
Abstract
Abstract
This study aimed to investigate a biocompatible, biomechanically functional, small-diameter (<6 mm) scaffold for tissue engineering a vascular graft using acellular porcine ureters. Porcine ureters were decellularized and sterilized using sequential treatment with hypotonic Tris buffer, sodium dodecyl sulphate 0.1% w/v (plus proteinase inhibitors), nuclease solution (RNase and DNase), and peracetic acid. The scaffold was compared with fresh ureter according to histology, immunocytochemistry, quantitative determination of alpha-galactosyl (α-Gal), and biochemistry. The biomechanical properties of the scaffold were compared with those of fresh ureters and human saphenous vein. The biocompatibility of decellularized ureters was assessed using in vitro contact and extract cytotoxicity tests. The in vivo biocompatibility was investigated using a mouse model. The histioarchitecture of the acellular ureteric scaffolds was preserved with some loss of basement membrane proteins while showing no evidence of cellularity. There was no evidence of residual α-Gal epitope present in acellular ureter. The ultimate tensile strength, compliance, and burst pressures of the acellular ureters were not compromised, compared with fresh tissues (p > 0.05), and the results compared favorably with fresh human saphenous vein samples (p > 0.05). The decellularized scaffolds were shown to be biocompatible with porcine smooth muscle and endothelial cells in vitro. One month after subcutaneous implantation in mice, explants were analyzed immunohistochemically using anti-CD3, Factor VIII, F4/80 (macrophage), and α–smooth muscle actin antibodies. The fresh tissue controls had a significantly thicker capsule (of inflammatory cells and fibrous tissue) than decellularized implants (p < 0.05). Decellularized explants were infiltrated with a combination of fibroblast-like cells and macrophages, indicating a healthy repair process. This study has demonstrated the potential of acellular porcine ureteric scaffolds in tissue engineering small-diameter living vascular grafts.
Introduction
The most extensively investigated allogeneic graft is the glutaraldehyde-treated human umbilical vein, which has shown excellent clinical application.1,2 However, the glutaraldehyde treatment prevents cell ingress or regeneration, and this may account for why reported results from clinical studies have been mixed, with failures related to aneurysm formation, calcification, and infection.3–5 Other allogeneic grafts include decellularized6,7 and cryopreserved 8 veins, but these have not been used extensively in the clinical setting. Xenogeneic scaffolds include bovine ureter (Synergraft SGVG 100, Cryolite, Inc., Kenneshaw, GA, 3 and Flonova, Bionova International Pty Ltd, Melbourne, Australia 4 ), bovine carotid artery (Artegraft; Artegraft Inc., North Brunswick, NJ), 9 and visceral veins (Procol; Hancock Jaffe Labs, Irvine, CA). 5 Currently, the bovine ureter Synergraft is used clinically to create arteriovenous (A-V) fistulas when autologous veins are no longer available. Mixed results have been reported, and a recent study demonstrated significant residual cellularity and antigenicity in these scaffolds. 10 With an ever-expanding population of patients with end stage renal failure awaiting renal transplantation, there is an urgent need to develop a suitable conduit for dialysis access. Results of the bovine ureter Synergraft when used for femoropopliteal bypass surgery have been unsatisfactory, raising further questions about its suitability for clinical use.
Weinberg and Bell 11 first reported upon the tissue engineering of an arterial graft using cell-seeded collagen gels. However, these constructs had poor burst strength (650 mmHg) when compared with human saphenous vein (HSV; 1680 mmHg).12,13 Others have investigated the utility of polymer scaffolds composed of polyglycolic acid, poly L-lactic acid, or poly D,L-lactic-coglycolic acid.14,15
Auger and colleagues 16 used sheets of human neonatal smooth muscle cells wrapped around porous tubular mandrels. Burst pressures were reported to be supra-physiological (2594 ± 501 mmHg). 13 L'Heureux and colleagues 17 further developed sheet-based tissue engineering by producing a scaffold from human fibroblasts, a decellularized internal membrane, and a functional endothelium. These constructs had burst pressures of 3688 ± 1865 mmHg and remained biologically and mechanically stable for 8 months when implanted into nude rats as abdominal aortic interposition grafts (internal diameter of grafts = 1.5 mm). Although all these strategies show promise in the experimental setting, they have not been translated to the clinic.
Tissue-engineered vessels have the potential to improve clinical outcome when autologous tissue is not available. The aim of tissue engineering is to create new functional autologous tissue using biocompatible scaffolds that regenerate in vitro or in vivo. The choice of an appropriate scaffold is crucial for tissue engineering of blood vessels; the ideal scaffold should not elicit a specific immunological or nonspecific inflammatory response, should possess sufficient biomechanical properties to withstand hemodynamic stresses, and hould allow for endothelialization and re-population with autologous recipient vascular cells.
Porcine ureter is particularly attractive as a potential scaffold because of its anatomical similarity to blood vessels. It is a tapered valveless tubular conduit possessing a strong matrix and small internal diameter. 18
The aims of this study were to evaluate the characteristics of an acellular, porcine ureteric scaffold for use in tissue engineering a small-diameter blood vessel that could be used as a medical device for creating A-V fistulas for dialysis access in patients with end-stage renal failure.
Materials and Methods
Tissues
Porcine tissues (ureters, thoracic aorta, carotid artery and ileofemoral artery) were harvested from 35-kg Large White female pigs (Sus Scrofa) after Schedule (I) killing within the University of Leeds Central Biomedical Services Facility. HSV samples were obtained as discarded tissues during surgery after informed consent and local ethical approval.
Preparation of fresh and decellularized porcine ureters
Porcine ureters (40 in total) were immediately transported to the laboratory in Hanks' balanced salt solution (Sigma-Aldrich, UK) at room temperature.
Fresh ureters (n = 20) were washed in phosphate buffered saline (PBS; pH 7.4; Invitrogen, UK) containing antibiotics (penicillin 60 mg/L, streptomycin 100 mg/L, Invitrogen) several times, including luminal instillation. Fresh ureters were then used immediately or stored at −40°C on moist sterile filter paper. Decellularized ureters (n = 20) were prepared using a modification of the method described by Booth et al. 19 All steps in this process were performed at 37°C, with agitation at 320 rpm and required instillation of solutions into the lumen of the ureter unless stated otherwise. Ureters were transferred to a Class II cabinet and washed several times in PBS containing 10 KIU (v/v) aprotinin (Tyrasol, National Health Service supplies) and then incubated for 16 h in hypotonic Tris buffer [10 mM (w/v) Tris, Sigma, Poole, UK, 0.1% (w/v) ethylenediaminetetraacetic acid (EDTA), VWR International, Lutterworth, UK, and aprotinin (10 KIU/mL), adjusted to pH 8.0] at 4°C. Ureters were then incubated for 24 h in hypotonic Tris buffer containing 0.1% (w/v) sodium dodecyl sulfate (SDS, Calbiochem, Beeston, UK). Tissue samples were then washed in PBS containing no protease inhibitors and incubated in nuclease solution containing 1 U/mL RNase (Sigma) and 0.5 U/mL DNase (Sigma) in buffer [50 mM Tris-hydrochloric acid (HCl), 10 mM magnesium chloride, and 50 μg/mL bovine serum albumin, pH 7.5; Sigma] for 4 h, followed by thorough washing using PBS. Ureters were sterilized in 0.1% (v/v in PBS) peracetic acid for 3 h, followed by thorough washing in PBS, and then processed for experimentation or stored at −40°C.
