
Abstract
Background:
There is increasing recognition of extensive crosstalk between programmed cell death pathways (PCDPs), such as apoptosis, pyroptosis, and necroptosis, resulting in a highly redundant system responsive to a breadth of potential pathogens. However, because pyroptosis and necroptosis propagate inflammation, these redundancies also present challenges for therapeutic control of dysregulated hyperinflammation seen in cytokine storm (CS) generated organ dysfunction.
Hypothesis:
We hypothesize that the conversion of existing knowledge regarding apoptosis, pyroptosis, and necroptosis into a computational model can enhance our understanding of the crosstalk between PCDPs via simulation experiments of microbe interactions and experimental interventions.
Materials and Methods:
Literature regarding apoptosis, pyroptosis, and necroptosis was reviewed and transposed into an agent-based model, the programmed cell death agent-based model (PCDABM). Computational experiments were performed to simulate the activation of various PCDPs as seen by differing microbes, specifically: influenza A virus (IAV), enteropathic Escherichia coli (EPEC), and Salmonella enterica (SE). The potential protective value of PCDP crosstalk was evaluated by silencing either pyroptosis, necroptosis, or both. Computational experiments were also performed simulating the effect of potential therapies blocking tumor necrosis factor (TNF) and interleukin (IL)-1.
Results:
The PCDABM was implemented in the agent-based modeling toolkit NetLogo. Computational experiments of infection with IAV, EPEC, and SE reproduced cross-activation of PCDPs with effective microbial clearance. Simulations of anti-TNF and anti-IL-1 did not reduce the aggregated amount of inflammation-generated system damage, the surrogate for CS-generated tissue damage.
Conclusions:
Redundancies have evolved in host PCDPs to maintain protection against a wide range of pathogens. However, these redundancies also challenge attempts at dampening the pathogenic hyperinflammatory state of CS using therapeutic immunomodulation. Integrative simulation models such as the PCDABM can aid in identifying potentially targetable inflection points to mitigate CS while maintaining effective host defense.
Cellular death as a host defense against infection is well described. 1 Programmed cell death pathways (PCDPs), such as apoptosis, pyroptosis, and necroptosis, are critical components of this defense. 2 Although initially thought to operate distinctly, there is increasing recognition that pathways exhibit extensive crosstalk.3,4 This contributes to a highly redundant system whereby the host can respond to a breadth of potential infectious agents in an ongoing arms race between host and invader. In the host, PCDPs act to defend against infection by reducing available cells for replication/infection, and by inducing a robust inflammatory response against invading organisms. Pathogens, on the other hand, develop strategies to evade these systems to maintain their ability to infect human cells.5–7 The clinical cost of this arms race is perhaps most well demonstrated by pathologic inflammation. As pyroptosis and necroptosis propagate inflammation, the redundancies that are beneficial from purely an antimicrobial standpoint, e.g., the recruitment and amplification of antimicrobial inflammatory processes, present challenges in the modern intensive care setting in terms of dysregulated hyperinflammation seen in cytokine storm (CS) and subsequent organ dysfunction.
The burden of disease that this entity imposes on patients is substantial and severe, with clinical consequences represented by septic shock and associated multisystem organ failure, acute respiratory distress syndrome (ARDS), and death.8,9 It is recognized that CS is driven by the immune response to infection in general, with PCDPs specifically implicated. 8 Attempts at treating CS with interruption of individual pathways or branch points in inflammatory cascades have not shown the clinical benefit that would be expected based solely on the mechanistic understanding of its drivers.8,10
We posit that the general lack of success in CS treatment is due in great part to the redundancy present between PCDPs. The relatively recent recognitions of the degree of crosstalk between PCDPs is an example of how system-level interactions can be difficult to parse with traditional experimental biology, suggesting a need for adjunctive methods to represent and examine the dynamics of the aggregated behaviors of such systems. Biology heavily relies on schematics and diagrams to represent the components and interactions present in a particular system in an integrative fashion, but as the amount of knowledge regarding a system increases, the complexity of these representations can make it difficult (if not impossible) to intuit their behavior. We, and others, have previously proposed that agent-based models can be used for dynamic knowledge representation to evaluate the behaviors represented in these commonly used diagrams. 11
In this work, we present the conversion of static diagrams that embody existing knowledge regarding apoptosis, pyroptosis, and necroptosis into a computational model, the programmed cell death agent-based model (PCDABM), to enhance our understanding of the dynamics of the crosstalk between PCDPs. Notably, various pathogens have evolved different means of engaging and interdicting PCDPs. We simulate infections by three well-described and studied pathogens, namely influenza A virus (IAV), enteropathic Escherichia coli (EPEC), and Salmonella enterica (SE). To demonstrate the extent of interconnectedness between PCDPs, we simulate experiments that knock out key components of the pyroptosis and necroptosis pathways. To demonstrate the difficulty of therapeutically intervening on CS generated by PCDP activation, we simulate anticytokine interventions that block tumor necrosis factor-α (TNF-α) and interleukin (IL)-1β.
