
Abstract
Taking rapid and efficient formation of functional tissues as our long-term goal, we discuss in this study a new and generic approach toward formation of multilayered three-dimensional (3D) tissues using nanofibers. 3:1 poly (epsilon-caprolactone) (PCL) (8% w/v)/collagen (8.0% w/v) solution was electrospun into nanofibers with an average diameter of 454.5 ± 84.9 nm. The culture of human dermal fibroblasts (NHDF) on PCL/collagen nanofibers showed a high initial cell adhesion (88.1 ± 1.5%), and rapid cell spreading with spindle morphology. Three-dimensional multilayered cell–nanofiber constructs were built with alternating NHDF seeding (1 × 105cells/layer) and PCL/collagen nanofiber collection on site of electrospinning, where almost all the seeded cells retained in the constructs. The formed construct showed layered structure with uniform cell distribution in between layers of PCL/collagen nanofibers. In the 3D constructs, cells continuously proliferated and deposited new extracellular matrix. By culturing either fibroblast/fiber layered constructs or keratinocyte/fibroblast/fiber layered constructs, dermal-like tissues or bilayer skin tissues (containing both epidermal and dermal layers) were consequently produced within 1 week. Taken together, the present study reports a novel approach to 3D multilayered tissue formation using a bottom-up, on-site layer-by-layer cell assembly while electrospinning. This approach has marked potentials to form functional tissues composed of multiple types of cells, heterogeneous scaffold composition, and customized specific microenvironment for cells.
Introduction
Our previous study 7 has indicated that initial uniform cell distribution throughout the entire scaffold with a high cell seeding density is critical to facilitate the new tissue formation and regulate the integrity of formed tissue. Extensive efforts have been made to improve the cell ingrowth into scaffolds by carefully selecting the scaffold materials used, optimizing the scaffold structure, and exploring new cell seeding and culturing techniques.8–13 Significant progress has been made; however, most of the formed tissues are still far from ideal.
Most tissues and organs in our body are usually composed of multiple types of cells and varying extracellular matrix (ECM). The cells together with ECM are arranged/organized in an elaborate and hierarchical order to achieve multiscale functions, and to mutually regulate the cellular activity by soluble bioactive molecules, cell–cell direct contact, or cell–ECM interaction.14–17 The elaborate structure also provides individual cells with a defined microenvironment where the cells experience specific imparted cues and show corresponding responses. To maintain the proper cell phenotype, biomimetic design to microscopically recapitulate the cell residing environment is considered as a superior approach.
Recent development of microfabrication technology enables us to fabricate and modify scaffolds on a micro/nanometer scale. Electrospinning, a high-voltage driven spinning technique, receives a particular attention due to its ability to produce nanofibrous scaffolds, having not only similar dimensions as collagen fibrils in natural tissue matrix, 18 but also various fiber attributes in spatial arrangement (alignment or random) and mechanical properties. 19 A variety of polymers such as poly(lactic-co-glycolic acid) (PLGA), poly-L-lactide (PLLA), poly(epsilon-caprolactone) (PCL), poly(ethylene oxide terephthalate)-poly(butylene terephthalate) (PEOT-PBT), collagen, chitosan, or hybrid20–26 have been successfully electrospun into micro/nanosize fibers. The advantages of these fibrous scaffolds in promoting cell growth and maintaining proper cell phenotype, as well as the differential manipulation of cell attachment by the nanofiber orientation have been demonstrated in a number of studies.21,27–31 However, some challenges in use of nanofibrous scaffolds are also recognized, especially the difficulty of infiltrating cells into the nanofibrous meshes. In response to this challenge together with the consideration of microscale manipulation of cell spatial arrangement and allowing coculture of multiple cell types in the same scaffold, we have developed an on-site layer-by-layer (L-b-L) tissue generation procedure using electrospun fibers. In this approach, cells can be uniformly assembled into multilayered three-dimensional (3D) structure with the assistance of electrospun nanofibers (Fig. 1). In addition, during this L-b-L tissue rebuilding, it would be flexible to vary cell seeding density and cell type for each cell layer and the composition for each nanofiber layer. With the opportunity to precisely control fiber layer thickness, fiber diameter, and fiber orientation, as well as to include bioactive molecules into the fibers for each fiber layer, it is possible to create a specific 3D microenvironment for each cell type within the same construct.