Histology
Tissue specimens were fixed using 10% (w/v) neutral-buffered formalin (Triangle Biomedical Supplies, Skelmersdale, UK), dehydrated, and embedded in paraffin wax. All specimens were serially sectioned for microscopic evaluation. Cellular distribution and histioarchitecture were assessed using hematoxylin and eosin (H&E) staining. The presence of glycosaminoglycans, elastin, and collagen was evaluated using the elastin Van Gieson and Masson's Trichrome stains. To assess removal of nuclear material, sections were incubated with Bis-Benzamide H33258 fluorochrome (1 μg/mL, Calbiochem, Chandlers Ford, UK) for 15 minutes before viewing with a 4',6-diamidino-2-phenylindole filter using a fluorescence microscope (Olympus BX51, Southall, UK). Immunohistochemical analysis of the basement membrane proteins was performed using monoclonal antibodies specific for collagen IV (mouse immunoglobulin (Ig)G1 isotype, Sigma), laminin (mouse IgG1 isotype, Novocastra Laboratories, Orton Southgate, UK), and fibronectin (mouse IgG1 isotype, Novocastra Laboratories). Amplification, using the Envision DAKO K5007 kit (DakoCytomation, Ely, UK) was employed. Sections were then incubated in diaminobenzidine substrate solution (Sigma). IgM and IgG isotypes were included as negative controls. All histological sections were viewed using an Olympus BX51 light microscope. Positive control tissue included porcine femoral artery.
Biochemical analyses
The DNA content of fresh and decellularized tissue was assessed using gel electrophoresis after a papain digestion process previously described. 20
The hydroxyproline (an indication of collagen content) and sulfated sugar (an indicator of glycosaminoglycans (GAGs)) content of fresh and decellularized ureter was determined and compared with that of fresh porcine arterial tissue, carotid artery, femoral artery, and aorta. Before performing assays, it was necessary to remove the periadventitial fat from fresh tissues and to hydrolyze the fresh and decellularized tissues. Delipidized (chloroform 2:1 methanol solution) fresh tissues and decellularized ureters (n = 4 of each) were washed in PBS and freeze-dried to a constant dry weight. Samples were then hydrolyzed in 5 mL of 6 M HCl (VWR International) in an autoclave for 4 h and then neutralized with 6 M sodium hydroxide.
Hydroxyproline content of the digests was measured using the method described by Edwards & O'Brien. 21 Briefly, 50 μL of test solution or hydroxyproline standard (2–8 μg/mL) was placed in a 96-well plate. Oxidizing solution (100 μL; 1.41 g chloramine T, Sigma, in 100 mL of distilled water) was added to each well and the plate shaken gently for 5 min. Ehrlich's reagent (100 μL; 7.5 g p-dimethylamino benzaldehyde, Sigma, 30 mL propan-1-ol and 13 mL 62% (v/v) perchloric acid, Sigma) was added to each well and mixed thoroughly. The plate was covered and incubated in a water bath at 60°C for 45 min. Absorbance was read at 570 nm and the hydroxyproline content of the digests interpolated from the standard curve.
GAGs were assessed in the hydrolyzed tissue digests using the di-methylene blue (DMB) assay first described by Farndale et al. 22 Briefly, 40 μL of test solution (chondroitin sulfate 0–200 μg/mL) and 250 μL of DMB (Sigma) were added to 96-well plates. Within 60 s after the DMB solution was added, the absorbance was read at 525 nm, and the sulfated sugar content of the digests was interpolated from the standard curve.
Analysis for the xenoantigen galactosyl (α1,3) galactosyl
Sections (5 μm) of fresh and decellularized ureter (n = 3) were stained for the alpha-galactosyl (α-Gal) epitope using anti-α-Gal antibody (Alexis, Nottingham, UK). Standard immunoperoxidase methods were employed to assess for the presence of the α-Gal epitope. Positive control tissue included porcine pericardium. For further confirmation that the decellularization process had eliminated the presence of the α-Gal epitope, an antibody absorption technique was performed on fresh, decellularized, and hypertonic treated ureter. Decellularized ureter that had been treated with alpha galactosidase was used as a negative control (no α-Gal). Tissue samples (0.1 g; n = 3) were finely chopped and mixed with 1 mL anti α-Gal (1:10) antibody (Alexis) in Tris buffered saline (TBS) azide bovine serum albumin (BSA) [0.1% (w/v) BSA, 0.1% sodium azide (w/v), Sigma, in TBS] in BSA-blocked Eppendorf tubes and incubated overnight at 4°C. The tissue samples were then centrifuged at 600 g for 15 min, and 750 μL of supernatant was removed and added to BSA-blocked Eppendorf tubes. A further 750 μL of TBS azide BSA was added to the tissue samples, followed by centrifugation and extraction of a further 750 μL of tissue extract. The levels of anti-α-Gal antibodies remaining in the tissue extracts were then compared using enzyme-linked immunosorbent assay using α-Gal-BSA (10 μg/mL in PBS, Dextra Laboratories, Reading, UK).
Biomechanical assessment of decellularized and comparator tissues
Low-strain-rate uniaxial tensile loading to failure
Uniaxial tensile testing to failure was performed using a Howden tensile testing machine (London, UK), according to the protocol described by Korrosis et al. 23 Circumferential and axial tissue strips (one of each) 1.5 and 4 mm wide, respectively, were dissected from nine fresh and nine decellularized porcine ureters and from six fresh HSVs. The wall thickness of all specimens was measured using a Mitutoyo thickness gauge with a resolution of 0.01 mm. Tissue strips were mounted on a purpose-built holder, which allowed for a gauge length of 3 and 8 mm for the circumferential and axial samples, respectively. This gave a length:width ratio of 2:1 in both cases. Specimens were mounted in their relaxed state. Subsequently, the samples were loaded on to the testing machine, which was set to produce a specimen preloading of 0.01 N. The specimens were then loaded to failure at a rate of 10 mm/min and a load-elongation response recorded. Data acquisition was performed using MTT.exe software (Cambridge, UK) and further interpreted using Microsoft Excel (Redmond, WA). The stress-strain behavior of the specimens was analyzed according to six parameters. These have been well described 23 and included the elastin and collagen phase slopes, transition stress and strain, ultimate tensile strength, and failure strain (ɛUTS).