Materials and Methods
Agent-based modeling
The PCDABM was implemented using NetLogo, 12 a programming language/platform for agent-based modeling (ABM) Supplementary S1. Agent-based modeling represents interactions between computational objects (agents) that are programmed with specific rules and tasks. Running the ABM produces behaviors of the various agents as they interact with each other and their environment in a virtual world. The benefits of using ABM for modeling complex biologic systems for translational purposes are well described. It is very intuitive to conceive of biologic systems as a series of interacting cells, which behave based on their cellular programming (rules). The output of biologic research focused on identifying cellular responses to various molecular signals lends itself to conversion into agent-level rules. Agent rules often incorporate some degree of stochasticity/randomness, as is noted in many biological systems. Last, there is an inherently spatial aspect to ABMs that maps well to recognizable tissue patterning seen in histologic data.13,14
Agent-based modeling can be considered computational instantiations of hypotheses (e.g., how one believes cells function in their environment) and simulation experiments using ABMs can provide insight into non-intuitive and potentially paradoxical behaviors; this process can be thought of as conceptual model visualization and evaluation. Once constructed, because of stochasticity incorporated into their behavioral rules, ABMs can be subjected to simulation experiments that vary their initial starting conditions and/or interventions applied during the course of a single experimental run.
Biologic representations in the PCDABM
All modeling requires some degree of abstraction. The focus of this study is to understand the interactions of various PCDPs with different pathogens as it relates to dysregulated inflammation. Thus, the model does not represent the entire pathophysiologic profile(s) of the infectious agents or its subsequent consequences to the host. For example, there is minimal utility in modeling the infectious diarrhea seen with EPEC and SE in this study. Instead, we focus on the initial infection and subsequent inflammatory events related to programmed cell death (PCD). For each component represented in the PCDABM, we reviewed the scientific literature to ascertain the established mechanisms and behaviors associated with that particular component.
We recognize that there are often ambiguous and potentially conflicting findings in the literature, and therefore we rely on knowledge that is presented in review articles as well-established consensus information. Although there are several recognized PCDPs, for this initial version of the PCDABM we focus specifically on three of the most commonly studied pathways: apoptosis, pyroptosis, and necroptosis. The mechanistic components obtained from the literature are summarized in a state diagram (Fig. 1), which was used to guide programming the model. The specific biologic representations and associated abstractions are described below and summarized in Supplementary Table S2.
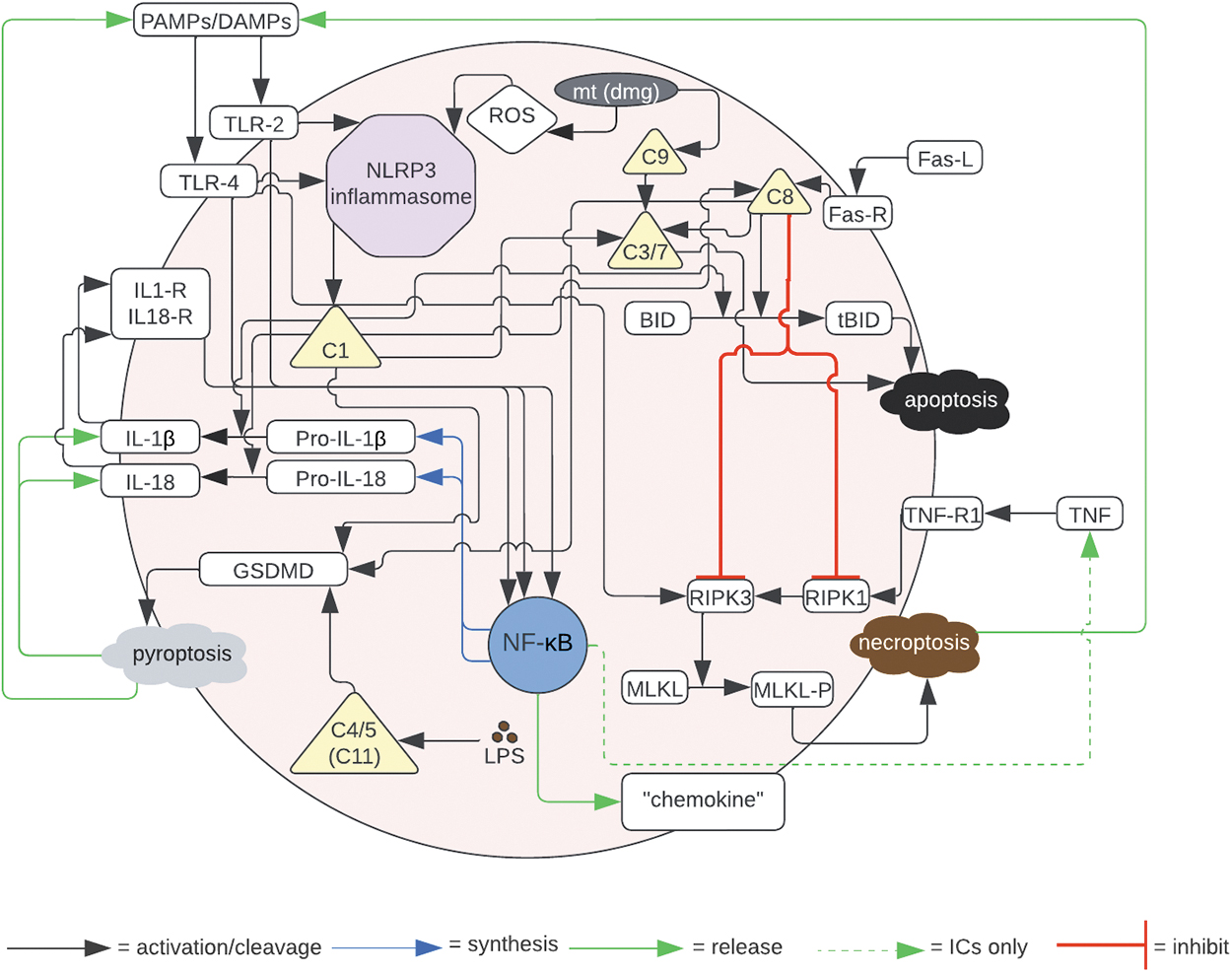
State diagram of incorporated PCDs based on current literature.
The PCDPs were implemented in two generic cell types in the PCDABM:
Generic tissue cells (TC). These agents are intended to represent tissue cells that are subjected to an initial infectious insult. They incorporate the various forms of PCD. The state of health of the individual TCs represent the health of the entire host system, which represents a primary outcome metric in terms of the simulation experiments (total system health). Generic immune cells (ICs). Immune cells represent an abstracted version of various subtypes of immune cells and, in this initial model, combine their key functions into a single entity to simplify the model structure. Immune cells can respond to stimulation, perform chemotaxis, and assist in eradication of infected TCs by simulating phagocytosis. Immune cells also incorporate the PCD rules and can produce select inflammatory cytokines.
We then implemented representations of three specific infectious agents: IAV, EPEC, and SE. These specific pathogens were selected, in part, because of their well-characterized interactions with PCDPs.4,15
IAV: given that viruses are known to interact with PCDPs in distinct ways from bacterial pathogens, we chose to include IAV as a model infection for viruses. In addition to requiring intact host cells for replication, IAV induces autocrine and paracrine signaling through the Fas pathway, which leads to apoptosis. 15
EPEC: chosen as a model of an extracellular bacterium. The pathophysiology of EPEC induced diarrhea is primarily through signaling, but also through T3SS proteins that inhibit NFκB and the NLRP3 inflammasome.
SE: chosen as a model of an obligate intracellular bacterium. Known to inhibit PCDPs using its type 3 secretion system (T3SS) proteins that inhibit NFκB and caspase 8 function(s). 15
Model representation
The PCDABM was implemented using NetLogo 6.2.2. It is a virtual world that consists of a 41 × 41 grid with 1,681 patches. Although the PCDABM uses a two-dimensional representation of an inherently three-dimensional system, for the purposes of this initial implementation, it is a sufficient abstraction. This is true due to the demonstrated ability of these values to capture the overall dynamics of the interactions present in the literature (see Fig. 1) as well as the lack of availability or specific need for three-dimensional histologic data given the current explicit goal of conceptual model visualization.
At initialization, TCs are superimposed on the patches (one TC per patch), for a total of 1,681 TCs. There are 100 ICs distributed randomly along the grid, and they move in a random pattern when there are no chemokines present. Each cell is programmed to follow preset rules about its interactions with the surrounding environment. Time is arbitrarily represented in the system with each cycle or “tick,” through the model (i.e., all rules and procedures are run once) set to represent 10 minutes of time. The model contains variables that represent total system health (represented by the sum of individual TC health) and total system damage (total individual TC health minus individual TC damage). The units for these variables are arbitrarily set, with each TC starting at a health of 100 and specific injuries or insults decrementing some of that health. As health value is decremented from any particular TC, this same amount is subtracted from the global total system health variable and added to the global total system damage variable. The setup of the PCDABM grid along with the user interface is represented in (Fig. 2).
