] randomly assigned 149 multi-trauma patients to receive a continuous infusion of hydrocortisone (200 mg/d for 5 d) or placebo (the HYPOLYTE Study). The primary end point of this study was hospital-acquired pneumonia within 28 days. In the intention to treat analysis, 35% of the patients treated with hydrocortisone and 51% of those treated with placebo developed pneumonia (p = 0.007). In the three recent large randomized controlled trials, low-dose short-duration treatment with glucocorticoids was not associated with an increased risk of secondary infections [124–126]. These data suggest that the combination of vitamin C and low-dose glucocorticoids may simultaneously and synergistically blunt both the excessive proinflammatory and immunosuppression response characteristic of sepsis and thereby prevent PICS syndrome.
Conclusions
The major pathogenetic mechanisms whereby patients with sepsis die include refractory vasodilatory shock, ARDS, MSOF, and PICS syndrome. Because of their overlapping and synergistic immunomodulating effects, HAT therapy may restore the dysregulated immune system and thereby prevent or limit vasoplegic shock, ARDS, and the bioenergetic failure that characterizes sepsis and reduce the risk of death.
Footnotes
Author Disclosure Statement
The author has no real or potential conflicts of interest concerning this manuscript.