
Abstract
Abstract
There is no effective treatment for the loss of functional salivary tissue after irradiation for head and neck cancer or the autoimmune disease Sjögren's syndrome. One possible approach is the regeneration of salivary glands from stem cells. The present study aimed to investigate whether small pieces of human submandiblar gland tissue contain elements necessary for the reconstruction of salivary rudiments in vitro via acinar and ductal cell differentiation. Primary submandibular gland (primary total human salivary gland; PTHSG) cells were isolated from human tissue and cultured in vitro using a new method in which single cells form an expanding epithelial monolayer on plastic substrates. Differentiation, morphology, number, and organization of these cells were then followed on basement membrane extract (BME) using RNA quantitation (amylase, claudin-1 (CLN1), CLN3, kallikrein, vimentin), immunohistochemistry (amylase and occludin), viability assay, and videomicroscopy. On the surface of BME, PTHSG cells formed acinotubular structures within 24 h, did not proliferate, and stained for amylase. In cultures derived from half of the donors, the acinar markers amylase and CLN3 were upregulated. The PTHSG culture model suggests that human salivary gland may be capable of regeneration via reorganization and differentiation and that basement membrane components play a crucial role in the morphological and functional differentiation of salivary cells.
Introduction
Tran et al. 2 have recently developed a method of obtaining primary salivary cell cultures enriched in epithelial cells from human submandibular glands (huSMGs) not exposed to radiation or chemotherapy and after excluding neoplastic transformation of the submandibular glands themselves. This cell culture was capable of forming a polarized monolayer with high trans-epithelial resistance and expressing occludin and claudin-1 (CLN1). Therefore huSMG culture is considered to display a ductal phenotype. 2
Physiological salivary secretion requires acinar cells; therefore, it is important to know how the acinar cells can be expanded from human salivary gland primary cultures. It has been demonstrated that the intercalated duct is the source of multipotent cells that may give rise to acinar or myoepithelial cells.3,4 Other data have shown that acinar cells may be renewed using autologous cell division. 5
In the last 2 decades, a human salivary gland (HSG) cell line was used to study acinar differentiation.6,7 Basement membrane extract (BME), such as Matrigel, can induce HSG cells to differentiate into acinar structures.6- 8 The advantage of the HSG differentiation model is the uniformity of the cells in the culture, although their neoplastic properties may complicate the interpretation of the experimental results. 6 The occurrence of complete functional differentiation of HSG cells after morphological changes induced by BME is uncertain. Amylase gene expression is frequently unrelated to the acinar morphology,9,10 and HSG cells are not capable of forming a polarized epithelial layer with tight junctions, which prevents initiation of the vectorial water transport process. 11 It is also questionable whether tissue renewal capacity in salivary glands can be modeled using a neoplastic cell line. The obvious next step in the study of salivary differentiation is to investigate whether primary HSG cells, such as huSMG cells, can be used as a progenitor cell source to develop acinar and ductal structures using BME. The use of primary salivary cells from adult patients may also elucidate whether adult salivary glands are capable of regeneration after application of effective stimuli through their progenitor elements. The aim of the present study was to modify the huSMG culturing method to obtain differentiated acinar cells using a basement membrane extract.
Materials and Methods
Source of huSMG
Portions of huSMGs were obtained from the Department of Oral and Maxillofacial Surgery of Semmelweis University (Budapest, Hungary). The samples were collected from patients undergoing head and neck tumor operations with permission from the Regional Human Research Ethical Committee (permission number: 67/2005). Patient care had priority over research use of the specimens. This article describes results from 13 individual salivary glands from 13 patients. Five other samples were used in our experiments to develop the methods described here (data not shown). Patients involved in this study received no prior radiation treatment or chemotherapy.
Primary culture
The culture method is based on the protocol of Tran et al., 2 with modifications. The submandibular salivary gland samples (approximately 2 g) were transferred on ice in RPMI-1640 medium (Sigma-Aldrich, Saint Louis, MO) supplemented with 5% fetal calf serum (FCS) and antibiotic/antimycotic solution (100 U/mL penicillin, 100 mg/mL streptomycin solution, and 2.5 mg/mL amphotericin-B, Sigma-Aldrich). Samples were washed twice in F12 medium (Gibco Invitrogen, Carlsbad, CA) supplemented with antibiotic/antimycotic solution. Larger blood vessels can be removed using tweezers; smaller ones can be eliminated using 70-mm filters before cell plating. Our method does not exclude all platelets and single blood cells, however. Platelets disintegrate in 3 days, other blood cells cannot attach to solid surfaces, and these cells are probably eliminated when the medium is changed.
Samples were then mechanically minced using scissors. The minced tissue was transferred into 30 mL of dissociation buffer and incubated in a water bath for 4 h at 37°C with vigorous vortexing every 30 min. The dissociation buffer consisted of 0.2 U/mL of liberase blendzyme 3 (Roche Diagnostics Corporation, Indianapolis, IN) and 0.1% trypsin (Gibco) in F12 medium. At the end of this incubation period, 200 μL of 50 mg/mL DNase I (Roche) was added, and the cell suspension was pipetted up and down for 2 min. The cell suspension was centrifuged at 230 g for 5 min, the supernatant was discarded, and 20 mL of cold Dulbecco's modified Eagle medium (Sigma) with 10% FCS was used to resuspend the cell pellet. The cell suspension was centrifuged again (230 g, 5 min) and the supernatant discarded. The cell pellet was suspended in 10 mL of HepatoSTIM medium (BD Biosciences, Franklin Lakes, NJ) supplemented with 10% FCS, with antibiotic/antimycotic solution and 1% glutamine (Sigma). This suspension was filtered through a 70-μm cell strainer (BD Biosciences) then plated onto four 60-mm Primaria tissue culture dishes (BD Biosciences). Culture medium was changed twice a week. Two distinct modifications were made from the method used by Tran et al.