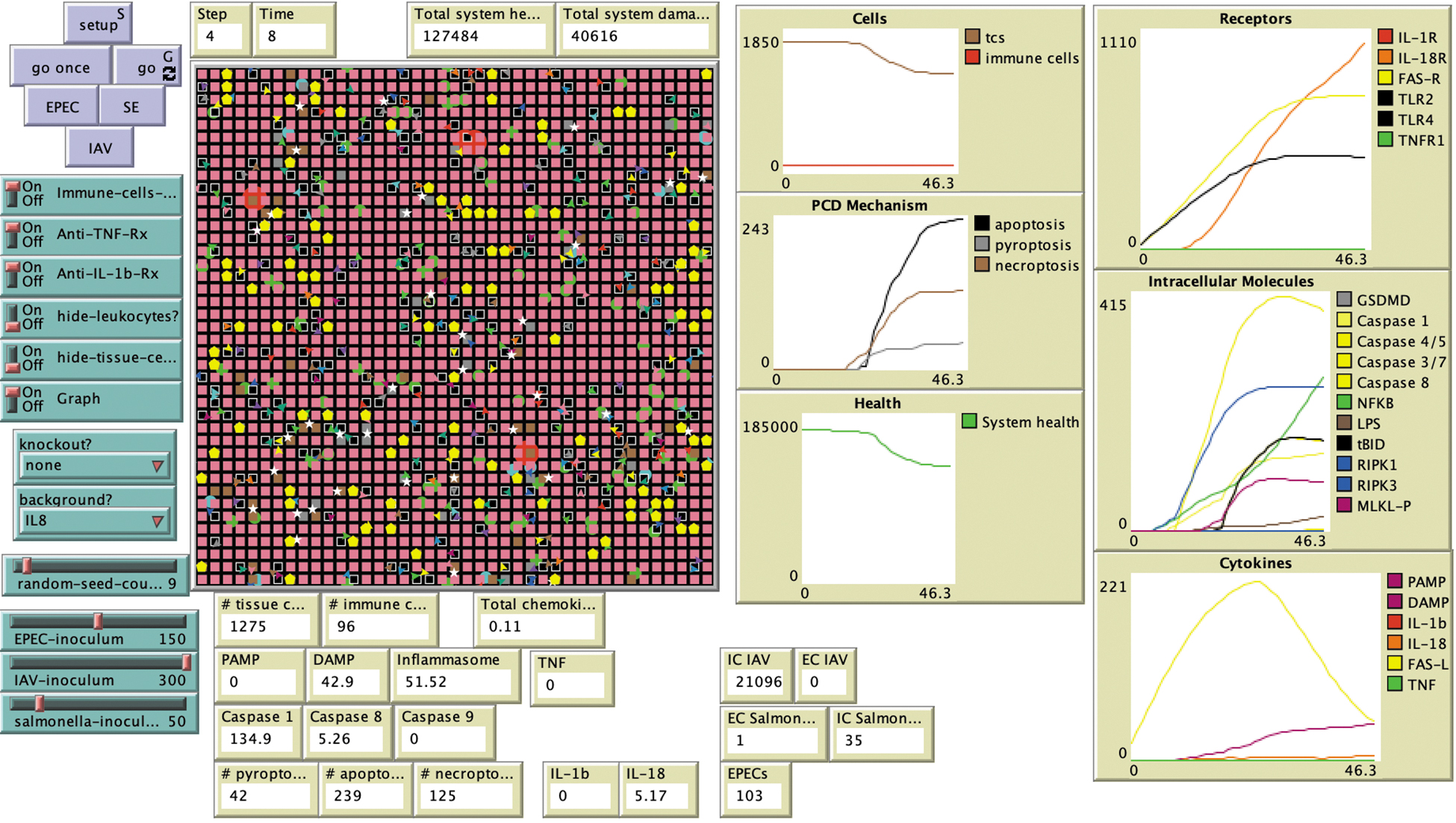
Programmed cell death agent-based model (PCDABM) two-dimensional grid and interface. IAV = influenza A virus; EPEC = enteropathic Escherichia coli; SE = Salmonella enterica; DAMP = damage-associated molecular pattern; GSDMD = gasdermin D; IL = interleukin; LPS = lipopolysaccharide; MLKL = mixed lineage kinase domain-like pseudokinase; NFκB = nuclear factor kappa-light-chain-enhancer of activated B cells; PAMP = pathogen-associated molecular pattern; TNF = tumor necrosis factor.
Simulation experiments
Simulation experiments involve setting initial experimental conditions and then letting the simulation model execute. We recognize that in the real world, biologic processes are already in motion, and that the “start-from-stopped” nature of the simulation is, to this degree, artificial. However, recognizing that all perturbations to biologic systems, be they experimental interventions or insults that result in disease, do also have an initiating point (their time 0), for purposes of evaluating the effect of these perturbations. Once the PCDABM was implemented, three distinct types of simulation experiments were designed: calibration, knockouts, and anti-inflammatory therapies.
Knockout and therapy simulations were implemented by setting the respective target variable(s) in the PCDABM to 0.
Calibration experiments simulating infection with IAV, EPEC, and SE, respectively, to ensure that the model demonstrated plausible behavior with respect to infection and the system's ability to clear it.