Compliance testing
Fresh and decellularized ureters (n = 8) were subjected to dilation testing to assess the effect of decellularization on compliance of the graft. Testing was conducted in a dilation rig as described previously. 24 During testing, each ureter was attached to the end of a syringe using a cable tie, and four reference points were marked along its axis. The dots were placed 1 cm apart, with the first and last dots marked 3 cm from the proximal and distal ends, respectively. The distal end of the ureter was then clamped using a Ligaclip applicator (M-11 AutoSuture, Leeds Teaching Hospitals, UK) and the syringe filled with saline and attached to a pressure manometer. Incremental pressure (0–160 mmHg) was then applied and an image of the ureter at each pressure increment recorded using a digital camera (Sony Cyber-Shot 5.1, Sony Corp., Tokyo, Japan). Images were then downloaded on to a personal computer (Gateway 2000, Pentium II, Irvine, CA) and analyzed using Image Pro Plus software (Media Cybernetics, Silver Spring, MD). All images had an aspect ratio of 1:1 and were calibrated with the aid of the scale on the syringe. At each pressure increment, the dilation of the ureter at the proximal and distal ends was measured as the external diameter halfway between the marked reference points. This ensured that the dilation was measured at the same point regardless of the axial elongation of the pressurized specimen. The results were averaged and presented as percentage change in ureter diameter with respect to increasing internal pressure.
Burst pressure testing
The burst pressure of the decellularized ureter was compared with that of fresh porcine ureter, fresh porcine carotid artery, fresh porcine femoral artery, and HSV. Tissues (n = 4 of each) were screened before testing by measurement with electronic calipers (Mitutoyo Absolute Digimatic electronic calipers, Leeds, UK) to ensure that all segments had an external diameter of 6 mm or less. During testing, each sample segment was attached to a custom-made burst pressure rig 25 using 4/0 Prolene (Ethicon, Livingstone, UK) sutures. The burst pressure rig comprised a fluid reservoir filled with 0.9% (w/v) normal saline solution, an air pressurization system with Joucomatic 400 air regulator (Leeds, UK), silicone tubing with Luer lock connectors, a Comark 3357 fluid pressure gauge (Leeds, UK) with “Record and Hold” function and a customized pipette for each test segment. Once mounted, each sample segment was flushed through with normal saline from the reservoir and the open end tied off with 4/0 Prolene. The segment was then pressurized at a rate of 100 mmHg/s until burst point. The pressure at which the segment failed was recorded on the digital pressure gauge and documented.
Biocompatibility of decellularized porcine ureters
Isolation and culture of vascular cells
Endothelial cells were isolated from porcine thoracic aortas using 0.1% collagenase (w/v) (Sigma-Aldrich). The resulting cell suspension was cultured in endothelial cell culture medium [medium M-199 (Sigma-Aldrich) supplemented with 1 mL (3 mg/mL) of endothelial growth factor supplement (Sigma), 20% (v/v) fetal calf serum (Biosera, Ringmer, UK), penicillin 60 mg/L, streptomycin 100 mg/L, 1 mL L-glutamine (2 mM, Invitrogen) solution, 0.4 mL pyruvate (5.5 mg/mL, Sigma), and 0.2 mL of heparin (5000 U/mL, Leo Laboratory, Bucks, UK), per 200 mL of culture medium]. Smooth muscle cells were isolated using the explant method, which involved dissecting the aortic wall into 1 mm3 pieces before transfer into a 25 cm3 cell culture flask to which 2 mL of smooth muscle culture medium [Dulbecco's modified Eagle medium (DMEM) supplemented with penicillin 60 mg/L, streptomycin 100 mg/L, 1 mL L-glutamine solution per 100 mL, and 10% (v/v) fetal calf serum] was added before incubating in an atmosphere of 5% (v/v) CO2 in air at 37°C. The culture medium was changed every 24 h for the first 3 days and every 48 h thereafter. Once sufficient numbers of smooth muscle cells could be seen growing from the aortic tissue segments, the flasks were flooded with culture medium and the tissue pieces removed. Smooth muscle and endothelial cells were characterized using immunofluorescent antibodies to α–smooth muscle actin and vimentin and von-Willibrand factor and CD31, respectively.
Contact cytotoxicity assay
Tissue samples (n = 3) measuring 0.5 cm2 were attached to the center of individual wells in a 6-well tissue culture plate using collagen gel (acid-solubilized rat-tail collagen neutralized with 0.1 M sodium hydroxide). Primary porcine smooth muscle and endothelial cells (passage 3) were seeded at appropriate seeding densities to allow the cells to become confluent in their respective culture media. Plates were then incubated at 37°C in 5% (v/v) CO2 and air for 48 h. The plates were viewed under an inverted microscope to determine the confluence and morphology of the cells. The cells were then fixed and stained with Giemsa solution (VWR International) for 5 min. A positive control consisted of cyanoacrylate adhesive (VWR International) and a negative control of collagen gel. All images were viewed under an inverted light microscope and digitally recorded after air-drying the plates.
Extract cytotoxicity assay
Tissue samples (n = 6) of fresh and decellularized ureter were finely minced and incubated in DMEM, 3 mL/mg, for 72 h at 37°C with gentle agitation. The supernatant was collected, tested for sterility, and stored at −20°C until required. Primary endothelial and smooth muscle cells were seeded onto 96-well plates at densities that would allow them to become confluent in the presence of their respective growth media. The plates were incubated for 24 h at 37°C in 5% (v/v) CO2 and air, after which the culture medium was aspirated, and 100 μL of DMEM [containing 20% (v/v) FBS and supplements] plus 100 μL of the test extract were added to six replicate wells. Controls included 40% (v/v) di-methyl sulfoxide in DMEM (positive control) and DMEM alone (negative control). The plates were incubated for a further 24 h at 37°C in 5% (v/v) CO2 and air. The effect of the extracts on the cell viability was determined using the ATPLite-M assay (Packard, Pangbourne, UK) following the manufacturer's instructions.
The in vivo biocompatibility was investigated by implanting decellularized and control tissue (n = 3) subcutaneously into α-galactosyltransferase gene-knockout mice (BresaGen Xenograft Management, Ltd., Immunology Research Center, St. Vincent's Hospital, Fitzroy, Victoria, Australia) under appropriate UK Home Office licenses for 1 month. Control tissue consisted of fresh porcine ureter acting as a positive control and α-galactosidase–treated porcine ureter as a negative control. The tissues implanted into the mice consisted of three groups: fresh, decellularized, and α-galactosidase treated. Three mice were used per group, and each mouse had two 0.5-cm2 sections of tissue implanted subcutaneously at two separate locations. After 1 month, the explants were investigated immunohistochemically for signs of inflammation, hyperacute rejection, and cellular infiltration. Serial sections of the explanted tissues were stained with H&E to evaluate the histioarchitecture and capsule thickness. Image Pro Plus™ software (Media Cybernetics, Bethesda, MD) was used to calculate the capsule thickness at four points around each explant (n = 3) and the mean value calculated. The primary antibodies used were von-Willibrand (Factor VIII) rabbit antihuman (DAKO), CD3 mouse antihuman (Vision Biosystems), Macrophage F4/80 rat antimouse (Invitrogen) and α–smooth muscle actin mouse antihuman (DAKO). Macrophage F4/80 and Factor VIII antibodies were retrieved enzymatically and CD3 retrieved using heat induction. The primary antibodies were amplified using polymer amplification (Factor VIII), an adapted ABC system for animal research (CD3) and the standard ABC system (Macrophage F4/80). Sections were then incubated in diaminobenzidine substrate solution. Control tissue included mouse spleen and lung.
