
Abstract
Background:
The quality of scientific literature is judged by study design, validity, and applicability to unique patient populations.
Methods:
We searched the available literature to explore the hierarchy of evidence, explain research fundamentals such as sample size calculation, and discuss common study designs employed in surgical research and the interpretation of trial designs.
Results:
Each unique study design has restraints created by some degree of systematic errors and bias. This article provides definitions for the scientific boundaries of case control, retrospective, before-and-after, prospective observational, randomized controlled designs, and meta-analyses.
Conclusion:
Critical thinking and appraisal of the literature is a skill that requires lifelong training and practice. Clinical research education and design need to garner more attention in the medical community.
Never before has the importance of interpreting and incorporating evidence-based medicine (EBM) been so crucial. Although most clinicians lack formal training in the interpretation and implementation of scientific evidence into practice, they want to offer the best evidence-based practice to patients. This type of medicine was defined in 1996 as “a systematic approach to clinical problem solving by the integration of best research evidence with clinical expertise and patient values” [1,2]. Substantial barriers exist in fully incorporating EBM into clinical practice. The sheer volume of published literature presents a major obstacle. Other obstacles are poor understanding of statistics, difficulty in literature access, and attitude/cultural barriers [3].
The quality of scientific literature is judged by study design, validity, and applicability to unique patient populations. Level I evidence typically represents high-quality studies in the form of randomized controlled trials (RCT) and meta-analyses of RCTs. Every subsequent decrease in level represents a higher likelihood of a threat to validity. In this article, we explore the hierarchy of evidence, explain the fundamentals of sample size calculation, and discuss the interpretation of trial designs.
Hierarchy of Evidence
The concept of levels of evidence began in 1979 with the Canadian Task Force [4]. David Sackett, a Canadian clinical epidemiologist, is considered the founding father of a systematic appraisal of the medical literature and the development of a grading system [5]. Since that time, the framework in which studies are ranked often has been represented as a pyramid (Fig. 1). The pyramid hierarchy is heavily dependent on the type of clinical research in question and the research design. The hierarchy does not always take into consideration whether studies are poorly executed or underpowered. For example, an observational study could be undervalued when in fact it could be more valid than a poorly executed or underpowered RCT [6]. In this article, we focus on common study designs utilized to guide clinical practice.
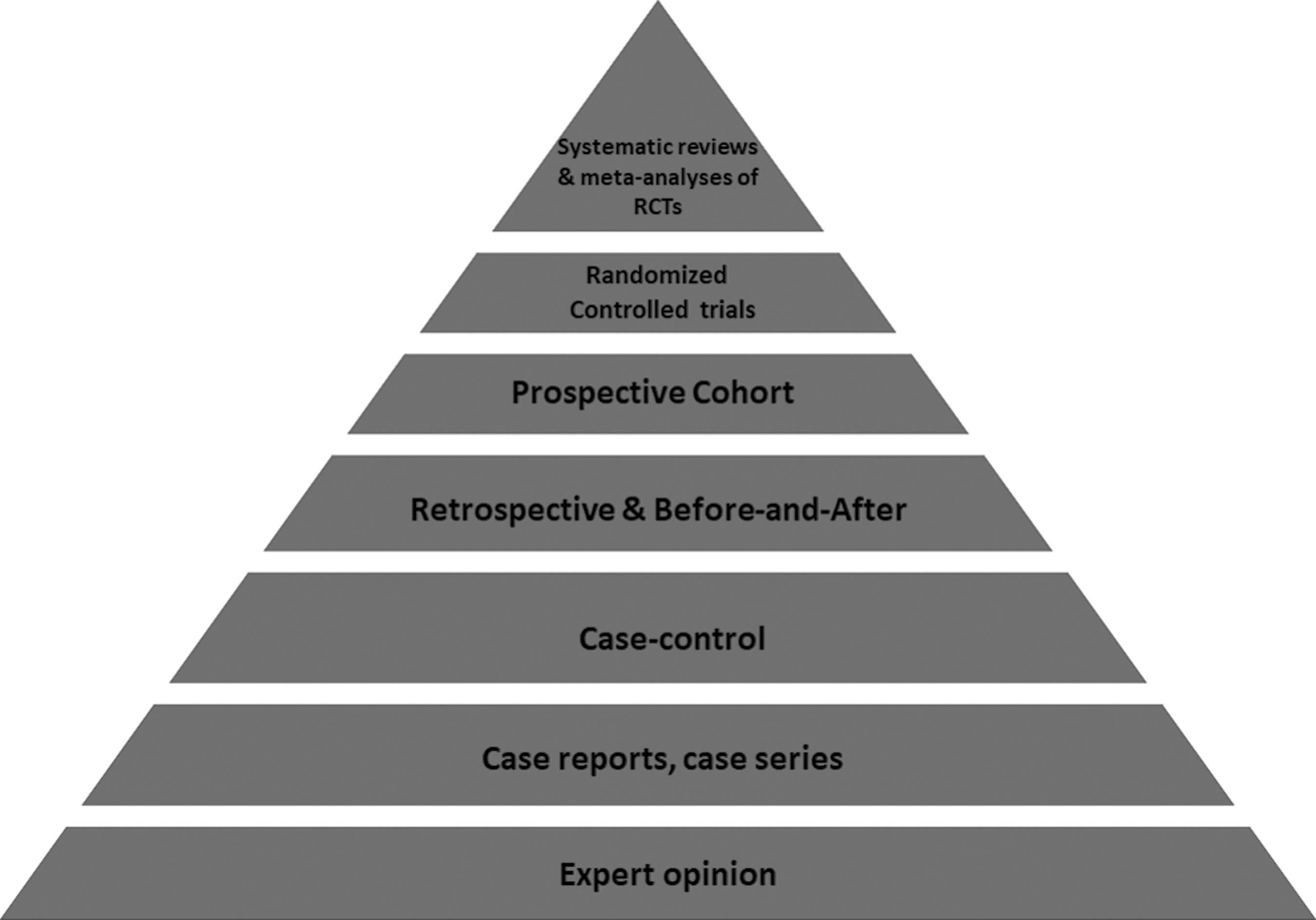
Hierarchy of evidence depicted by pyramid diagram. Pinnacle of the pyramid corresponds to highest level of evidence and thus more validity.
Case Reports and Case Series