Knockouts of NLRP3 and RIPK3, respectively, were implemented to assess the behavior of the system under those conditions in each infection. These were used to evaluate degree of crosstalk between the PCDPs.
Anti-inflammatory therapy with either anti-TNF or anti-IL-1β was simulated with each infection.
This schema leads to five distinct experiments for each of the three simulated infections: baseline, NLRP3 knockout, RIPK3 knockout, anti-TNF therapy, and anti-IL-1β therapy. Experiments for each of the infections were run with 10 repetitions using random seeds 0 to 10, and every experiment was allowed to run for a total of 1,000 time steps. The results of interest measured in the PCDABM are total system health/damage, pathogen inoculum amount, and number of dead cells, broken down by PCD type or fate.
Figures were plotted using the pandas and matplotlib Python packages. Statistical significance was calculated with Student t-test utilizing the scipy Python package. The model/code for the PCDABM is publicly available.
Results
Calibration experiments
Subjecting the PCDABM to IAV, EPEC, and SE infections demonstrated plausible behavior with respect to infection clearance, total system health, and PCDPs utilized (Fig. 3). In IAV infection, in keeping with known antiviral mechanisms, apoptosis is the primary pathway used to control infection. This led to the system having, in general, less collateral damage from dysregulated inflammation (apoptosis is notably not an inflammation-propagating PCDP); this is reflected in the results where total system health remained higher than in other infections, despite the same inoculum. For EPEC and SE infection, the system effectively cleared the insult, with system health decreasing as infectious inoculum increased. With respect to PCDPs in EPEC and SE infection, all three pathways were utilized for infection clearance, with necroptosis being the most utilized and apoptosis being the least utilized. Given that some authors have reported that SE downregulates NFκB and Caspase 8 expression 15 and EPEC has been described to inhibit or dampen NFκB and NLRP3 inflammasome expression, 15 one might expect that EPEC and SE infection would therefore not have pronounced activation of the pathways when affected by the aforementioned proteins, but this was not found to be the case in our experiments.

This figure shows the response of the PCDABM when subjected to three infectious agents in terms of total system health and count of dead cells by PCDP type. Panel
Knockout experiments
Figure 4 shows the effect of simulated NLRP3 and RIPK3 knockouts in all three infections with respect to total system damage and PCDP fate. Infection clearance and system health at varying inoculum values were not significantly changed from baseline. Total system health was not altered by the knockout of either NLRP3 or RIPK3. Although there appears to be some differences from baseline because of the knockout of RIPK3 for both EPEC and SE, these did not turn out to be statistically significant: for EPEC p = 0.30 and for SE p = 0.26 (note the degree of overlap in the stochastic replicates present). Similarly, no significant differences were found from baseline due to knockout of NLRP3 in either EPEC or SE infection: for EPEC p = 0.30, for SE p = 0.59. As for PCDP fate: in the NLRP3 knockouts, there was little effect on their distribution in IAV; this is as expected given the predominance of apoptosis with this pathogen. However, PCDP distributions were altered in EPEC and SE infections, with necroptosis being the only PCDP activated. Conversely, in the RIPK3 knockouts, EPEC and SE simulations resulted in pyroptosis being the only PCDP present, while IAV remained by and large unaffected. The results show that although pyroptosis and necroptosis could be suppressed by interruption at key points in their pathways, crosstalk resulted in alternative PCDPs compensating for these interruptions, with little effect on overall system damage.