FCS was used for coating the Primaria dishes (1 h incubation at 37°C before cell plating) and for supplementing the HepatoSTIM medium. All of the attached cells were used for further passage, in contrast to the original huSMG culturing method, in which the floating aggregates of the 1-day-old culture were used. Therefore the primary cell culture obtained by the modified method is called primary total human salivary gland (PTHSG) in our study.
Subcultures and differentiation
Typically, PTHSG cultures reached confluence in 7 days on plastic culture dishes when cells were passaged using 0.25% Trypsin-ethylenediaminetetraacetic acid (Gibco). PTHSG cells from passages 1 and 3 were used for the differentiation assay. The PTHSG cells were grown on Primaria plastic tissue culture dishes or 12-well dishes pre-coated with basement membrane extract (BME). BMEs were purchased at protein concentrations of 12.8, 13.2, and 17.1 mg/mL (Cultrex BME, Trevigen Inc., Gaithersburg, MD) and 9.1 mg/mL (Matrigel, BD Biosciences). BME was thawed on ice and used to coat plates at 60 μL/cm2. The coated plates were then incubated at 37°C for 2 h before cell seeding.
Cell proliferation and viability assays
Wells of a 96-well plate were coated with 20 μL of BME or FCS (control) and then incubated for 2 h at 37°C. FCS was then removed from the wells, and approximately 3500 first-passage cells were seeded per well in 100 μL. Three days after plating, the cell number was estimated according to metabolic activity using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay (Sigma). The proliferative capacity of PTHSG cells on plastic from different passages was also compared using cell counting, as described below. Two × 105 cells were plated on 35-mm plastic dishes in duplicate. After 3 days, cells were trypsinized and centrifuged at 800 g for 5 min. The cell pellet was resuspended in 1 mL of culture medium, and the cell number was counted from three independent aliquots using a hemocytometer.
Immunohistochemistry
PTHSG cells were plated on 12-mm-diameter microscope glass coverslips (Paul Marienfeld Gmbh, Lauda-Königshofen, Germany) with or without BME. Cells were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 20 min at room temperature and then permeabilized with Triton X-100 (5 min, 0.1% V/V in PBS). Nonspecific antibody binding was blocked using incubation with 5% FCS in PBS at room temperature (90 min). The antibodies for amylase (10 μg/mL polyclonal antiserum, rabbit, Sigma), vimentin (5 μg/mL monoclonal antibody, mouse, Calbiochem EMD Biosciences, Darmstadt, Germany), occludin (0.6 μg/mL monoclonal antibody, mouse, Zymed, San Francisco, CA), CD34 (0.4 μg/mL monoclonal antibody, mouse, FITC-conjugated, Santa Cruz Biotechnology, Heidelberg, Germany), and C-kit (0.4 μg/mL monoclonal, antibody, mouse, phycoerythrin-conjugated, Santa Cruz Biotechnology) were diluted with this solution and incubated overnight at room temperature. Alexa 488- and Alexa 568-conjugated secondary antibodies against rabbit and mouse immunoglobulin G (IgG; Molecular Probes, Carlsbad, CA) were added to samples at a dilution of 1:500 and incubated for 90 min at room temperature. Nuclei were stained with propidium-iodide (0.025 mg/mL, Sigma) in 0.01% Triton X-100 for 30 min at room temperature or 0.01 mg/mL bisbenzimide (Hoechst 33258, Sigma). The stained preparations were evaluated using a confocal laser scanning microscope (Olympus BX61-FV300, Olympus America, Center Valley, PA) or an inverted fluorescent microscope (Axiovert 200M, Zeiss, Jena, Germany). Photomicrographs were taken using an AxioCam camera (Zeiss).
Sections were made for light microscopy. The excised submandibular gland tissue samples were fixed in 4% paraformaldehyde for 48 h. The PTHSGs cultured on BME for 3 days were fixed in 4% paraformaldehyde for 30 min. Both were dehydrated in 70%, 96%, and 99% ethanol followed by xylene and finally embedded in paraffin. Sections were cut at 3-μm thickness on a rotary microtome and then dewaxed and rehydrated. The sections were washed in PBS and counterstained for 1 min in Mayers hematoxylin with EZ Mount mounting medium (ThermoElectron Corporation, Pittsburgh, PA). Photomicrographs were taken using a SPOT camera (Diagnostic Instruments, Sterling Heights, MI) connected to an Eclipse E-600 TMS (Nikon, Tokyo, Japan) phase contrast microscope.
Videomicroscopy
Cell cultures were kept at 37°C in a humidified 5% carbon dioxide atmosphere in 35-mm Greiner Petri dishes within a CellMovie microscope stage incubator (Cellmovie, Budapest, Hungary). The incubator was attached to a computer-controlled Leica DM IRB inverted phase-contrast microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Märzhäuser Scan IM 120 × 100 motorized stage (Leica Microsystems). Images were obtained at 5-min intervals over 1-, 3-, and 6-day periods from three to five neighboring microscopic fields of three parallel cultures using the 10× objective of an Olympus DP70 camera (Olympus, Tokyo, Japan). The automated time-lapse imaging was controlled using software developed at the Department of Biological Physics, Eötvös University (Budapest, Hungary).