Data analysis
Data are expressed as means ± 95% confidence limits. Data were analyzed by one-way analysis of variance. Individual group means were then compared using the T-method to calculate the minimum significant difference at p < 0.05 and p < 0.01.
Results
Histological evaluation of fresh and decellularized ureter
Fresh and decellularized ureteric tissue samples (n = 6) were serially sectioned and stained using H&E and Hoechst 33258 dyes to investigate whether the decellularization process had removed all of the cells. There was no evidence of cellular or nuclear staining in the decellularized sections throughout the tissue samples (Fig. 1A–D). In addition, the histioarchitecture remained preserved. Histological evaluation of the ureter revealed the presence of collagen, elastin, and GAGs after decellularization, although these appeared less abundant in the decellularized tissue (Fig. 1E–H).

Histological characterization of fresh (
The presence of basement membrane proteins, collagen IV, and laminin, together with fibronectin, was investigated before and after decellularization. This revealed that the collagen IV and fibronectin were still present, although the staining appeared weaker in the decellularized sections, indicating that the decellularization treatment had removed some of the proteins (Fig. 2). Laminin staining in the decellularized sections was negligible, suggesting that the decellularization process removed this protein.

Immunohistochemical staining of fresh (
DNA content of decellularized ureters
DNA extracted and electrophoresed from fresh and decellularized tissues (500 mg samples, n = 3) revealed no evidence of DNA in the decellularized tissue samples.
Biochemical analysis of fresh and decellularized ureter
Fresh ureter was found to contain 45 μg hydroxyproline per mg of dry weight. After decellularization, the hydroxyproline content increased slightly, to 47 μg per mg, but this was not significant (Fig. 3). The decellularization process did not significantly alter the sulfated sugar content of the ureteric scaffold, which was 69 μg/mg before and 71 μg/mg after decellularization (p > 0.05). To gain an appreciation of the difference in collagen and sulfated sugar content between the scaffold and arterial tissues, the assays were also performed on porcine femoral artery, carotid artery, and aorta. This revealed that the sulfated sugar content of the ureteric scaffold was similar to that of the arterial tissues but that the hydroxyproline content was significantly lower, indicating a lower collagen content of the ureteric scaffold (Fig. 3).
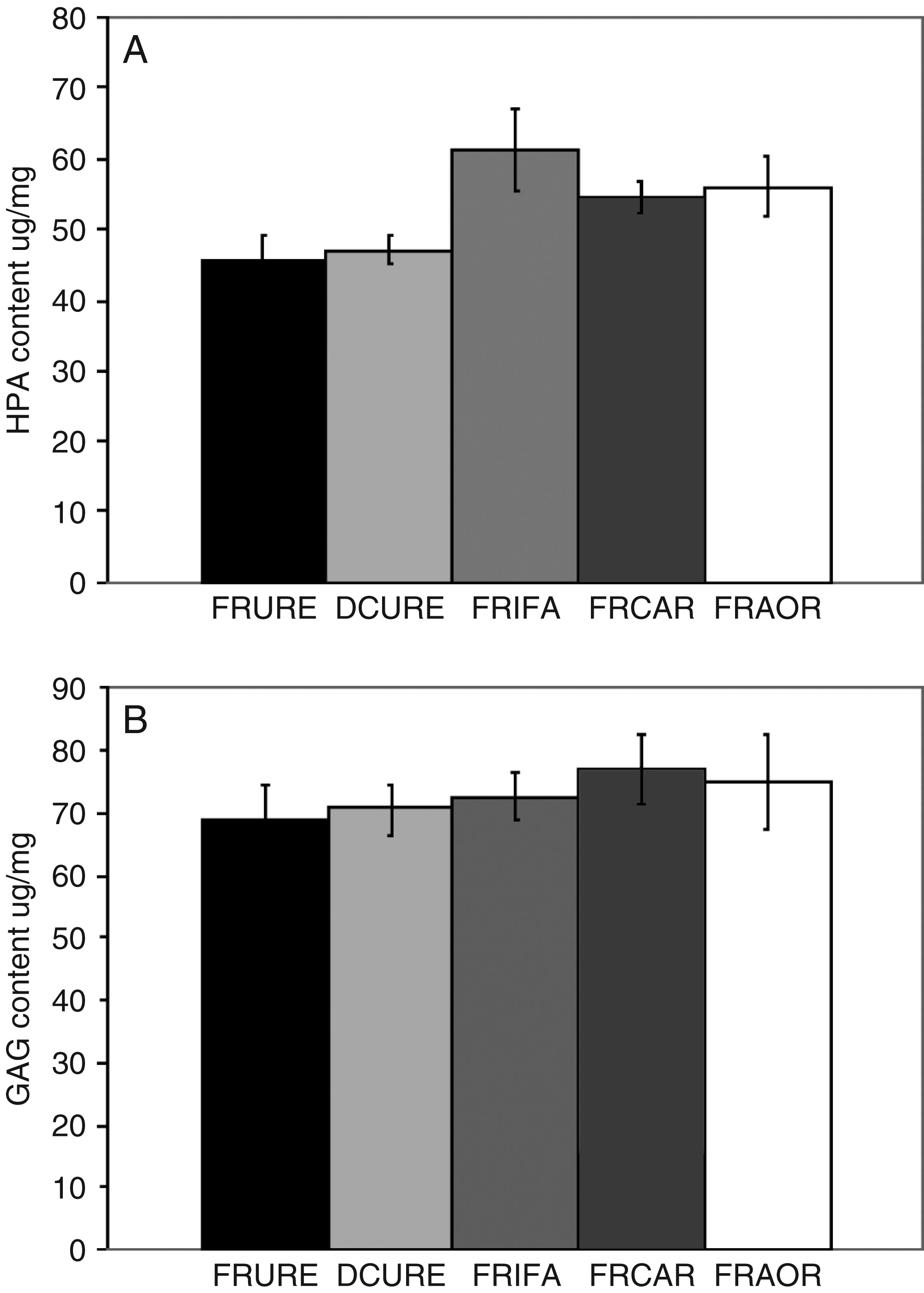
Hydroxyproline and sulfated sugar content of decellularized porcine ureter and fresh porcine ureter, femoral artery, carotid artery, and aorta. Data are expressed as the means (n = 6) ± 95% confidence intervals. FRURE, fresh ureter; DCURE, decellularized ureter; FRIFA, fresh iliofemoral artery; FRCAR, fresh carotid artery; FRAOR, fresh aorta.
Analysis for the xenoantigen Gal (α1,3) Gal
Immunoperoxidase staining of the decellularized ureter with antibody to gal (α1,3) gal showed no expression of the xenoepitope, which was evident in the fresh tissue (Fig. 4). Quantitative assessment using an antibody adsorption assay for the presence of the α-Gal epitope in decellularized ureter revealed no significant difference (p > 0.05) in the relative expression of the α-Gal epitope between galactosidase-treated ureter and decellularized ureter. In addition, there was no significant difference in the relative expression of α-Gal between HSV (negative control) and decellularized tissue. These results are summarized in Figure 5.