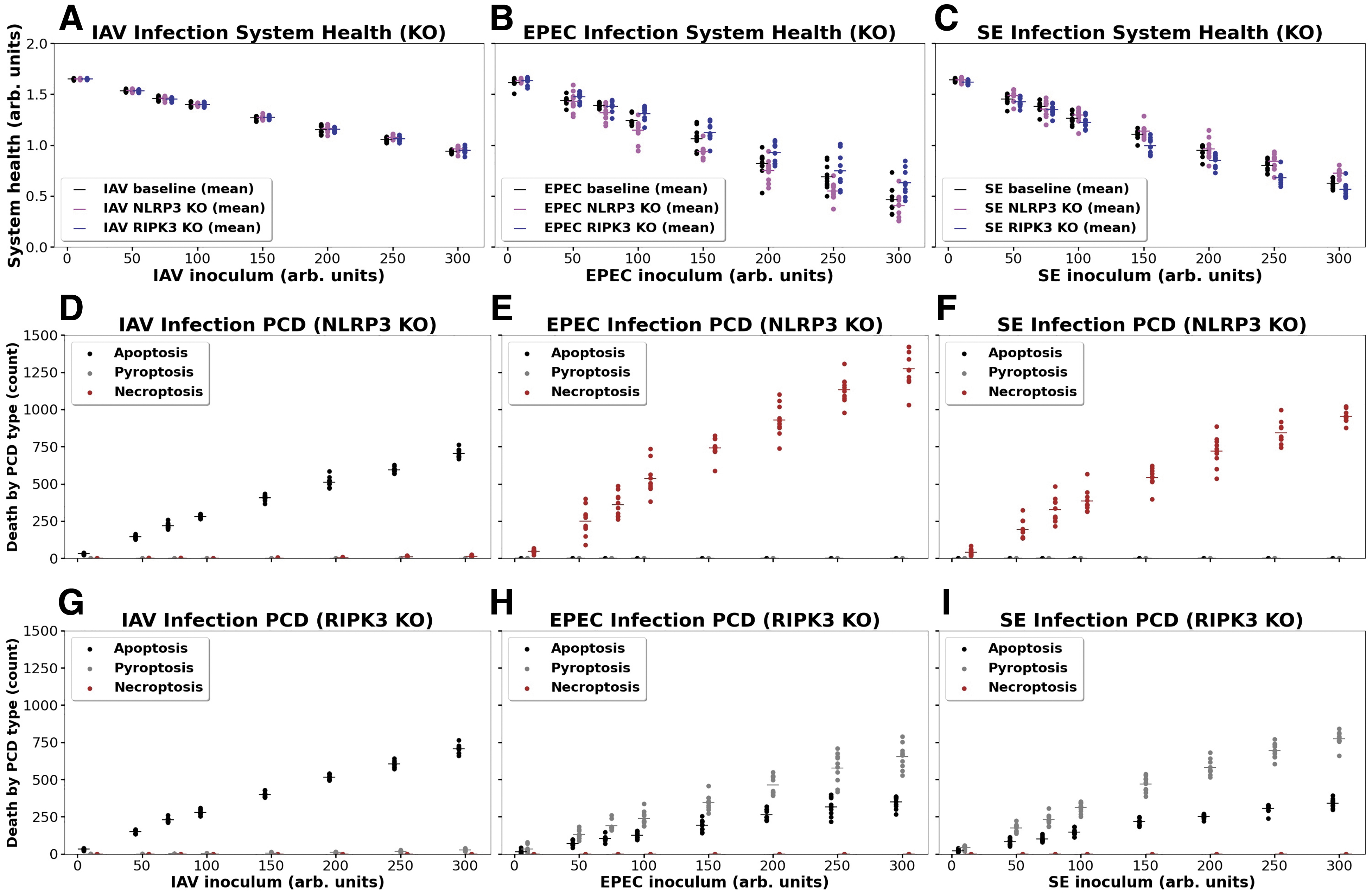
Effects on system health and PCDP fate in three infections with simulated NLRP3 and RIPK3 knockouts. Panel
Anti-inflammatory therapy experiments
Figure 5 shows the effect of simulated anti-IL-1β therapy and anti-TNF therapy in all three infections with respect to total system damage and PCDP fate. Total system health was not altered by the application of either of the anticytokine therapies. Although there appears to be some differences from baseline due to the addition of anti-IL-1β for both EPEC and SE, these did not turn out to be statistically significant: for EPEC p = 0.39 and for SE p = 0.48 (note the degree of overlap in the stochastic replicates present). For the distribution of PCDPs across the three pathogens; there were no substantive changes in these distributions (compared to baseline seen in Figure 3D–3F) in any of the different infections or anticytokine-applied conditions.
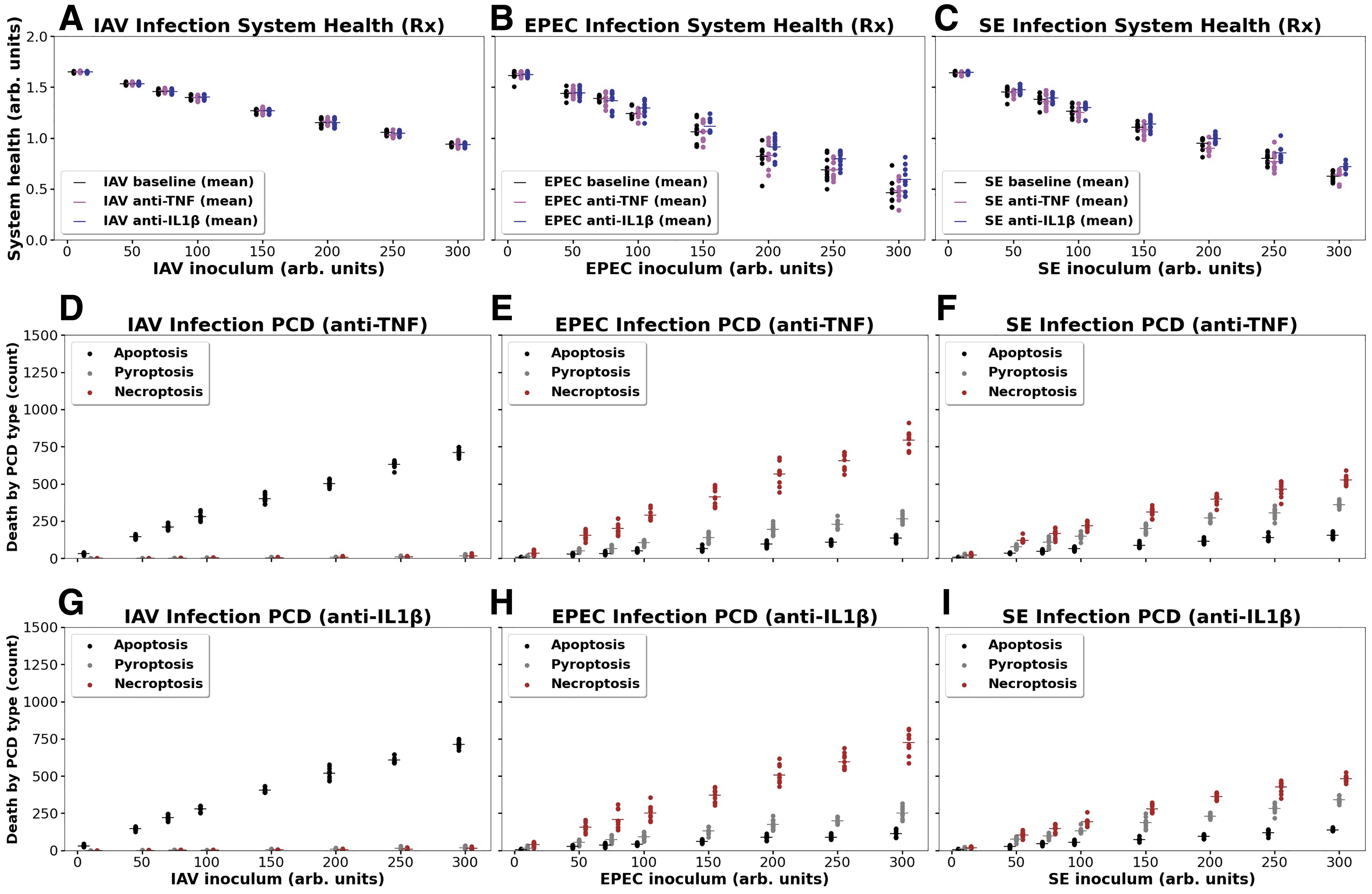
Effects on system health and PCDP fate in three infections with simulated anti-TNF and anti-IL-1β therapy. Panel
Discussion
Programmed cell death pathways represent critical pathways involved in tissue homeostasis and host response to infection, but also can, in certain circumstances, contribute to pathologic conditions such as cancer, cardiovascular disease, inflammatory bowel disease, ischemia reperfusion injury, and sepsis. Traditionally, these pathways were identified, characterized, and studied in isolation, but it is increasingly recognized that extensive crosstalk exists. This crosstalk makes it difficult to parse the relative contribution of each of these PCDPs to different disease states, and, consequently, to identify potential therapeutics targeting their components. We propose that computational modeling, and agent-based modeling in particular, is a useful adjunct to more traditional investigatory methods in being able to integrate and represent the spectrum of behaviors that arise from such crosstalk to better understand the robustness of these systems.