RNA quantitation
Total RNA was isolated from PTHSG cell cultures using the RNeasy micro kit (Qiagen Ltd, Crawly, UK) with on-column DNase digestion. The concentration of RNA was determined using Qubit (Molecular Probes). Total RNA (700 ng per sample) was reverse transcribed using the high-capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) with random octamers in 20 μL. For polymerase chain reaction (PCR) amplification, 5% of the cDNA was used with real-time PCR primers and 6-carboxy-fluorescein (FAM)-labeled minor groove binder probes (MGB). The probes and primers for amylase, CLN1, CLN3, vimentin, and the human acidic ribosomal phosphoprotein PO (RPLPO, used as an endogenous reference) were selected from the Applied Biosystems Assays-on-Demand database. PCR reactions (20 μL) were performed in duplicates using TaqMan Universal Master Mix (Applied Biosystems) on a Prism Sequence Detection System 7700 (Applied Biosystems) with the default settings (50° for 2 min, 95°C for 10 min, 45 cycles [95°C for 15 s, 60°C for 1 min]). Every culture experiment was repeated one to six 6 times depending on the amount of the tissue sample. Gene expression levels were calculated by normalizing the target RNA value to the value of RPLPO in the same sample. Results are expressed as fold changes in gene expression relative to a control (untreated) sample.
Statistics
Relative RNA levels and MTT measurements of samples were compared using analysis of variance (ANOVA) and the Bonferroni post hoc tests or Wilcoxon matched pair test where the parametric test was not applicable.
Results
Primary culture and subculture
The mechanically disrupted and enzymatically digested submandibular gland tissue consisted mainly of floating aggregates of cells. After single cells attached, clonal expansion of discrete epithelial fields was observed (Fig. 1A–K). On day 4 of culture, the PTHSG cells grew in discrete patches with a cobblestone-like epithelial appearance; elongated fibroblast-like cells were also observed sporadically (Fig. 1H). Seven days after seeding, PTHSG cultures reached 70% confluence, growing mainly in monolayer, although sporadic three-dimensional (3D) clumps were also found (Fig. 1J). PTHSG cells from passage 1 (Fig. 1L) had a similar appearance to those from passage 0 (Fig, 1A-J), but the 3D clumps diminished. Cells formed a monolayer at passages 2 and 3, and the abundance of fibroblast-like cells increased relative to epithelial cells (Fig. 1M). At all passages of PTHSG cell cultures, cellular senescence was observed after 25 to 30 days of cultivation; eventually, the cells lost their contact with the plastic substrate (Fig. 1K).
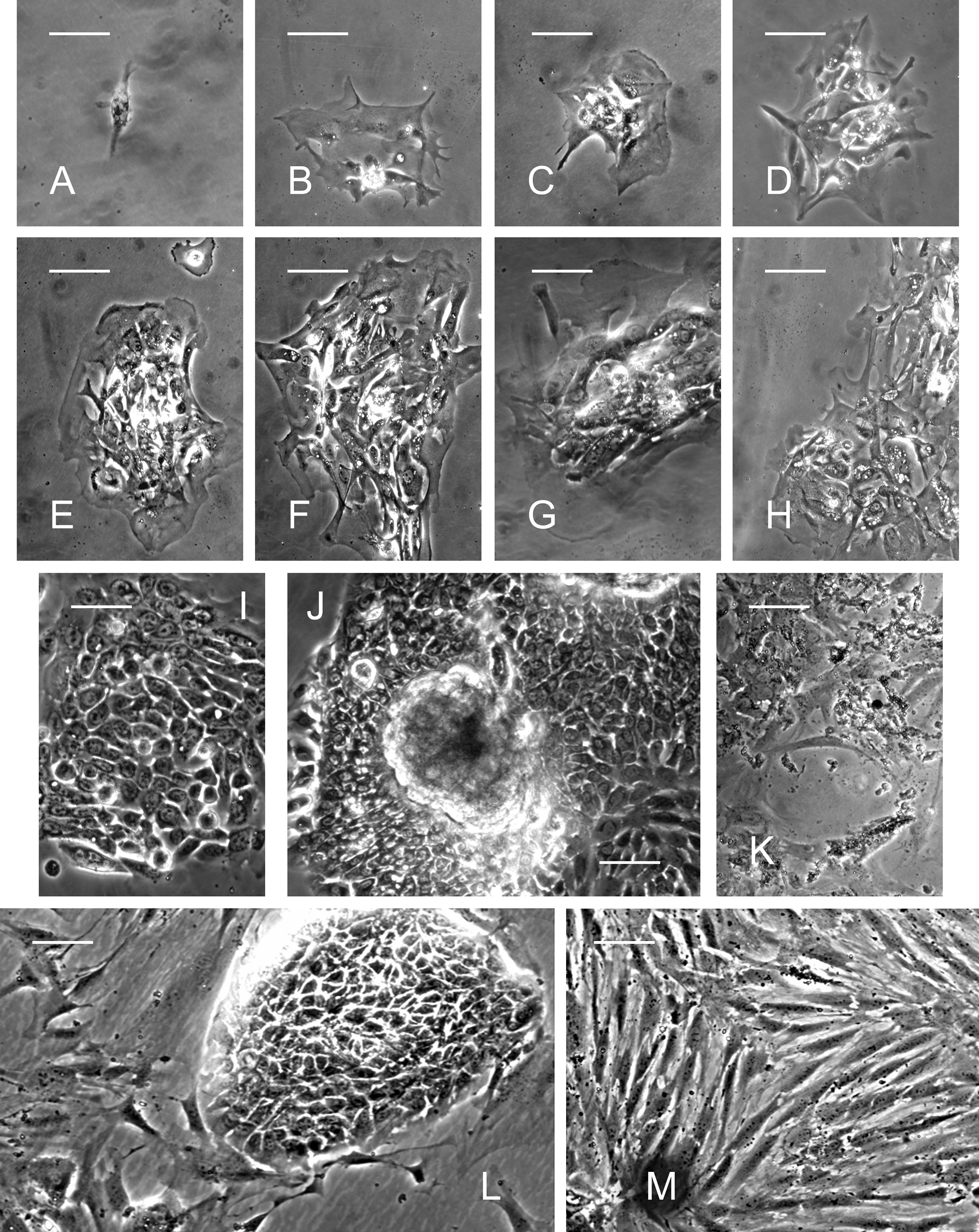
Phase-contrast micrographs of primary total human salivary gland (PTHSG) cells cultured on plastic. Expansion of a cell clone and formation of discrete epithelial fields during 7 days from passage 0 (Day 1,
Morphological changes on the surface of BME
On the surface of growth factor–reduced BME, PTHSG cells from passage 1 formed spherical, acinotubular structures within 24 h. Time-lapse microscopy revealed that cells migrated rapidly after plating, forming small aggregates that developed into acinotubular structures over the course of a day or less (Fig. 2, PTHSG_video_24h.mpg). Additional movement of the cells and some reorganization of the aggregates were observed in the following days (PTHSG_video_3days.mpg and PTHSG_video_6days.mpg). The cell aggregates appeared similar regardless of the type of BME used (Fig. 3A-D).