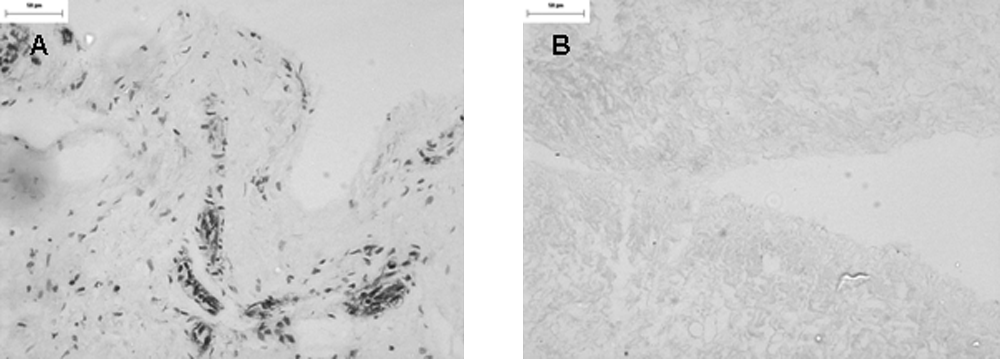
Immunoperoxidase staining of fresh (
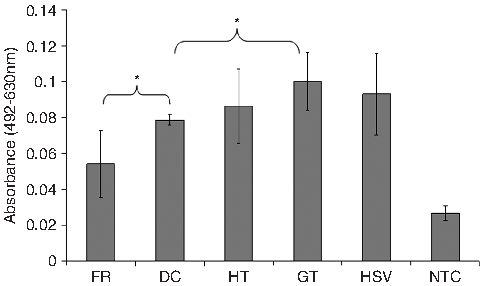
Antibody absorption assay for alpha-galactosyl (α-Gal) levels of anti-α-gal antibodies remaining in the supernatants after adsorption with fresh, decellularized, hypertonic-treated, and galactosidase-treated ureter; human saphenous vein (negative tissue control); and non-tissue negative control. Data are expressed as means (n = 3) ± 95% confidence intervals; *p < 0.05 (analysis of variance). FR, fresh ureter; DC, decellularized ureter; HT, hypertonic-treated ureter; GT, galactosidase-treated ureter; HSV, human saphenous vein; NTC, non-tissue control.
Biomechanical assessment of decellularized and comparator tissues
Ultimate tensile testing
The results for the ultimate tensile testing to failure of fresh and decellularized ureter are summarized in Table 1. Fresh HSV samples were used as controls. Decellularized samples of ureter showed no significant difference in the collagen or elastin phase slopes in the axial direction from that of fresh ureter and HSV. In addition, there was no significant difference in the ultimate tensile strength of decellularized ureter, fresh ureter, and HSV in the axial direction. Fresh ureter had a significantly higher ultimate tensile strength and ultimate failure strain (p < 0.05) than decellularized ureter in the circumferential direction, but there was no significant difference in these parameters between decellularized ureter and HSV.
*p < 0.05 decellularized vs HSV, **p < 0.05 decellularized vs fresh.
Compliance testing
No significant difference was observed in the compliance of the decellularized ureter and that of fresh ureter at the proximal or distal end (Fig. 6). This indicated that the decellularization process did not adversely affect the extensibility of the ureter.

Compliance characteristics (expressed as percentage strain) of fresh and decellularized ureter at the proximal (
Burst pressure testing
The mean internal diameter of the fresh and decellularized ureter and of porcine femoral artery, carotid artery, and HSV is shown in Figure 7A. The internal diameter of the decellularized ureter was similar to the arterial vessels and HSV. The absolute burst pressures of decellularized ureter were not significantly different from HSV (Fig. 7C; p > 0.05), but lower than porcine carotid artery (p < 0.05). The decellularized scaffold displayed burst pressures greatly in excess of physiological levels.
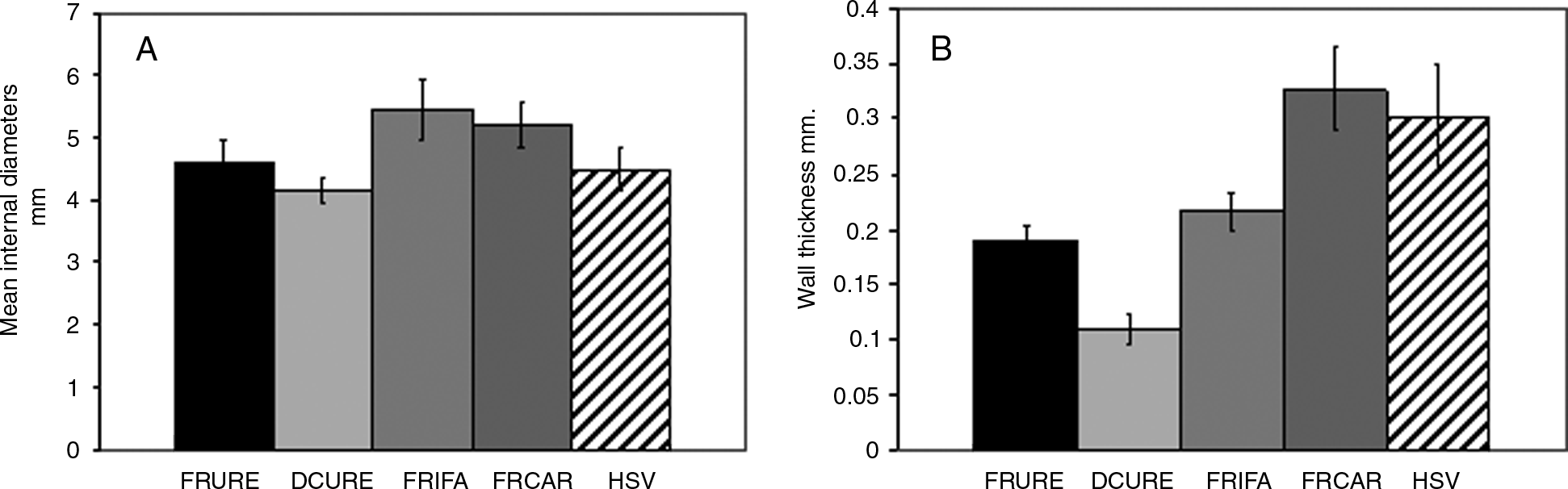
Burst pressure determinations of decellularized ureter and fresh porcine ureter, femoral artery, carotid artery, and aorta. Data are expressed as the means (n = 4) ± 95% confidence intervals. (
Biocompatibility of decellularized ureter
Contact cytotoxicity assay
Light microscopic analysis of the contact cytotoxicity plates revealed that smooth muscle and endothelial cells grew in contact with the decellularized ureter samples without exhibiting any signs of any change in cellular morphology or cell lysis from negative controls (collagen gel). Positive controls (cyano-acrylate) caused cell lysis to occur (Fig. 8).