The PCDABM represents an initial step towards this goal. The simulation experiments demonstrate how robust these pathways are to targeted interruption(s), be those interruptions arising from evolved pathogen strategies, simulated knockout experiments, or simulated pharmacologic interventions. However, we also recognize that there are limitations to the current iteration of the PCDABM:
Use of generic tissue and immune cell representation can miss differential expressiveness of various PCDPs in specific cell types. The current version implements the same PCDP structure and responsiveness across both cell agent types (with the notable exception that only immune cells can produce TNF); different tissue types and immune cell species may have differential preferences for PCDPs.
There are other PCDPs not represented in the current version of the PCDABM: the list of these additional PCDPs is extensive; some examples include ferroptosis, NETosis, entosis, and mitoptosis. 16
Non-comprehensiveness of pathway representation. As we previously noted, any modeling endeavor requires choices in terms of what to include and not include for a particular project. As a result, we recognize that we can only evaluate what has been included, and there is a perpetual risk that what we have left out may be important. For instance, the fact that there are non-canonical inflammasome activation pathways is well known, and the incorporation of these components may have an effect not seen in the current model. However, it is worth noting that the addition of more pathways usually leads to an increase in the parallelism and redundancy of the functions within a model, and the effects of such redundancies are already manifest in the current version of the PCDABM.