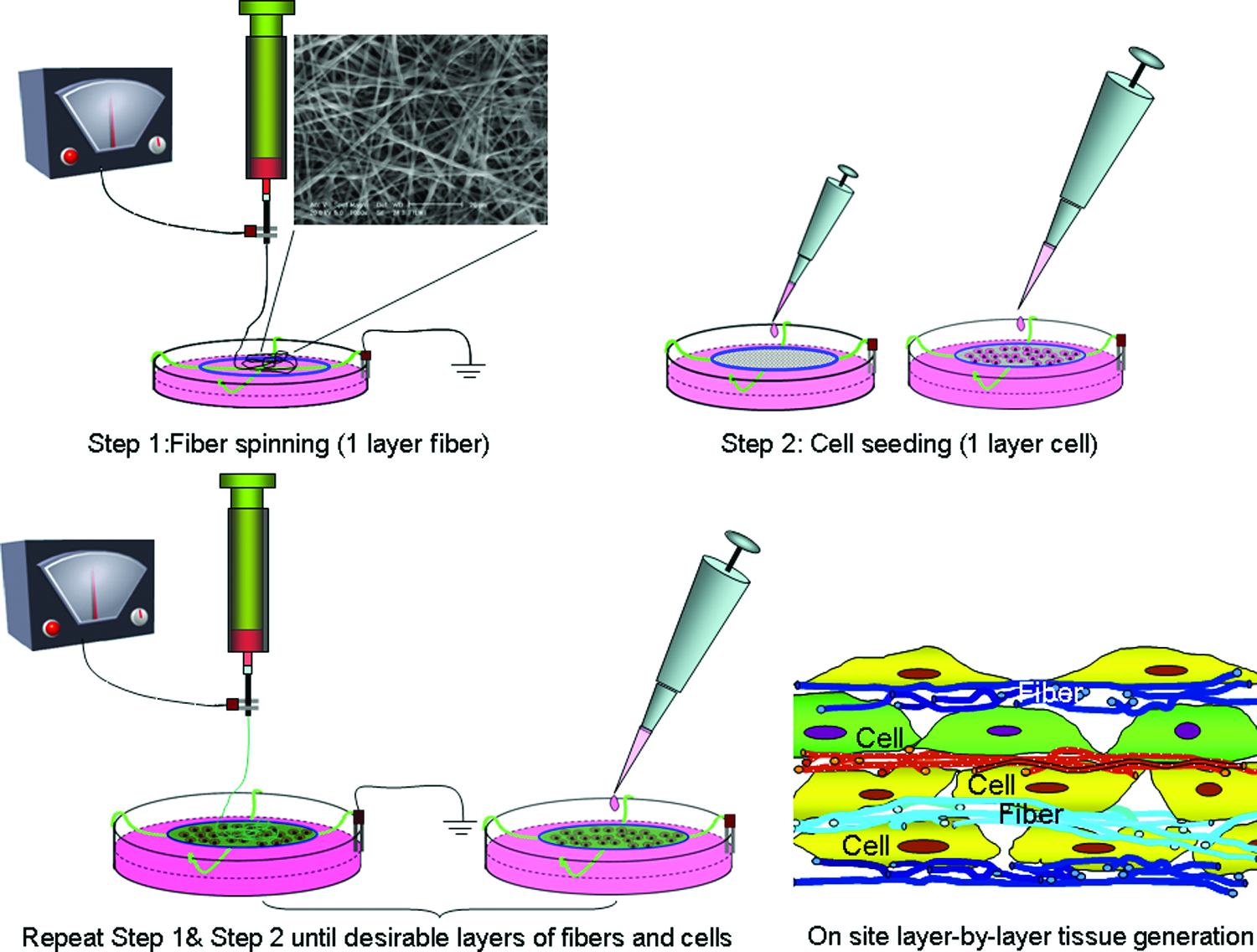
A schematic illustration of the on-site L-b-L cell assembly while electrospinning. As indicated by different colors, both fiber and cell layers can be varied during the cell assembly to create a customized final 3D construct according to the design. Color images available online at www.liebertonline.com/ten.
In this study, electrospun PCL/collagen nanofibers, human dermal fibroblasts (NHDF), and epidermal keratinocytes were used to demonstrate our proposed approach and then to create skin-like tissues using this technique as practical examples. PCL–collagen nanofibers were first deposited and then followed by cell seeding on top of the fibers. By repeating these steps, 3D cell–fiber multilayered structures were produced. Different aspects related to this approach such as cytotoxicity of trace solvent, fiber thickness, cell attachment, and distribution on the fibers were carefully studied. In addition, the 3D cell–fiber constructs were further cultured to understand the time-resolved tissue development.