Case reports and case series, along with expert opinion, form the base of the hierarchy of evidence. Although they remain an important form of communication for rare cases and accumulated wisdom, they represent the lowest quality of clinical “evidence” associated with the highest likelihood of bias and threat to validity. These study designs lack a comparator (“control”) and involve the description of one patient (case report) or a small aggregate of individuals (case series) [7]. Case reports and series can be helpful in unique circumstances when no higher level of evidence is available. There have been notable incidences where case reports have significantly altered the practice of medicine, but these are rare circumstances [7].
Case-Control Studies
Case-control studies can involve either the collection of retrospective or prospective data, and this study design often is chosen for rare diseases or events [8]. Prospective designs (nested case-control) are preferred but are infrequent. The study population or cases are “nested” in the prospectively collected cohort. Nested case-control studies have less bias but because of the rarity of the condition sometimes are not possible to perform because of the excessive time intervals or the resources required to complete them [9].
Retrospective Studies
Retrospective design is more commonly employed for case-control studies, as it is a more efficient method to evaluate rare diseases or events [8]. Retrospective studies, sometimes labeled “chart reviews,” often are beleaguered with biases, as they rely on datasets that were not designed originally to answer a specific research question. Nonetheless, there are several advantages to this study design, particularly for the investigator. These studies are relatively quick to perform, as the data are available already and are affordable and often exempt from institutional review, as patient contact is obviated. As a study design, they are subject to a significant risk of confounding, as this type of analysis is plagued with missing or incorrect data. Selection and sampling bias are important contributors to limitations in these study designs. To mitigate this, controls and comparators should be sought within the same population as the study cohort [10]. Retrospective studies can report only associations but cannot support conclusions of causality. However, the information they provide may be useful in planning future studies, for example, by offering estimates of event rates and study cohort characteristics [8].
Before-and-After Studies
Before-and-after studies evaluate outcomes after an intervention has been introduced into a study population. Most of these types of studies rely on historical controls, which introduce several potential biases to the results. Patient characteristics, medical diagnostics, and other technologies change over time, which will confound the results [11]. Additionally, data collection is not blinded, and physician/provider-dependent factors (e.g., familiarity with new surgical techniques such as laparoscopy) are not closely monitored [11]. The Hawthorne effect is a major concern in these studies and occurs when investigators (or study participants) are aware of an intervention and its desired goal, thereby biasing the study to yield a difference that might not actually exist [12].