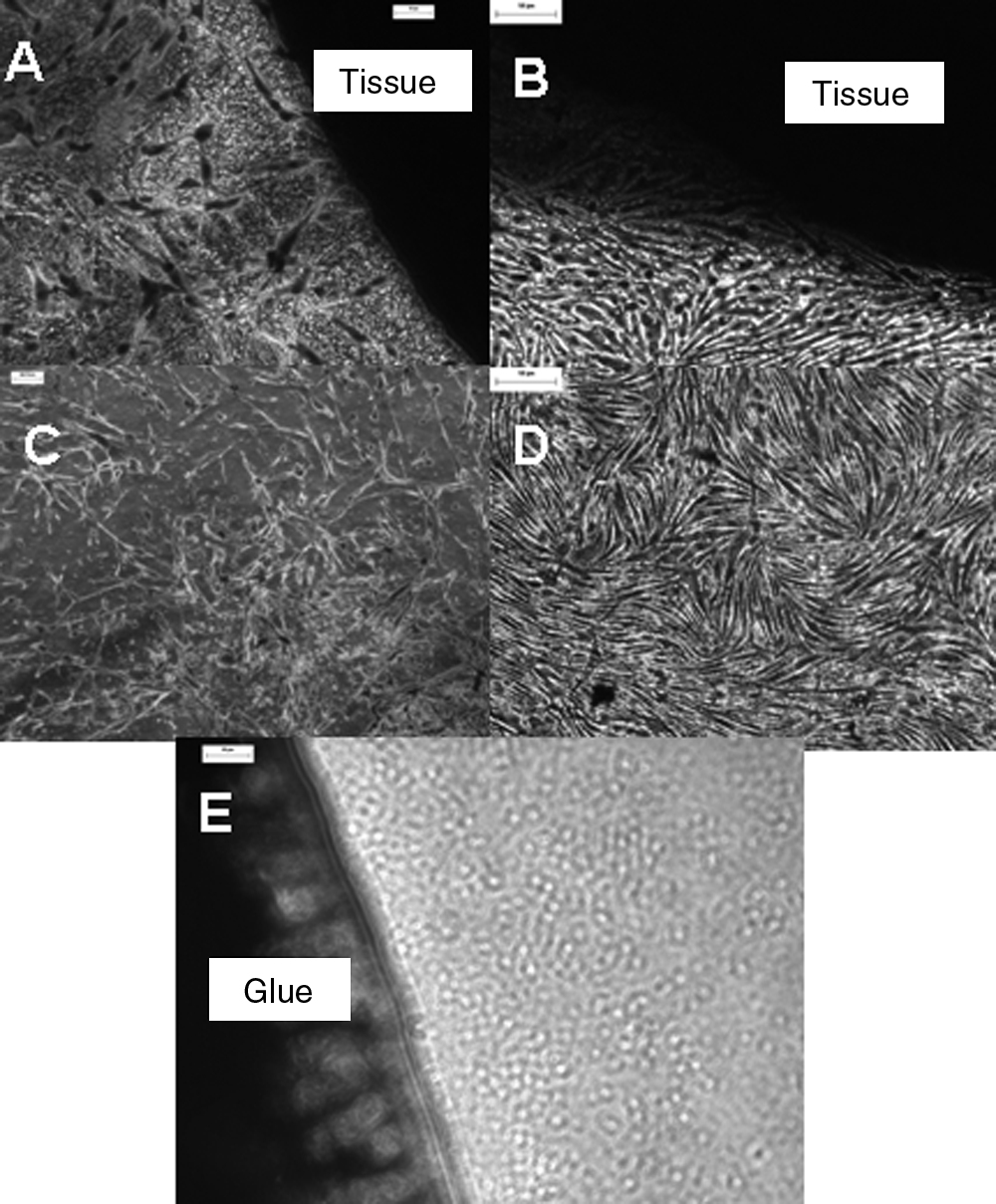
Contact cytotoxicity assays of decellularized ureter with porcine smooth muscle (
Extract cytotoxicity assay
There was no significant decrease in relative cellular adenosine triphosphate (ATP) content (used as a measure of cell viability) of smooth muscle and endothelial cells following incubation with the extracts of decellularized ureters compared to that of DMEM only negative controls (Fig. 9), as determined by the ATPLite assay (analysis of variance, p > 0.05). There was a significant reduction in relative ATP content produced by cells cultured in fresh tissue extracts and smooth muscle cells cultured with hypertonic-treated ureter (p < 0.05).
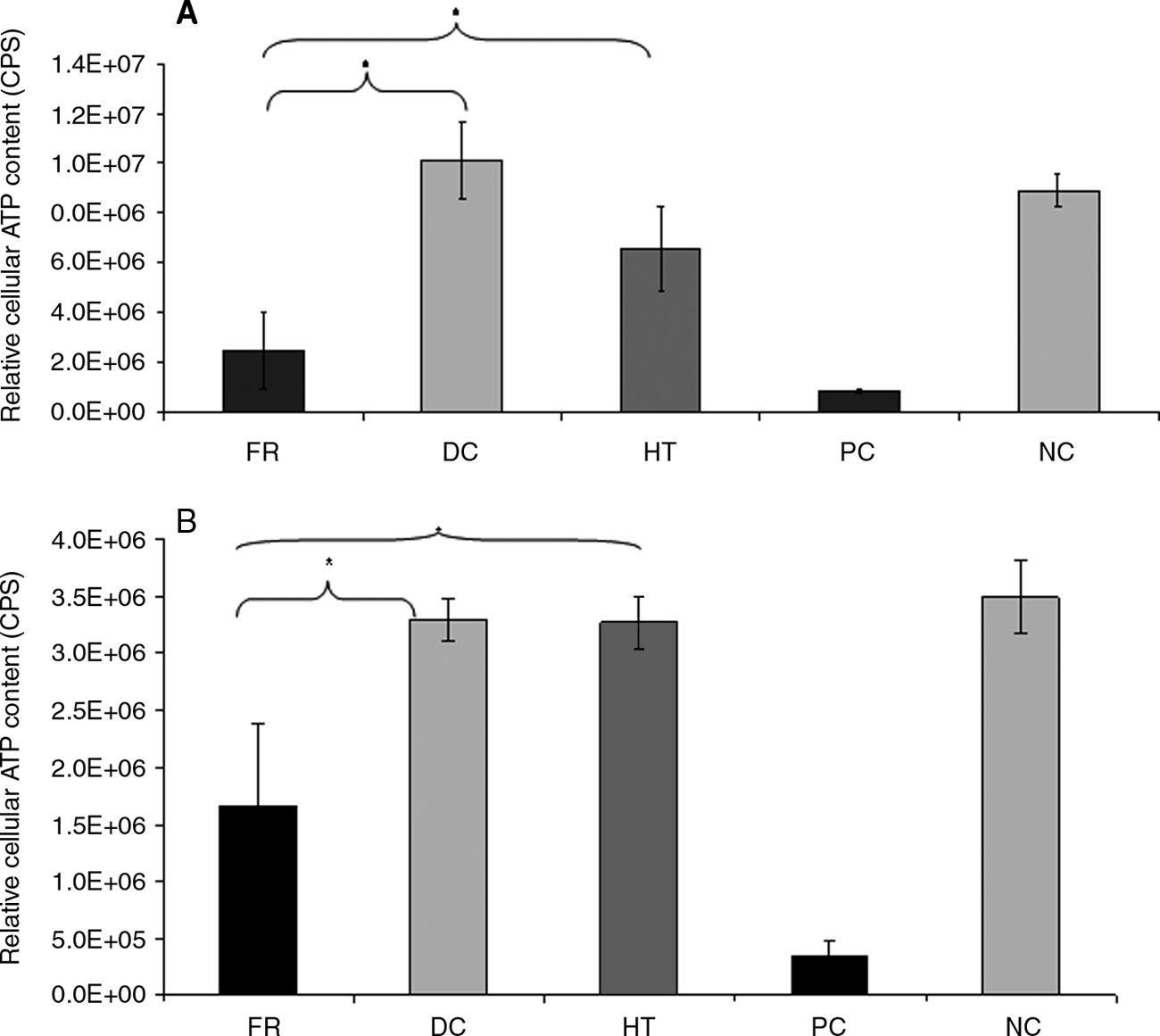
Extract cytotoxicity assay of fresh and decellularized ureter cultured in vitro with smooth muscle (
In vivo biocompatibility
Immunoperoxidase staining was used to phenotype the inflammatory and infiltrating cells in and around the explanted tissue samples (Fig. 10). H&E staining of the explanted tissue was used to investigate the capsule thickness of the various tissue groups implanted. Fresh tissue had a significantly thicker capsule (242 μm ± 122) than decellularized (74 μm ± 33) and α-galactosidase–treated (56 μm ± 22) tissue groups (p < 0.05). In addition, fresh explants exhibited vacuolation within the core of the tissue. Fresh tissue explants exhibited a greater percentage of CD3 staining than decellularized explants. This was particularly evident in the capsule. Fresh and decellularized tissue explants exhibited a higher number of FVIII-positive cells within the capsule and periphery of the tissue. Macrophages were more abundant in the decellularized explants than in fresh tissued and were seen throughout the tissue. Staining for α–smooth muscle actin appeared weak for fresh and decellularized explants, although more fibroblast-type cells appeared to be present in the decellularized tissue than in the fresh. Negative control samples were similar in appearance to decellularized explants.