Materials and Methods
Materials
PCL (Mw = 80,000), fluorescein isothiocyanate (FITC), tetramethylrhodamine isothiocyanate (TRITC)-conjugated phalloidin, and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). Collagen type I was purchased from Elastin Products (Owensville, MO). 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) was obtained from Oakwood Products (West Columbia, SC). Fetal bovine serum (FBS) was purchased from American Type Culture Collection (ATCC, Manassas, VA). All the other reagents and solution were obtained from Invitrogen (Carlsbad, CA) except as indicated.
Electrospinning and characterization
Collagen solution (8% w/v) and PCL solution (8% w/v) were prepared separately by dissolving in HFIP. For collagen–PCL blended solution, one volume of collagen solution and three volumes of PCL solution were thoroughly mixed to obtain a homogeneous mixture. The solution was transferred to a 5 mL syringe attached with a tip-blunt capillary (inner diameter = 0.9 mm). A steady flow of the solution from the capillary spinneret was achieved using a syringe pump from KD Scientific (Holliston, MA) at a flow rate from 5 to 15 μL/min. A high-voltage power supply from Gamma High Voltage Research (Ormond Beach, FL) was used to create electric field strength from 0.8 to 2.0 kV/cm between the tip-blunt capillary spinneret and the collection surface. The typical electrospinning distance between capillary spinneret and collection surface was between 7 and 10 cm. Electrospun fibers were collected on cover slips or aluminum foil for further use.
To characterize the electrospun nanofiber using a scanning electron microscope (SEM), fibers were collected on Si wafer and sputter coated with gold. The coated fibers were examined with a LEO 982 FEG SEM. To determine the diameter of nanofibers, images of five randomly selected areas were captured and analyzed by analysis software (NIS-elements BR 2.30 from Nikon).
FITC–BSA conjugates
FITC–BSA conjugates were prepared according to the manufacture's manual. Briefly, 3.0 mg of FITC was dissolved in 30 mL Hanks' balanced salt solution (HBSS) to make 0.1 mg/mL solution. About 0.8 g BSA was dissolved in 10 mL HBSS. The two solutions were mixed in the dark for 24 h at a pH 7.4 at room temperature to form FITC–BSA conjugates. After dialyzing against D.I. water to remove free FITC, solution was lyophilized. The obtained FITC-conjugated BSA was then stored in the dark at 4°C for future use.
Cell culture and seeding
Normal human dermal fibroblasts (NHDF; passage 1–2) and epidermal keratinocytes (NEK, passage 1–2) were purchased from Cambrex Bio Science Walkersville (Walkersville, MD). Fibroblasts were subcultured in fibroblast growth medium-2 (Cambrex) till passage 4–6 for use in the experiments. Keratinocytes were subcultured in keratinocyte serum-free medium (KSFM; Invitrogen), containing 25 μg/mL bovine pituitary extract, 0.2 ng/mL epidermal growth factor, and 1% penicillin and streptomycin. Cell passaging was performed at a splitting ratio of 1–3 when the monolayer culture reached 70–80% confluence.
To seed the fibroblasts onto electrospun fibers, cells were trypsinized, centrifuged, and resuspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and 1% penicillin and streptomycin at a final concentration of 1 × 105 cells/mL. After seeding of NHDF cells onto the electrospun fibers, cell–fiber constructs were cultivated in DMEM with 10% FBS and 1% penicillin and streptomycin in a humidified incubator at 37°C with 5% carbon dioxide (CO2) for designated times.
Cytotoxicity measurement by the tetrazolium salt (MTT) assay
Cytotoxicity of trace HFIP in the medium was performed using MTT assay. 32 Briefly, conditioned medium samples (1 mL) were added into NHDF cells precultured in a 24-well plate overnight and further cultured for another 24 h. Then, cells were incubated with thiazolyl blue tetrazolium bromide (MTT; Sigma, St. Louis, MO) solution (500 μg/mL) in HBSS for 60 min at 37°C in a CO2 incubator. The dye solution was discarded, and 500 μL dimethyl sulfoxide (DMSO; Sigma) was added to extract the formazan product. Then, 100 μL of the extract was transferred to a 96-well plate. The well plate was agitated for 5 s before reading, and the absorbance was measured at 570 nm with Synergy™ HT Multi-Detection Microplate Reader (BioTek Instruments, Winooski, VT).