Before-and-after studies can be “controlled” and “uncontrolled” [13]. A stepped wedge cluster randomized trial represents an example of a controlled before-and-after study. This study design often is characterized as “phased implementation” whereby an intervention is introduced to distinct clusters of patients over time [14]. Every cluster eventually transitions from control to intervention, but the exact time of transition is randomized. This design often is used in the setting of resource constraints, whereby introducing the intervention to a larger cohort is too expensive or logistically not possible. Although imperfect, before-and-after study designs frequently are relevant in policy decisions [15].
Prospective Observational Review
Prospective observational studies follow study subjects longitudinally over time. The key difference between prospective and retrospective studies is that in the former, the primary research question and data fields are defined before the first subject is enrolled, and the data are collected expressly for the purpose of conducting research. This has the added advantage of minimizing the risk of missing or unreliable data. Typically, these studies investigate the effects of exposures on outcomes [16]. Prospective observational studies often are termed prospective cohort studies, the most famous of which is the Framingham Heart Study [17]. In fact, the validity of large prospective cohort studies is considered just below randomized controlled trials in the hierarchy of EBM. These study designs evaluate risk factors for diseases or health outcomes. The aim of this design is to measure the strength of the relation between an exposure and the outcome of interest. Classically, these results are described as a risk ratio (i.e., relative risk) or hazard ratio. The ratio represents the rate of an outcome or event observed in the cohort exposed to the risk compared with that in patients who were not exposed. Major challenges to this study design are the large sample size required and the long period of longitudinal follow-up needed to observe events. Also, loss to follow-up remains an important obstacle.
Randomized Controlled Trial
An RCT is considered the “gold standard” clinical study design because it theoretically eliminates bias and confounding through randomization of participants into groups exposed or not exposed to an experimental intervention. If performed correctly on a large enough sample, randomized assignment to trial arms ensures equal distribution of both known and unknown confounders, thus enhancing the internal validity of the trial. In other words, any difference in outcome between the groups can be ascribed confidently to the effect of the intervention.
Another critical component of the RCT to reduce bias is concealment of allocation whereby the subject is assigned to a study arm without the influence of the investigator. This differs from blinding, which means that some or all involved parties are unaware if they are receiving an intervention during the trial. Frequently, RCTs actually are “open-label” trials, especially trials of surgical interventions. In an open-label study, the investigator is aware of the study arm assignment, although he or she should not have the ability to choose to which arm the patient is allocated. Knowing the assigned treatment arm increases the risk of response bias; i.e., the investigator or subject's responses to subjective outcomes (such as pain) are biased because of conscious or unconscious pre-existing beliefs. Investigators can mitigate response bias by choosing objective outcomes (such as death) and using blinded outcome assessors separate from the research team. These strategies are especially pertinent to surgical interventions. Much of the criticism of the RCTs performed during the 2020 COVID-19 pandemic have been attributable to their open-label designs and the addition of co-interventions that likely resulted in confounding [18,19].
All subjects enrolled in an RCT should be included in the analysis [8]. The term “intention-to-treat analysis” maintains that all subjects randomly assigned to particular groups are represented in the final analysis, regardless of whether they actually received the intervention [20]. This practice accounts for study drop-outs. On the other hand, per-protocol analysis evaluates only subjects who actually received the intervention under investigation (and sometimes specifies a level of protocol compliance) in order to be considered in the analysis [20]. This type of analysis excludes inadvertent dropouts. This is especially problematic when the reason for dropout is side effects, disease progression, or even death while waiting for the intervention. Loss to follow-up introduces a serious risk of selection bias and creates a scenario that restricts the analysis only to those who tolerated the intervention, benefited from it, or both.
Despite being well-planned and well-conducted, an RCT can lack generalizability. A single-center RCT can lack external validity for a variety of reasons such as a homogenous patient population or rigid protocols that prove impossible to implement at other institutions. Multicenter RCTs address this shortcoming by recruiting subjects from different hospitals frequently located in distinct geographic sites.