Captured phase-contrast microscope photographs from a 24-h video showing the dynamics of acinotubular structure formation on the surface of basement membrane extract.
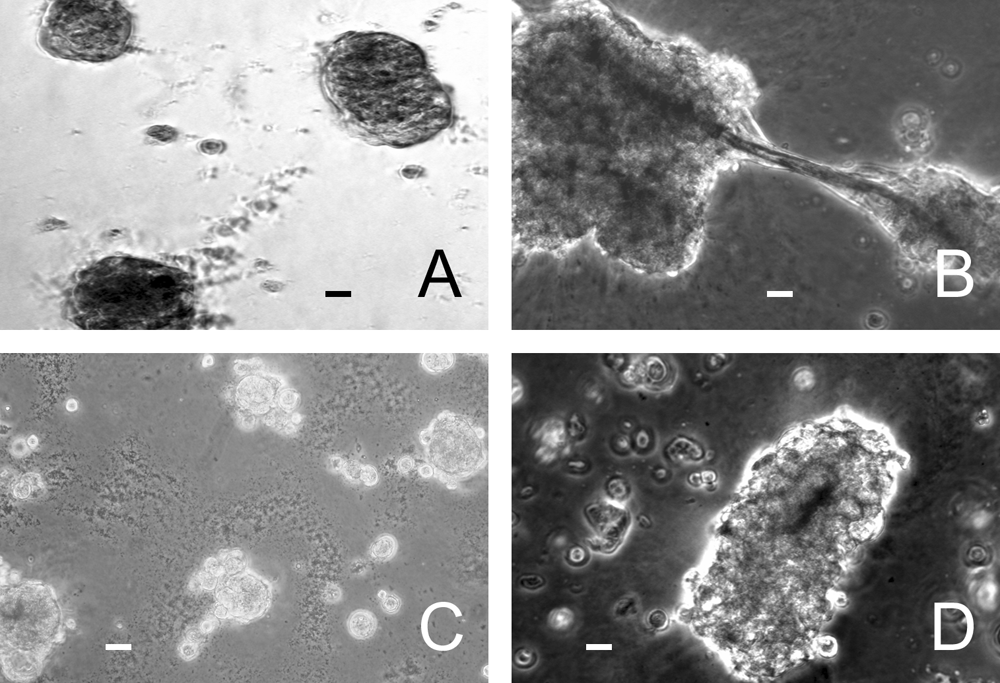
Acinotubular structures on the surface of various basement membrane extract (BME) preparations: Matrigel (9.1 mg/mL,
Sections of paraffin-embedded BME cultures showed ductal networks with lumens and acinus-like structures (Fig. 4B, C), whereas the cells on plastic formed monolayer (Fig. 4A). The acinotubular-type structures detected in most of the sections of cultures derived from 46- to 81-year-old patients appeared more weakly organized (an example is shown in Fig. 4B) than intact submandibular glands (Fig. 4D), although a well-organized salivary gland–like structure was detected in a section of a culture derived from a 20-year-old man (Fig. 4C), with an appearance similar to normal SMG tissue (Fig. 4D).

Hematoxylin-eosin stained sections of paraffin-embedded primary total human salivary gland cells. Monolayer of cells on a plastic surface (
Cell proliferation and viability of PTHSG on BME
Cell viability of PTHSG cultures from passage 1, grown on a plastic surface or on the surface of BME, was evaluated using MTT assay. Five individual experiments were performed on samples originating from different patients. There were significantly fewer active cells after 3 days of cultivation on BME than on plastic (Fig. 5). In a different experiment, the proliferation of PTHSG cells on a plastic substrate over 3 days was estimated using direct cell counting. The changes in cell number PTHSG passages 1 and 3 were compared. The cell number increased similarly in 3 days at passage 1 (126 ± 12%) to passage 3 (112 ± 13%).