Cell viability staining
Live and dead cells were stained with a Live/Dead Reduced Biohazard Viability/Cytotoxicity Kit according to the manufacturer's manual. Briefly, PCL–collagen nanofibrous membranes collected on glass coverslips were seeded with NHDF (2 × 104 cells/cm2). After 24-h culture, cells were washed with HBSS and then stained with a fluorescent dye solution containing both SYTO 10 and ethidium homodimer-2 for 15 min. Excessive dye was removed by washing with HBSS, and the cells were fixed with 4% glutaraldehyde in HBSS. Dead and live cells were examined under a Nikon fluorescent microscope. Nuclei of live cells were stained green by SYTO 10, and those of dead cells were stained red by ethidium homodimer-2.
Cell morphology on electrospun fibers by laser scanning confocal microscopy
About 0.5 mL of NHDF cell suspension (2 × 104 cells/mL) was seeded onto electrospun nanofibers collected on coverslips. After 24-h culture at 37°C in a humidified atmosphere containing 5% CO2, cells were washed with phosphate-buffered saline (PBS) and fixed in 3.7% formaldehyde in PBS for 5 min, and then washed extensively in PBS. Cells were dehydrated with ethanol for 5 min, permeabilized with 0.1% Triton X-100 in PBS, and washed again in PBS. Cells were stained with a 50 μg/mL TRITC-conjugated phalloidin (Sigma) solution in PBS for 40 min at room temperature, and then washed three times with PBS to remove unbound phalloidin conjugate. Then, one drop of Vectashield mounting medium with DAPI was dispensed onto the coverslip. Cells were viewed at ex360nm/em460nm (DAPI) and ex540nm/em570nm (TRITC) using LCM 5 PASCAL with Axiovert 200 confocal microscope from Carl Zeiss.
L-b-L cell assembly and 3D tissue formation
To achieve on-site cell seeding along with electrospinning of nanofibers, particularly to keep the seeded cells hydrated, the electrospinning approach was accordingly modified by directly spinning fibers onto the grounded medium surface, as shown in Figure 1. A high voltage was applied between the tip-blunt needle and grounded culture medium (DMEM with 10% FBS). To assist cell seeding and define the shape of the multilayered cell–fiber construct, a platinum circular wire loop (3 cm in diameter) was used, which was placed right above the medium surface. Using this modified approach, constructs with only fibroblasts (dermal constructs) or with both fibroblasts and keratinocytes (bilayer skin constructs) were created. To create dermal constructs, a layer of collagen–PCL was first electrospun onto the wire loop, and then 1 mL of fibroblast suspension (1 × 105 cells/mL) was evenly seeded onto the fiber mesh. Then, a second layer of collagen–PCL nanofibers was electrospun onto the cell-seeded layer after the medium drained. By repeating the above steps, fibroblast–fiber alternated multilayered constructs were formed. The multilayered constructs were further cultured in DMEM with 10% FBS in a humidified incubator at 37°C with 5% CO2 for designated time. Medium was refreshed every 2–3 days.
To form bilayer skin constructs with both fibroblasts and keratinocytes, 18-layer fiber–fibroblast alternated construct was first similarly prepared as mentioned above. Then, a thin layer of PCL/collagen fiber was electrospun right on top of the fibroblast–fiber layer. One milliliter of keratinocyte suspension (1 × 106 cells/mL) was seeded onto the fibers. By repeating the same procedure, another layer of fiber/keratinocyte was added. Medium was refreshed after electrospinning with coculture medium (3:1 DMEM/F-12 containing 5% FBS, 1% penicillin and streptomycin, 5 μg/mL insulin, 0.4 μg/mL hydrocortisone, and 1 μM isoproterenol). The multilayered cell–fiber construct was cultured in a humidified incubator at 37°C with 5% CO2 for designated times, and medium was refreshed every 2–3 days.
Contraction
Contraction of cultured cell–fiber constructs (both dermal constructs and bilayer skin constructs) was quantitatively determined as the follows. Small punches (Ø = 6 mm; n = 4) were obtained from the assembled cell–fiber constructs (dermal, 10 layers of fibroblasts; bilayer skin, 18 layers of fibroblasts and 2 layers of keratinocytes) after overnight culture, and then continuously cultured. To calculate the contraction of the cultured construct, images were taken at days 1, 3, 7, and 14 using a NIKON SMZ 1500 stereo microscope, and areas were measured using Nikon NIS-element BR software. Contraction was calculated as follows: area at specific time divided by original area (day 1) × 100.