Meta-Analysis
A meta-analysis of the available literature, specifically the merging of data from multiple RCTs, is considered the pinnacle of EBM. Often, a meta-analysis will accompany a systematic review, but it is not necessarily required. Meta-analysis is a statistical method to pool data from multiple studies (not necessarily RCTs) that has the advantage of increasing the sample size to detect small differences and to determine the magnitude of the effect of an intervention [21]. The results are presented as forest plots (Fig. 2). The weight of each trial usually is represented by the size of a figure within the plot. Trial size contributes to the weight observed in the forest plot analysis.

Forest plot depiction of results of meta-analysis. Reprinted with permission [33].
A major limitation of meaningful meta-analysis is heterogeneity of studies. Individual studies may investigate similar interventions, but these can differ substantially with regard to the setting, severity of disease, and study methodology [25,26]. In these circumstances, investigators may attempt to assess heterogeneity with a Q test or I2 index [22]. The Q test can detect the presence of heterogeneity, whereas the I2 index represents the percentage of variability. Also, I2 can determine to what extent this variability is attributable to true heterogeneity. For example, I2. = 0 signifies that all variability between studies is attributable to sampling error, and there is homogeneity within a set of studies. In turn, an I2 = 75% means that a high degree of heterogeneity exists between studies, and differences cannot be ascribed solely to sampling error (i.e., within-study variability) [23]. In the event there are no similar trials that compare interventions directly, or when more than two interventions need to be compared, a traditional meta-analysis cannot be performed. In these situations, indirect or network meta-analyses may be used [24].
Sample Size Calculation
When designing a superiority study, the size, or number of participants, must be considered carefully in order to avoid observing differences that do not really exist (Type 1 error) or not observing differences that in fact do exist (Type II error). In a 1978 New England Journal of Medicine study, 71 “negative” RCTs were analyzed to evaluate whether the sample sizes were large enough to detect a difference in outcome between the groups [25]. The authors found that 67 of 71 studies were at risk for Type II error, meaning that even if a significant difference existed, nearly all the studies were underpowered to detect that difference.
In sample size calculations, there are some central concepts that deserve further description. These include: The null hypothesis (H0), the probability of reaching a level of significance (α), and the probability of not observing statistical significance (β) In a typical superiority trial, the null hypothesis postulates that a difference between treatments does not exist. Although an investigator may be studying whether drug A is better than drug B, the null hypothesis supposes that no difference will be observed. Thus, if the investigators reject the null hypothesis, there actually exists a meaningful difference. Type I errors are designated by α and can be considered a false-positive error. In medical research, the alpha often is defined as equivalent to 0.05 or 5%. In order for a difference to be considered “statistically significant,” it has to be observed less frequently than α. In the real world, randomness is reality. Differences occur by chance, so α sets a limit.
The β was the central problem plaguing the RCTs described in the 1978 NEJM article. In order to determine a sample size, one has to know the β A power analysis needs to be performed to initiate a study, and statistically, this is defined as 1 – β Power is the probability of obtaining a statistically significant result when a real difference exists [21]. In general, a β < 0.20 is the accepted standard for medical research.
Another important concept when calculating power is the baseline rate of either the disease under investigation or the known therapy established in the literature [21]. Researchers have some liberty in determining the effect size of the intervention; this has been described as the minimum clinically important difference (MCID) [21]. The essential principle in MCID determination remains the clinical significance of the expected difference. Obviously, a tiny percentage difference, such as a decrease of 0.2%, likely is not clinically justifiable to study and will require an enormous sample size to detect. Estimating a large MCID can reduce the required sample size to detect but will risk a Type II error [25].
Superiority, Non-Inferiority, and Equivalence Trials
Randomized controlled trials may be further classified as superiority, non-inferiority, and equivalence trials. The nuanced differences between the assumptions, execution, and interpretation of these trials merit further discussion.
Superiority trial: The new antibiotic compared with placebo
Suppose a new antibiotic named Cefmendoza/Yehbactam is under investigation to treat a new pandemic xenoinfection caused by cross-species transmission of porcine herpesvirus. At present, there is no effective treatment; and the investigators want to design a trial that will show a better outcome for these patients when using this new antibiotic compared with supportive care alone. In this trial, the α is 0.05 and the β is 0.2; and the investigators hypothesize that Cefmendoza/Yehbactam will decrease the mortality rate from 50% to 30%. A power calculation is performed, and the investigators aim to disprove the null hypothesis and show a difference in outcome.