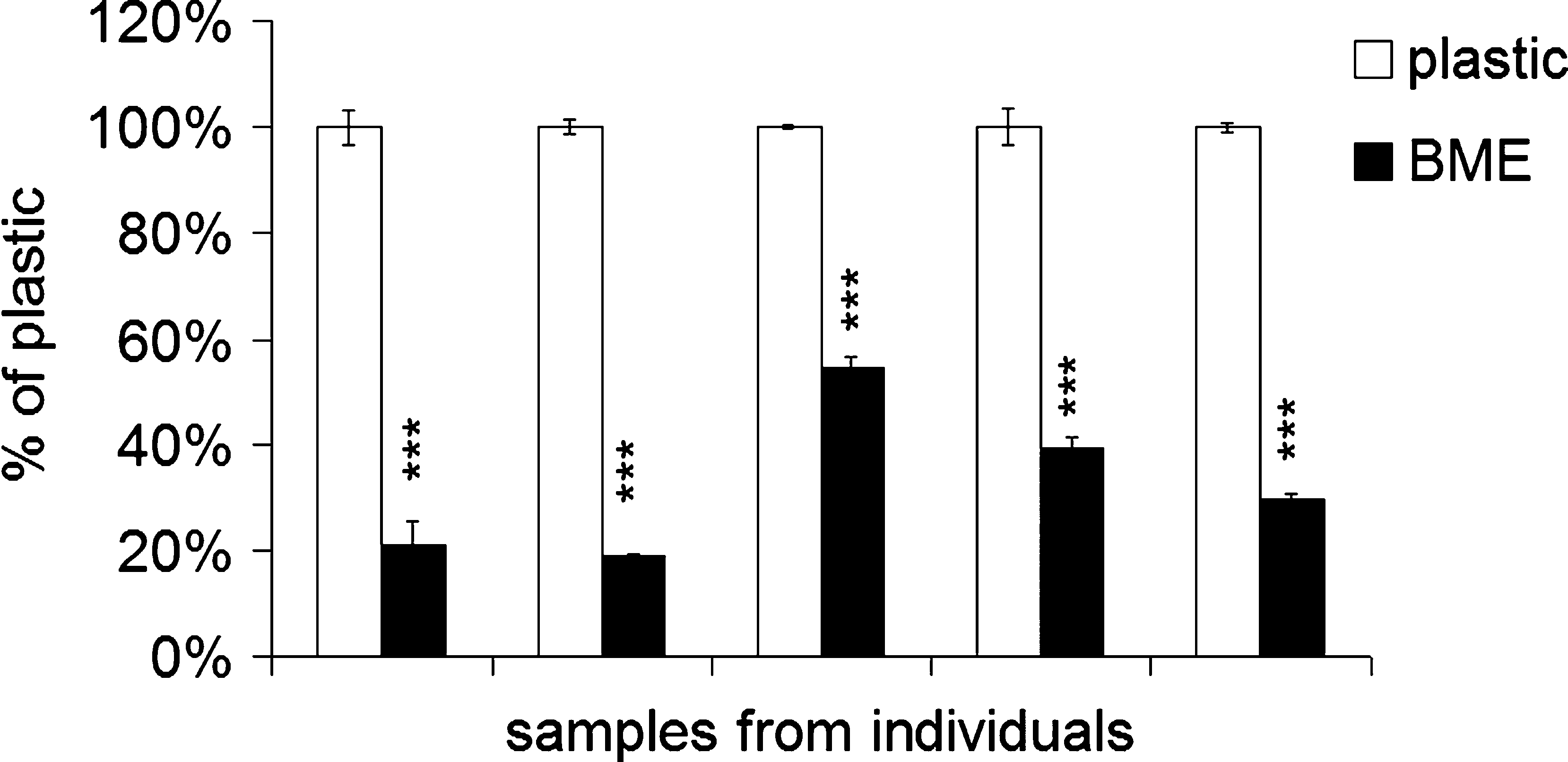
Cell viability of primary total human salivary gland cells cultured on basement membrane extract and plastic surfaces from five independent samples at passage 1 estimated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Results are reported as means ± standard errors expressed as a percentage of the growth on plastic. Data were statistically analyzed using analysis of variance followed by Bonferroni test. ***p < 0.001
Immunocytochemical analysis of amylase, vimentin, C-kit, CD34, and occludin expression
PTHSG cells from passage 0 contained epithelial fields with strong amylase immunoreactivity (Fig. 6A) but also a notable number of weakly amylase-immunoreactive cells (Fig. 6B) on the plastic substrate. First-passage cells grown on plastic showed weak and sporadic amylase immunoreactivity (Fig. 6C), whereas cells with an acinar structure on BME were strongly positive on average (Fig. 6D). Confocal microscopy revealed that the acinar structures consisted of amylase-positive and -negative cells (Fig. 6E). The amylase immunoreactivity of cells on BME was less intense if the cells had first undergone seven passages on plastic (Fig. 6F).
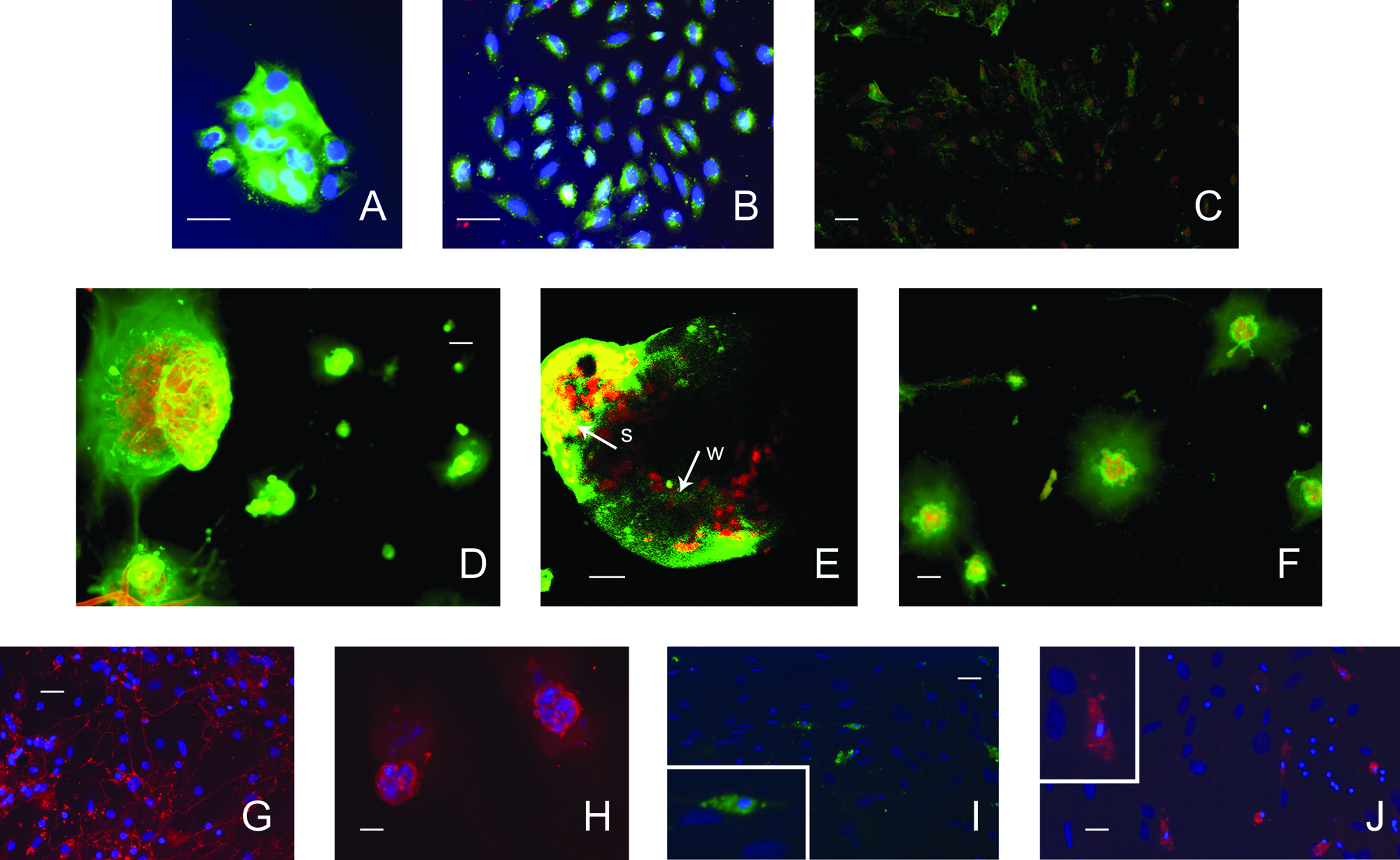
Immunofluorescent staining of amylase protein (Alexa488-labeled secondary antibody) of primary total human salivary gland cells cultured on basement membrane extract (BME) or plastic. Nuclei were counter-stained with Hoechst 33258 (4',6-diamidino-2-phenylindole, blue,
First-passage PTHSG cells displayed occludin immunoreactivity on plastic and BME (Fig. 6G, H). First-passage cultures grown on plastic also contained vimentin-positive cells (data not shown), whereas globular acinar structures on BME were vimentin negative. Primary (passage 0) PTHSG cell cultures contained occasional 3D clumps, which resembled stem cell niches. Some cells within the niche-like 3D clumps had small, dense nuclei and showed C-kit and CD34 immunoreactivity (Fig. 6I, J).
Reverse transcriptase PCR analysis of gene expression in PTHSG