Histological analyses
To visualize the distribution of cells in the fibers, tissue samples were harvested and fixed in 1.5% glutaraldehyde in Dulbecco's phosphate-buffered saline (D-PBS) for overnight at 4°C. Then, the samples were embedded in sample freezing medium (Richard-Allan Scientific, Kalamazoo, MI) and plunge frozen at −50°C. The frozen tissue was sectioned into thin slices (7–10 μm thick) at −25°C with a HM 550 cryostat from Richard-Allan Scientific. The slices were collected onto glass slides, air-dried at room temperature, and stained with either hematoxylin or stained with DAPI (Vector Laboratories, Burlingame, CA). The stained slides were examined under a light microscope or Nikon Eclipse E1000 fluorescence microscope.
For general histology, tissue specimens fixed in 1.5% glutaraldehyde/D-PBS were dehydrated in a graded series of ethanol solutions until 100% ethanol, and then embedded in paraffin and cut into thin sections (3–4 μm thick). The sections were then stained with hematoxylin and eosin (H&E) (Sigma). The stained slides were examined under a Nikon light microscope, and representative images were digitally documented.
Cell proliferation and new ECM deposition
Cultured constructs of both dermal (10 layers of fibroblasts) and bilayer skin (18 layers of fibroblasts and 2 layers of keratinocytes) constructs (small punches Ø = 6 mm; n = 4) harvested at days 3, 7, and 14 were analyzed for cell number and new ECM deposition as previously described. 7 Briefly, constructs digested in proteinase K solution were performed for DNA assay using a commercially available assay kit (CyQuant cell proliferation assay kit) according to the manufacturer's instructions. Glycosaminoglycans (GAGs) content was measured with dimethyl-methylene blue dye. Bare scaffolds were similarly digested and used as controls.
Statistical analysis
Each experiment was repeated at least three times, and data were expressed as the mean ± SD. All the viability and cell attachment measurements were collected in triplicate for each group. Unpaired Student's t-test was used to evaluate the significance between experimental groups. A value of p < 0.05 was considered to be statistically significant.
Results
Collagen/PCL nanofibers and cellular response
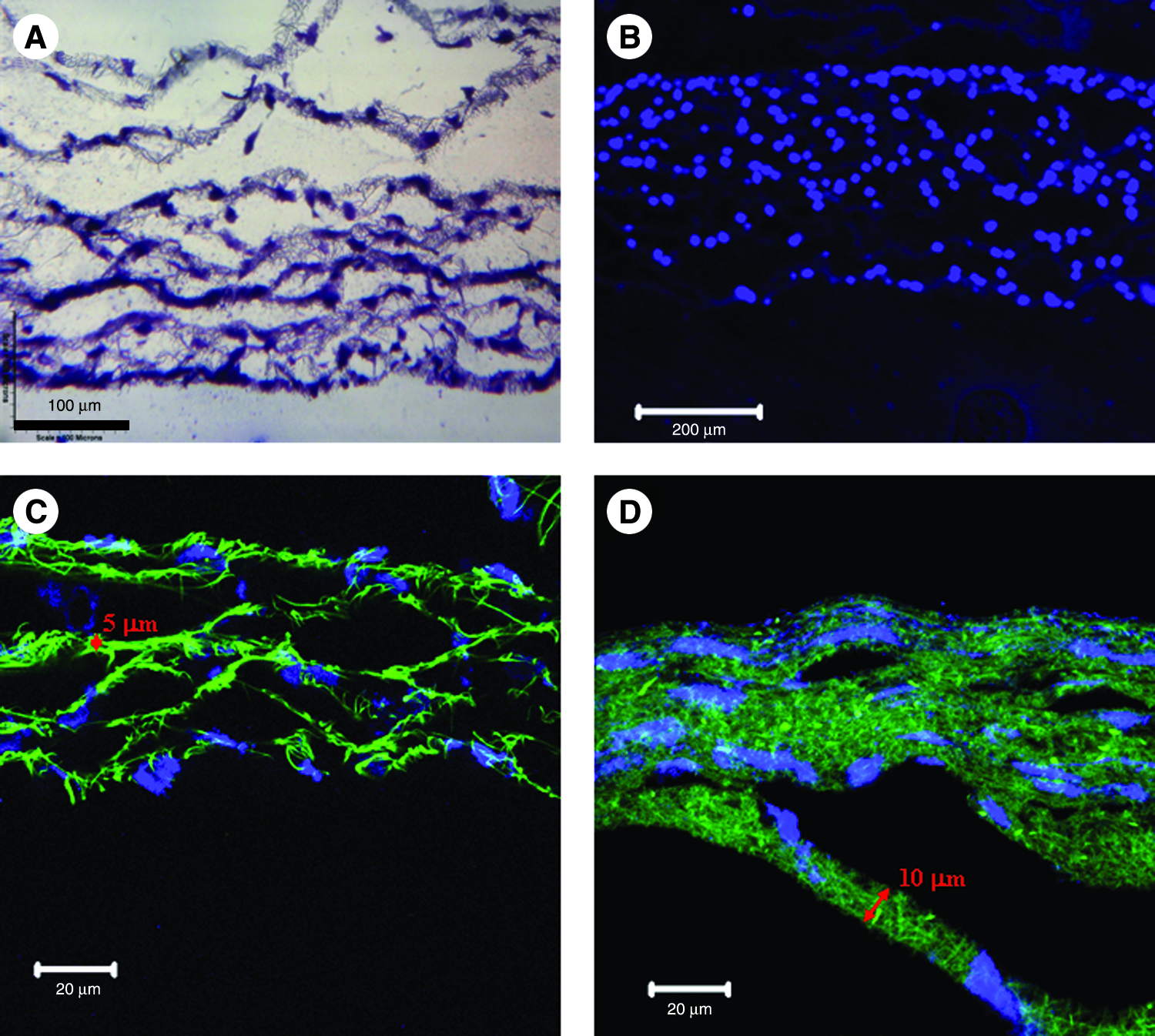
Collagen–PCL blended solution (1:3 in volume ratio) was successfully electrospun into nanofibers using the experimental setup (voltage = 10 kV, collecting distance = 10 cm, and flow rate = 10 μL/min) at the concentration of 7.5–8% w/v, as shown in Figure 2. To determine the surface topography and the average diameter of electrospun fibers, nanofibers collected on Si wafer were examined under SEM after gold sputter coating. The average