When performing this trial, the investigators are hoping to establish a new standard of care by demonstrating better (superior) outcomes (Fig. 3). Superiority trials demand both an intention-to-treat (ITT) analysis and a per-protocol (PP) analysis, require reporting of protocol violations, and should account for all patients even if they were lost to follow-up [26]. All of these factors are important when considering the internal validity of a superiority trial. For example, suppose that our new antibiotic is associated with a high incidence of adverse effects causing a substantial proportion of patients to discontinue taking the drug early and cross over to the standard care group. Selective reporting of the PP analysis describing the success only of those subjects able to complete the therapy course would present a distorted picture of how effective the drug is in treating patients, leading to a Type 1 error (i.e., falsely rejecting the null hypothesis when there really is no difference). On the other hand, excessive protocol violations and significant amounts of missing data will cause the groups to appear more similar; this overlapping of groups will detract from any signal of benefit and increase the risk of a Type 2 error (i.e., falsely accepting the null hypothesis when a true benefit exists). It must be emphasized that a “negative” superiority trial does not mean that the two treatments are equivalent or non-inferior.
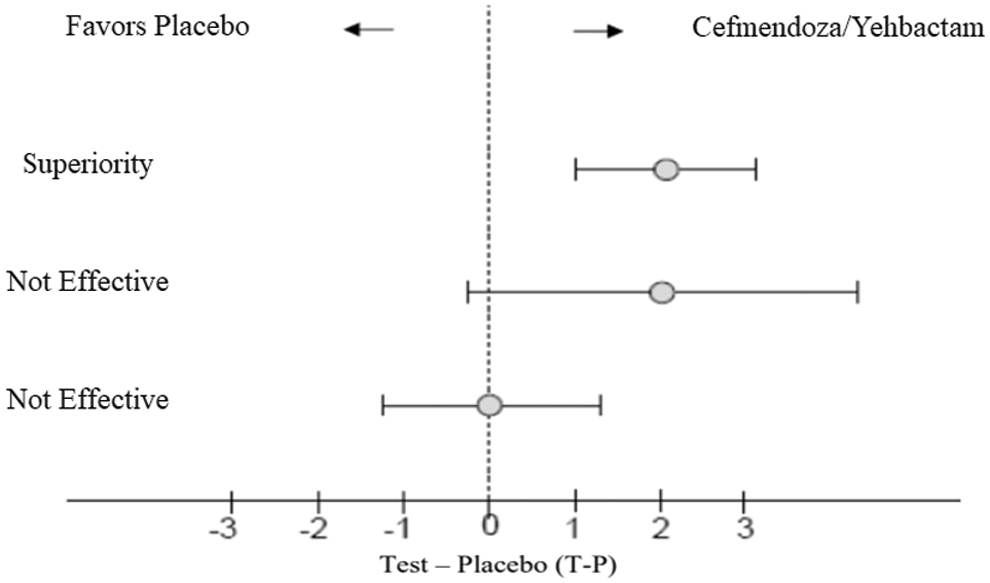
T-P indicates the test drug when compared with placebo. Null hypothesis is T-P < 0, demonstrating no effect of test drug. When T-P > 0, null is rejected, and the test drug demonstrates an effect [29].
Equivalence trial: An even newer antibiotic compared with standard treatment
In an equivalence trial, the null hypothesis is that a difference does exist between treatments and the investigator aims to dispel this null hypothesis, thus proving that they are equivalent [26]. Equivalence trials are necessary when an effective treatment is already used in practice, so it would be unethical to assign a subject to placebo. Suppose Cefmendoza/Yehbactam is now the standard of care for our new xenoinfection. However, a competitor pharmaceutical company has developed another antibiotic, Crippsomycin, and desires to enter the market. With this design, they want to show that treatment with Crippsomycin is not meaningfully better or worse (-Δ to +Δ) than Cefmendoza/Yehbactam, the active control substance.
Non-inferiority trial: “Good enough” and maybe even better?
A non-inferiority (NI) trial is a one-sided equivalency trial whereby the investigators are interested only in proving that the new treatment is not materially worse than the standard treatment [26]. Similar to equivalence trials, these trials compare a new treatment with a known effective active control (Fig. 4). The motivation for an NI trial usually is to demonstrate that the intervention efficacy is not only “good enough” [27], but is more attractive for other reasons such as lower treatment cost, greater convenience, or a better side-effect profile. A crucial design element is the non-inferiority margin, as this determines how to define “good enough.”