In five patients, the quantity of salivary gland tissue samples was enough for harvesting RNA directly from the tissue and culturing the remaining cells. Amylase mRNA expression in first-passage PTHSG cells cultured on plastic was compared with expression in the tissue. In one case, there was little difference in expression between the primary tissue and the first-passage plastic culture, but in the other four cases, amylase expression decreased dramatically (78- to 2033-fold) between harvest and first-passage culture. Expression in the first-passage cultures varied by more than orders of magnitude between different patients.
The effects of BME, and of the protein concentration of the BME preparation used, on the expression of salivary gland–associated amylase mRNA was examined in first-passage PTHSG cells cultured from 12 different patients, with statistical analysis using two-way ANOVA. Protein concentration of the BME had no effect on the outcome within the range examined (9.1-17.1 mg/mL; p = 0.99, Table 1), whereas cells from different patients displayed a wide range of amylase induction (p < 0.001), and great variability in the expression of other genes (Fig. 7A–E). To estimate the trend in change of gene expression, the data from replicate cultures, from various BMEs, and from the different patients were grouped together, and the average change in expression for each gene was calculated (Fig. 8A). The expression of amylase increased significantly on BME, but no significant changes were found in the mean expression values of the other genes (CLN1, CLN3, kallikrein-1, and vimentin). Half of the samples (cells from 6 patients) showed no substantial increase (fold change was less then 1.5) on BME, and the other half (n = 6) had more than a 2.5-fold increase. Therefore, BME-treated samples were divided into two groups: one with amylase expression considered to be upregulated (at least 2.5-fold increase, Group A) and the other with no change in amylase expression (less than 1.5-fold increase, Group B). In Group A, the CLN3 gene was also upregulated, and the expression levels of other genes were unchanged (Fig. 8B). In Group B, CLN1 was downregulated, and the expression levels of the other genes were unchanged (Fig. 8C).