diameter of PCL–collagen nanofibers was 454.5 ± 84.9 nm, and the surface of electrospun fibers was smooth by close examination (Fig. 2A, inset). As shown in Figure 2A, the pore size, that is, the interfiber space of the PCL/collagen nanofiber meshes randomly collected on Si wafer for 1 min, was noticeably below 5 μm, smaller than the typical detached cell size 6–10 μm. To visualize the distribution of collagen in electrospun fibers, an immediately available FITC-conjugated BSA was used as a substitute model. The direct examination of BSA–FITC/PCL (1:20 w/w) nanofibers collected on a glass coverslip showed a strong and uniform green fluorescence distributed throughout the whole surface of nanofibers (Fig. 2B inset). To determine whether BSA only distributed on the fiber surface, cross sections of FITC–BSA/PCL nanofiber mesh collected on aluminum foil were carefully examined. It was found that green fluorescence was also evenly distributed across the full-diameter of fibers (Fig. 2B), indicating a well mixing of BSA in PCL solution and no separation after electrospinning.

Electrospun 3:1 PCL (8% w/v)/collagen (8.0% w/v) nanofibers and the cell attachment. (
Adhesion and spreading of fibroblasts on the randomly collected nanofiber meshes were analyzed by MTT assay and actin staining. MTT assay results revealed that a high initial cell attachment (88.1 ± 1.5%) to PCL/collagen meshes was achieved after incubation for 12 h, in comparison to tissue culture plastic surface (taken as 100%). Fluorescence staining of cytoskeleton protein, F-actin, showed that HDFB spread nicely on the surface of PCL/collagen fibrous meshes (Fig. 2C, D) and remained spindle-like morphology. Cells were mainly retained on the mesh surface (Fig. 2C), and no migration into nanofibrous meshes was observed. Intracellular actin filaments and contact points between cell and nanofiber were also observed at a high magnification (Fig. 2D). Necessary to mention, electrospun PCL/collagen fibers also showed weak fluorescence though they were not fluorescently labeled.
Wet electrospinning and cytotoxicity of trace solvent in electrospun fibers
One key step toward the formation of 3D tissue using the approach as shown in Figure 1 was to collect the electrospun fibers on medium surface and to hydrate the fibers. Thus, a wet electrospinning was set up. Instead of using a solid metal surface, grounded medium was used to collect electrospun fibers. It was found that the fibers could deposit directly onto the medium surface. The morphology of meshes was similar to those collected on the metal surface (data not shown). In addition, the PCL/collagen fibers rapidly hydrated after depositing on the medium surface and remained on the medium surface.