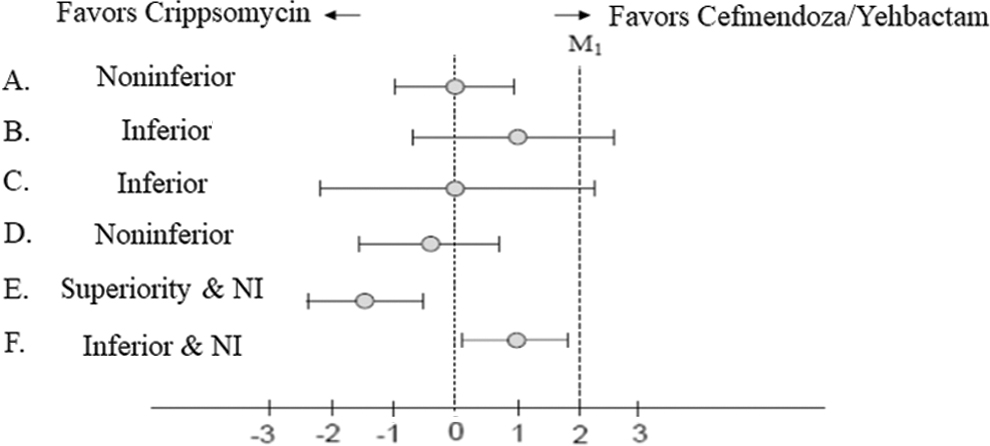
In a noninferiority trial, test drug should have effect equal to active control. There is no placebo in this study design. Effect of active control is assumed. Purpose of this design is to show that test drug is not inferior to active control within a certain margin, which is referred as the NI margin indicated by M1 [29]. (
Suppose, for example, that Cefmendoza/Yehbactam is effective in lowering the mortality rate to 30%, but costs $200,000 USD per treatment course and causes a 40% incidence of hirsutism and psychosis. Crippsomycin may not be as effective in reducing the mortality rate; however, it costs only US$10,000 with no side effects. Therefore, the investigators decide to define the non-inferiority margin as 8%, meaning that if the upper limit of the 95% confidence interval (CI) is no higher than the 8% higher mortality rate compared with Cefmendoza/Yehbactam, then the trial will declare that Crippsomycin is “good enough” or non-inferior in treatment efficacy but is desirable for other reasons. The non-inferiority margin often is subjective and based on expert opinions about the degree of MCID [28].
Note that it is possible that a new treatment is both inferior AND non-inferior, depending on how the margin is defined and the 95% confidence intervals of the findings (Fig. 5). Also, it is possible for a non-inferiority trial to prove the superiority of the new treatment compared with the active control. One important caveat is that because there is no placebo, the internal validity of an NI trial relies on the active control having its expected efficacy (“assay sensitivity”). For example, if in this NI trial, the mortality rate in the Cefmendoza/Yehbactam arm is 50% (as opposed to 30% in our pivotal superiority trial described above) and the upper 95% CI of the mortality rate for Crippsomycin is 56%, we cannot declare non-inferiority because the active control lacked assay sensitivity. Similarity of new treatment to the active control can mean that either both treatments were effective or that both were ineffective [29].
Another important caveat about NI trials is that the selective reporting of the ITT analysis is more biased toward Type 1 error (falsely accepting non-inferiority), which is opposite to a superiority trial whereby selective PP analysis reporting increases the risk of falsely accepting superiority. Similarly, excessive protocol violations and loss to follow-up in an NI trial will cause the groups to appear more similar and thus lead investigators and readers to conclude falsely that the new treatment is non-inferior when in fact the new treatment is indeed inferior. Unfortunately, the quality of reporting of NI trials has been inconsistent and sometimes misleading [30]. In all cases, when interpreting both superiority and NI trials, the reader should evaluate whether the investigators have reported their trial design and results adequately according to best practices [31-32].
Conclusions
Critical thinking is a lifelong process, and critical appraisal of the literature undoubtedly is a skill that requires training and practice. Clinical research education and design continues to garner more attention in the medical community. There are several excellent educational articles that focus on developing this skill [8,21,26]. The goal of this article is to familiarize the reader with important concepts to inspire a better understanding of the available literature. We hope readers can appreciate the vast quantity of available knowledge but understand that limitations exist in nearly every type of study design and investigation.
Author Disclosure Statement
No competing financial interests exist.