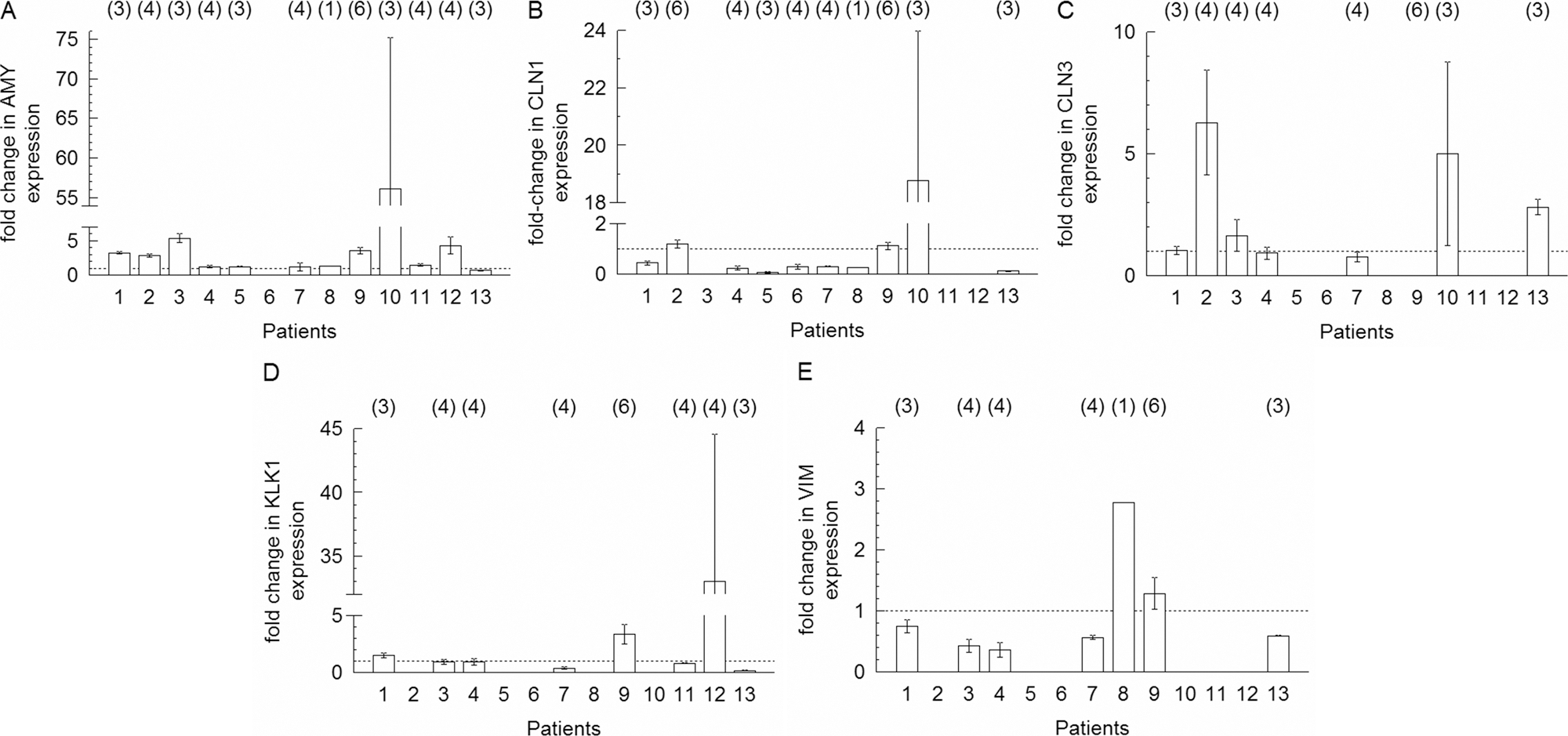
Fold changes in gene expression in cells cultured on basement membrane extract relative to plastic, measured using real-time reverse transcriptase polymerase chain reaction. Fold changes in amylase (

(
The effects were statistically analysed by two-way ANOVA.
Discussion
This study describes a method of culturing isolated huSMG cells to investigate the mechanism of differentiation and morphogenesis of salivary tissue and the possibilities of directing regeneration of salivary gland tissue. Salivary cell populations were initially isolated and cultured according to the protocol of Tran et al. 2 ). The original protocol was changed here because the huSMG cell culture, enriched for ductal phenotype epithelial cells, proliferated slowly on plastic and failed to survive on BME (data not shown). The huMSG method used no FCS in the medium, and the first passage (1 day after isolation) originated only from the floating cell population. This method is supposed to prevent overgrowth of fibroblast cells and resulted in an epithelial cell–rich culture capable of forming a closed monolayer with tight junctions on filters. 2 However, huSMG cell culture may lose its progenitor cell content during the enrichment process or the long-term in vitro cultivation because of the slow growth rate. The PTHSG passage 0 cultures were grown for approximately 1 week on an FCS-coated plate and passaged at 70% confluence. The culture medium consisted of HepatoSTIM with 10% FCS, and all attached cells were used for the following passages and experiments. These conditions, and the observed morphology of the cells in the resulting cultures, suggest that PTHSG cultures contain a number of different cell types derived from the original gland samples. Heterogeneity was observed in the expression of the acinar marker protein, amylase, within PTHSG passage 0 culture, where strong and weak amylase immunoreactivity was seen in neighboring epithelial fields. However, similar to huSMG, 2 the expression of occludin was also observed in PTHSG cells, suggesting that the two cultures contain similar epithelial cell populations. The fibroblast-like cells in PTHSG cultures expressed vimentin, which is a component of intermediate filaments and is characteristic of mesenchymal cells. 12
The PTHSG culture was maintained for a maximum of 1 month. After that, cells began to lose their attachment to the substrate and die, implying that cell aging and mortality may also limit the extent to which these cells can be expanded before terminal differentiation on BME.
In vitro culture appears to cause de-differentiation of PTHSG cells, as suggested by the decrease in amylase immunoreactivity from salivary tissue to PTHSG culture and from passage 0 to 7. De-differentiation of cultivated cells has been observed in other systems (e.g., primary hepatocyte culture) in which number of liver-specific functions was progressively lost with time after isolation. 13 De-differentiation has also been observed when cells extracted from cartilage are expanding in vitro for reimplantation in chondral defects.14–16
The passage 0 cultures formed mostly monolayers, although 3D clumps developed sporadically; these clumps bore a resemblance to stem cell niches (Fig. 1J). 17 Some cells within the niche-like 3D clumps of the PTHSG primary cultures had small, dense nuclei and showed C-kit and CD34 immunoreactivity, which are reported to be stem cell and progenitor cell markers (e.g., adult stem cells from lung have been identified). CD34 + 18 and C-kit-expressing liver progenitor cells from adult mice showed ductal formation after seeding them into Matrigel. 19 Other tissue systems (hair follicle, gastrointestinal tract) support a model in which stem cells must reside in a specialized niche that provides an essential balance of regulatory cell types and factors, enabling division and differentiation.17,20–22 The stem cell pool gradually decreases with passage number in various primary cultures.23–25 Multiplication and differentiation of multipotent salivary stem cells could represent a direction for engineering regeneration of salivary tissues. Salivary gland stem and progenitor cells seem to be present in submandibular glands of neonatal and adult rats; 1 they may be localized to the intercalated ducts.1,3,4 It is our future objective to isolate and identify salivary stem and progenitor cells in adult huSMGs in PTHSG culture usng flow cytometry and to explore the limits to which they can be expanded before differentiation.