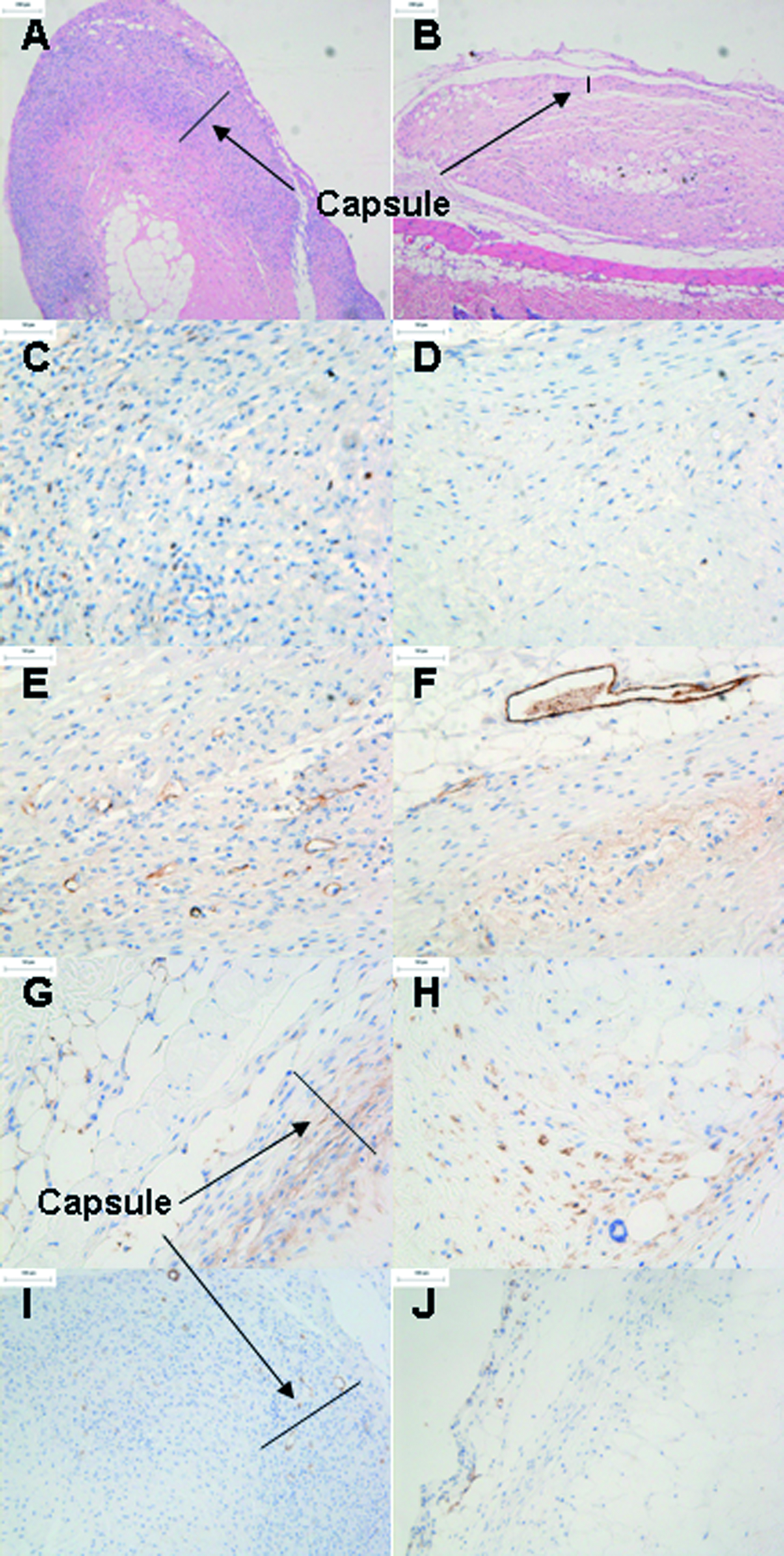
In vivo biocompatibility. Hematoxylin and eosin staining of fresh (
Discussion
There exists a pressing need to develop a small-diameter vascular graft to treat the complications of cardiovascular disease that meets the physiological demands of native arteries. The purpose of our study was to evaluate the characteristics of an acellular, porcine ureteric scaffold for potential use as a medical device for creating A-V fistulas in patients requiring hemodialysis access. Although autologous conduits remain the gold standard for revascularizing ischemic areas, patients undergoing bypass surgery face the added burden of harvesting these vessels, leading to potential wound complications. Other alternatives include allogeneic vascular grafts, including cryopreserved cadaver femoral vein allografts and the decellularized, cryopreserved Synergraft (CryoLife, Marietta, GA). These grafts are at greater risk of developing aneurysmal dilatation than polytetrafluoroethylene grafts.8,10 This occurs because the processed tissue is not acellular and the residual cells harbor antigenicity, which imparts an immune response upon implantation, causing destruction and weakening of the scaffold matrix. In most cases in which biological materials are being investigated, the tissue source is xenogeneic in origin. 26 Published work suggests that biological tissue-engineered scaffolds may be superior to prosthetic material in the face of infected fields. 27 A potential problem arising from pig-to-human transplantation is the porcine cell-specific disaccharide galactose (α1 → 3) galactose (the α-Gal epitope), responsible for hyperacute rejection occurring in xenotransplantation because of natural preformed antibodies. 28 The presence of this epitope in a xenogeneic matrix may cause an early inflammatory response after implantation, leading to graft failure.