Most of HFIP would evaporate during electrospinning, but trace amounts might still remain. To test if this would cause any adverse effect to cells, cells were seeded and cultured on electrospun nanofibers right after they were collected on liquid surface or on the glass coverslips without further vacuum treatment as previously reported. 22 The staining of cells with a live/dead kit showed negligible cell death, and most cells remained viable (Fig. 3A). To further study the potential toxicity of trace HFIP in fiber, especially those collected on the medium surface, nanofiber meshes collected on medium surface were further incubated in the same collecting medium. The medium was collected at different times and tested for cytotoxicity by MTT assay. The MTT result showed that no significant cytotoxicity was observed in all collected medium samples (Fig. 3B).

Cytotoxicity of electrospun nanofibers using live/dead staining kit (
L-b-L on site fabrication of layered fiber–cell structure
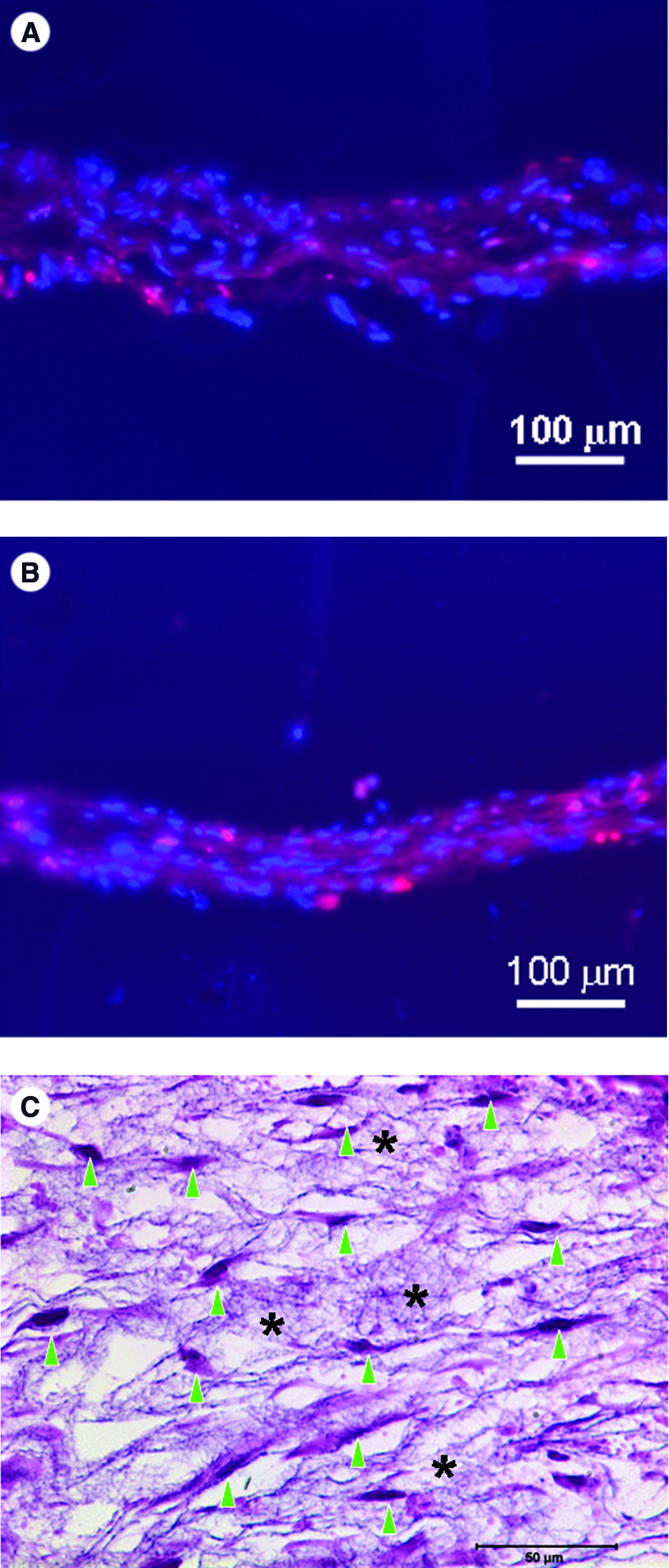
To test the feasibility of seeding cells onto electrospun fibers during the wet electrospinning, two-layer construct with alternating NHDF (1 × 105 cells/layer) and PCL/collagen/BSA–FITC fibers was first built on the surface of medium (DMEM containing 10% FBS) following the procedures as illustrated in Figure 1. After overnight culture, cells attached to PCL/collagen fibers and clearly showed two separate layers with uniform spatial distribution (Fig. 4A). Interestingly, in another experiment to layer dermal fibroblasts using alternative PCL/collagen and PCL nanofibers, selective attachment of fibroblasts to PCL/collagen nanofibrous layers was observed (Fig. 4B), indicating that the presence of collagen favors the cell adhesion. To determine if all the seeded cells remained on the nanofiber meshes, medium used for wet spinning was collected at the end of the experiment and counted for cells. Cell counting results indicated that cells did not leak out of the fiber mesh and remained on nanofiber layers.