In summary, the PTHSG culture appears to contain a mixed population of cells: salivary progenitors, differentiated epithelial cells (acinar, ductal), and mesenchymal cells (such as fibroblasts). It seems the fibroblasts proliferate the fastest, the epithelial cells de-differentiate, and the progenitor cells are the source of the expanding epithelial fields.
According to the phase-contrast microscopic observations, the PTHSG cells organized themselves into salivary-like structures on the surface of BME, regardless of the donor or of the BME protein concentration. In contrast, when ultra-thin (6 mL/cm2) BME was used, no morphological changes were observed, despite the fact that amylase was induced (data not shown). This suggests that the morphological changes of PTHSG culture were based mainly on the mechanical properties of BME, whereas amylase gene induction is more related to the chemical properties of the BME, such as the presence of growth factors. 26 In contrast to PTHSG cultures, the immortalized clonal HSG cell line formed different structures on the surface of BME preparations with different protein concentrations, 10 The multiclonal property of PTHSG culture, which includes fibroblasts, may adapt better to the various BME preparations, possibly via modification of the gel itself.
Reduction in the proliferative rate of PTHSG cultures grown on BME is consistent with the findings of studies on the HSG cell line6,7,10 but conflicting with the organogenesis of mammalian salivary glands, where notable growth happens in all stages, even after the formation of a well-defined lumen at the terminal bud stage. 27 The PTHSG culture is capable of rapid reorganization into salivary gland–like structures with little proliferation. Re-aggregation of dispersed cells to 3D spheres of epithelial layers has also been reported for neural cells, 28 hepatocytes, 29 prostate, 30 and respiratory 31 cells. The present in vitro observation may resemble a possible mechanism of rapid regeneration in vivo, in which the salivary cells are capable of rapid migration to the damaged area followed by the reconstruction of the normal tissue.
The level of organization of the acinotubular structure and the changes in cell type–specific markers (amylase, CLN1, CLN3, kallikrein, and vimentin) on BME varied between cultures derived from different patients. The occludin immunoreactivity of PTHSG cultures on BME was observed on plastic and BME, localized in the acinar-like structure, suggesting the formation of tight junctions. Overall gene expression data and morphological observation showed that BME tends to direct the cells into an acinar form, although it is possible that 3 days is not enough time to clearly define the direction of differentiation. The 6-day-long time-lapse observations suggested that further studies are needed to understand the mechanism of differentiation in PTHSG cells on the surface of BME. Furthermore, a large-scale experiment would be useful to determine the effect of clinical factors such as age, disease, symptoms, and sex on regeneration capacity.
The 3D acinotubular structures were vimentin-negative, suggesting that they consisted mainly of ephithelial-type cells, because vimentin is characteristic of mesenchymal cells, 12 although PTHSG culture on BME contained vimentin-positive cells from time to time. Indeed, the survival of fibroblast cells on BME might be a prerequisite for acinotubular formation. Fibroblast cells product a number of proteins (e.g., fibronectin). 32 An earlier study showed the expression pattern of fibronectin in the teratocarcinoma-embryo model indicates that fibronectin may be a key factor in morphogenetic movements. 33 It is highly likely that fibroblast-like cells epithelial cell activity, as has been clarified in in vivo and in vitro salivary, 34 intestinal, 35 and dental 36 differentiation, as well as in many other investigations. However, the exact molecular identities of growth and differentiation factors, as well as the potential direct cell-to-cell interactions, need to be determined in further studies.
Cultured submandibular gland cells have been transplanted to atrophic and normal salivary glands of rats, where they were selectively attracted to, and remained in, damaged areas without affecting normal tissue. 37 Better cell culture techniques are important to generate sufficient amounts of cells with the capacity to differentiate for replacing damaged salivary tissues. The PTHSG model system, with its capacity to generate acinotubular structures on BME, might have applications in regenerative medicine and progenitor-cell-based tissue engineering because it enhances cell growth due to the FCS treatment and maintains a complex cell population mixture that is suitable for generating differentiated cells. The culture conditions supported by BME probably induce the beginning of tissue regeneration and basic structure formation; PTHSG cells grown in a BME environment might be implanted into immuno-compromised animals to provide an in vivo model for further investigation of the complete regeneration and differentiation of the salivary acinar and ductal system.
Footnotes
Acknowledgments
Contract grant sponsor: Hungarian National Scientific Research Fund (OTKA); Contract grant numbers: NI69008, F49795, T61543, Hungarian Ministry of Education; grant number: OM18657/2005, Semmelweis University Research Grant; grant number: 12117/12055, Semmelweis University Research Fund. The authors wish to thank Elaine M. Byrne (Trinity Centre for Bioengineering, Trinity College, Dublin, Ireland) for her valuable critical comments, Bruce Baum for his helpful suggestions during the development of the project and in the preparation of the manuscript, and Tordainé Szabó Hedvig (II. Department of Pathology, Semmelweis University, Budapest, Hungary) for technical assistance in making sections of the cultured cells. Matt Hoffmann (National Institute for Dental and Craniofacial Research, Bethesda, MD) generously provided BME. We thank Markó Károly (Institute of Experimental Medicine, Hungarian Academy of Sciences, Budapest, Hungary) for help with confocal and fluorescent microscopy.