In this study, decellularized porcine ureters were prepared using a modification of the decellularization method described by Booth et al. 19 In brief, this consisted of hypotonic cell lysis, a low-concentration ionic detergent, SDS (0.1% w/v) to solubilize the fragments, protease inhibitors to inhibit autolysis, and nucleases to digest nuclear material. This is similar to the method adopted by Clarke et al. 3 in developing the bovine ureter Synergraft, with the exception of SDS treatment. This may explain why the bovine ureter Synergraft is not acellular. Our results demonstrated successful removal of cells from the porcine ureter, as demonstrated histologically and by the lack of DNA in the matrix. In addition, histological samples provided evidence for the preservation of the histioarchitecture. The decellularization process did not lead to a reduction in collagen or GAG content. The slight increase in hydroxyproline and GAG content post-treatment was possibly due to the loss of cell mass. The final collagen content of the tissues may be obtained by multiplying the hydroxyproline values by a conversion factor of 7.14, calculated from the relative proportion in collagen molecules. 29 The derived values of collagen for porcine ureter were similar to those found in other published work. 30 The hydroxy prolene assay (HPA) content for the ureteric scaffold was significantly lower than that of the arterial controls, but the differences were not great.
The ultimate tensile strength of the decellularized ureter was comparable with that of HSV in the axial and circumferential directions. There was no significant difference in the elastic or collagen phase slopes of decellularized and fresh ureter, indicating that the decellularization process did not adversely affect the scaffold. In addition, the elastic and collagen phase slopes of decellularized ureter were similar to those of fresh HSV in the axial direction, and decellularized ureter had a better elastic phase slope in the circumferential direction than fresh HSV. These findings are relevant and encouraging when considering the clinical application of these grafts.
The compliance of fresh and decellularized ureters was compared at the proximal and distal ends of the tissue, and there was no significant difference in compliance, indicating that decellularization did not affect the extensibility of the ureter.
The acellular scaffold also displayed supraphysiological burst pressures and was comparable with HSV and porcine iliofemoral artery controls, although the absolute burst pressure for decellularized ureter was significantly lower than porcine carotid artery. Recent published work 31 has suggested that burst pressure results can be dependent on the rate of pressurization, with higher rates of pressurization producing significantly higher readings and potentially overestimating these values. However, this study compared burst pressures of only a single graft type across different infusion rates. Hence, it is not an issue in controlled studies such as this one in which a comparison was made with native artery and the current gold standard, HSV.
Extensive biocompatibility testing was performed on the ureter to determine whether the decellularization process rendered the matrix cytotoxic because of residual SDS 32 and whether cells would survive and proliferate in direct contact with the tissue. Results of the contact cytotoxicity assay indicated that smooth muscle and endothelial cells proliferated normally and maintained their morphology when cultured in direct contact with decellularized ureter. The soluble components of the decellularized ureter were also nontoxic to both cell types. Fresh ureteric extracts exhibited cytotoxic effects on both cell types, as the significantly lower ATP levels than in negative controls and decellularized extracts indicated. This was probably related to the presence of autolytic enzymes produced by the dying cells in the tissue. The hypertonic-treated ureter was also found to exert a cytotoxic effect on smooth muscle cells; incomplete removal of hypertonic solution during the washing period may explain this. There are no reports of in vivo biocompatibility testing of decellularized ureter in the literature, and this represents the first study to investigate the inflammatory, cellular infiltration, and regenerative potential of this type of scaffold. We have demonstrated that fresh ureter elicits a predictable immune response in vivo, as seen by the development of a thick capsule comprising CD3-positive cells and macrophages with CD3-positive cells extending into the tissue core. In addition, the fresh tissue explants exhibited a vacuolated appearance consistent with necrosis. In contrast, decellularized tissue explants possessed thinner capsules and had less CD-positive T-cell infiltration and a more-uniform distribution of macrophages throughout the tissue than fresh samples, indicating a more-favorable immune response. Fresh and decellularized explants exhibited capsular and peripheral infiltration of FVIII-positive cells, suggesting that neovascularization may have been occurring. It is believed that the fibroblastic-type cells are part of the regeneration process, and although not many of these cells were seen in the decellularized explants, the short period of time that the tissues were implanted for can explain this. More importantly, the decellularized tissue explants showed no evidence of necrosis or signs of hyperacute rejection. The in vivo results are complimentary to the in vitro biocompatibility studies performed and provide further evidence to support the use of decellularized ureters in the clinical setting. In vivo remodeling of extracellular scaffolds as a function of macrophage phenotype has recently identified the importance of macrophage activity in remodeling tissue (unpublished data). 33 Our data show that the decellularized explants showed macrophage staining around (capsule) and inside the tissue (core). The galactosidase-treated explants also exhibited positive macrophage staining throughout the capsule and tissue core. These data are extremely encouraging and will provide a platform for longer-term studies to be undertaken.
Histological results demonstrated no residual α-Gal staining in the acellular scaffold, which was reflected in the antibody absorption technique, which showed no significant difference in the absorption levels for the α-Gal epitope between decellularized ureter and the HSV, which has no α-Gal epitope. There was, however, a significant difference in α-Gal epitope absorption between fresh and decellularized ureter. No studies have examined the expression of the α-Gal epitope in xenogeneic vascular graft matrices, and this is the first to report on such data. The failures seen in other xenogeneic grafts may be related to the presence of residual antigens, such as the α-Gal epitope, which can lead to antibody-mediated inflammatory reactions causing weakness in the matrix structure.
In conclusion, this study has demonstrated proof of concept for using the decellularized porcine ureteric scaffold in tissue-engineering a small-diameter living vascular graft that could be used as a medical device for creating A-V fistulas.
Footnotes
Acknowledgments
Financial support: Leeds Charitable Trusts Bridging award (CD) and EPSRC Portfolio (EI, JF). The authors would like to acknowledge Mrs. Sally Grey for her help with the immunohistochemistry for the in vivo data.
Author Disclosure Statement
EI and JF are nonexecutive directors and share holders of Tissue Regenix Ltd.
Parts of this work were presented at the Tissue and Cell Engineering Society meeting, Sheffield, United Kingdom, July 2006, and at Society of Academic and Research Surgery (SARS), Cambridge, United Kingdom, January 2007.