This last point is worth expanding upon, as it points to the central utility of this type of modeling: the ability to make explicit a non-intuitive behavior. For example, the negative effects of undesired inflammasome activation in the generation of CS and sepsis (particularly noted in severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2] infection) has led to an interest in NLRP3 antagonists as a therapeutic option. However, if there is any main lesson to be learned from roughly 50 years of attempting to modulate inflammation in sepsis, it is that what initially appears to be a reasonable linear assumption, breaks down when applied in a context that incorporates the breadth of complexity present in the real-world.
Toward this end, an area where dynamic computational models—such as the PCDABM—may provide crucial capabilities, is in their potential ability to deal with the heterogeneity and variability seen in biologic systems and in clinical data. These models help to highlight the impact of the aforementioned heterogeneity on the development of effective therapeutic control modalities. The variability seen in clinical presentations and associated clinical biomarker data is readily evident, and one major factor invoked as a reason for the difficulty in finding efficacious drugs able to pass clinical trials is patient heterogeneity.
Stochasticity and variable responsiveness: The current PCDABM relies on stochastic processes to generate the run-to-run variability in the simulation experiments. Although stochasticity in biologic systems is well recognized, we realize that this is not the only source of variability seen in biologic and clinical data. We assert that much of this behavioral heterogeneity arises from differential responsiveness of cellular-molecular pathways due to a plethora of genetic, epigenetic, and comorbidity-related factors, and have developed a set of computational and mathematical methods to incorporate these factors when attempting to reproduce clinical data.17,18
Impact of clinical heterogeneity on the search for effective therapeutics: It follows from the concept of differential biologic responsiveness as a cause for clinical heterogeneity that this would necessarily affect the search for effective molecular interventions (e.g., drugs) to steer diseased trajectories back to a state of health. In short, finding the right intervention for the right patient at the right time will require the ability to effectively classify an individual patient as suitable for a particular intervention while also having some means of determining what intervention should actually be applied. It is readily evident that the complexity of this task is beyond the boundaries of human intuition; it is for this reason that our group has developed a computational method that applies machine learning (ML) and artificial intelligence (AI) methods to address this complex control discovery task. In short, this method applies the approach used to train the game-playing AIs to the task of “winning” a game of survival against our simulations of sepsis, both in the traditional context, 19 and in hypothetical conditions that mimic a future pandemic or emerging antimicrobial resistance 20 .
Whereas a comprehensive discussion of these methods is beyond the scope of the current paper (as the PCDABM is too preliminary a model for these methods to be applied, designed to perform conceptual model visualization and evaluation at this stage), they do point to an acknowledgment of the difficult tasks ahead in order for models of this type to reach their translational promise. With respect to the PCDABM, we recognize its current limitations and see areas of refinement to bring it to a level where the above-mentioned translational methods can be applied. We believe, however, that even prior to the achievement of such a refined PCDABM, there is potential benefit as a means of hypothesis verification/conceptual model checking in other infections and conditions where PCDPs play an important pathophysiologic role, such as in surgical diseases like sepsis, soft tissue/wound infections, wound healing, atherosclerosis, and nosocomial infection.
Conclusions
We developed the PCDABM to examine the dynamics arising from individual PCDPs and their crosstalk. Experiments with the PCDABM simulated the effect of three well-described pathogens known to affect PCDPs, each representing a particular class of infectious agent: viral, obligate intracellular bacteria, and extracellular bacteria. The effect of two of the PCDPs, pyroptosis and necroptosis, were notable for their inflammation-propagating features, which led to increased system damage—simulating the dynamics of CS. Simulations demonstrated the consequences of the extensive crosstalk that exists between the PCDPs as reflected by the consistency of system behavior enabled by these redundancies.
We found that anti-inflammatory therapies targeted at singular points in these pathways did not affect the extent of tissue damage, nor did it appreciably affect the types of PCDPs activated under those conditions. Knockout of either RIPK3 or NLRP3 under infectious conditions affected the types of PCDPs utilized for infection clearance but importantly, did not affect the overall amount of tissue damage imposed on the system by remaining pathways.
Future work will focus on expanding the capabilities of the PCDABM to model additional pathogens, such as Staphylococcus aureus, represent surgically relevant disease processes, and to link the model with real-world experimental platforms to test predictions and aid in the design of additional experiments.
Footnotes
Authors' Contributions
Dr. Feuerwerker conceived of the project, implemented the PCDABM, performed the simulation experiments, and is the primary author. Dr. An aided in the conception of the project, the development of the model and performance of the simulation experiments and edited the manuscript. Dr. Cockrell aided in the implementation of the model and the performance of the simulation experiments.
Acknowledgment
Recipient of the 2023 SIS-Crely Best Use of Technology Award.
Funding Information
This work was supported in part by the National Institutes of Health Award UO1EB025825. This research is also sponsored in part by the Defense Advanced Research Projects Agency (DARPA) through Cooperative Agreement D20AC00002 awarded by the U.S. Department of the Interior (DOI), Interior Business Center. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred.
Author Disclosure Statement
The authors declare that there are no conflicts of interest related to this work.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.