Confocal microscopic images of cell–fiber constructs cultured for 24 h after seeding. (
In separate experiments, NHDF and PCL/collagen fibers were similarly L-b-L built into a 15-layer cell–fiber constructs and cultured for 2 days. Optical microscopy examination of the cross sections stained with hematoxylin clearly showed the presence of multiple cell layers among electrospun fibers (Fig. 5A), and the cells were elongated and embedded among fibers. To better visualize the cell spatial distribution across the full thickness of the multilayered constructs, frozen cross sections were stained with DAPI for cell nuclei and examined under a fluorescent microscope. Figure 5B shows 3D and yet homogeneous distribution of cells across the full thickness of the construct. Figure 5C and D are the cross sections of seven-layer fibroblast–fiber constructs but with different electrospinning time for the PCL/collagen/FITC–BSA fiber layers. When the electrospinning time was 30 s, around 5-μm-thick fiber layer was yielded (Fig. 5C). When the electrospinning time was increased to 1 min, the thickness of nanofiber layer was correspondently increased to around 10 μm (Fig. 5D). The final shape of the cell–fiber construct was defined by the metal wire loop (circular, and square loops were tested) and remained unchanged during the extended culture period (Fig. 6).
Microscopic images of multilayered cell–fiber constructs. (

Images of multilayered cell–fiber constructs cultured for 7 days on metal wire loops. Circle diameter and square length = 3 cm. Color images available online at www.liebertonline.com/ten.
3D skin tissue formation from L-b-L–built cell–fiber construct
As shown above, fibroblasts could be uniformly L-b-L assembled into the electrospun fibrous meshes along electrospinning and form a 3D construct. To further study the tissue formation of these multilayered constructs, 10-layer fibroblast–PCL/collagen fiber constructs were further cultured for 3 and 7 days. The cross sections of cultured constructs were stained with DAPI and examined under a fluorescent microscope. It was found that the cell–fiber construct became compact over a prolonged culture (Fig. 7A, B). To better visualize the cell distribution and morphology, cross sections of the constructs cultured for 7 days were stained with H&E and examined under a microscope. Fibroblasts showed elongated morphology and evenly distributed among fibers (Fig. 7C). No clear layers could be recognized any more at this time. The cultured constructs were also studied for the contraction, and the result revealed that continuous contraction occurred in the cultured dermal constructs (Fig. 8a). Compared to its original size, more than 40% contraction was observed by 14 days. Quantitative analyses of cultured constructs for both cell number and new ECM (GAG) clearly showed that fibroblasts continuously proliferated and deposited new ECM in the cultured dermal constructs (Fig. 8b, c), but the largest increase was observed by 14 days.
Formation of dermal tissue from fibroblasts/fiber layered constructs. Fluorescent images of 10-layer fibroblasts/PCL–collagen fiber constructs (see “Materials and Methods” section) cultured for 3 days (

Quantification of the contraction (
Using the similar L-b-L approach, fibroblasts and keratinocytes were built into the PCL/collagen nanofibers with 18 layers of fibroblast/fiber in the lower part and two layers of keratinocyte/fiber on the top. After culturing for 3 days, a bilayer skin construct with both epidermal layer (E) and dermal layer (D) was clearly seen on the H&E-stained cross sections (Fig. 9A). Seeded keratinocytes remained on the construct surface and formed a continuous epidermal layer, and fibroblasts retained in the lower part with a uniform distribution. A tight binding between epidermal layer and dermal layer was observed. At this time, two layers of keratinocytes still could be recognized by careful examination, but it became invisible by day 7 (Fig. 9B). The bilayer structure remained the same even after culture for 7 days, except that both the epidermal layer and dermal layer became thicker (approximately 50% increase for both layers). This could be partially attributed to the contraction observed (Fig. 8a). The area of the cultured bilayer skin constructs remained unchanged between 7 and 14 days (Fig. 8a). Quantitative analyses for total DNA and GAG were also performed on the cultured bilayer skin constructs to study the new tissue formation. It was found that cells continuously proliferated and new ECM (GAG) was deposited in the constructs (Fig. 8b, c). The individual contribution from fibroblasts and keratinocytes was not discerned.
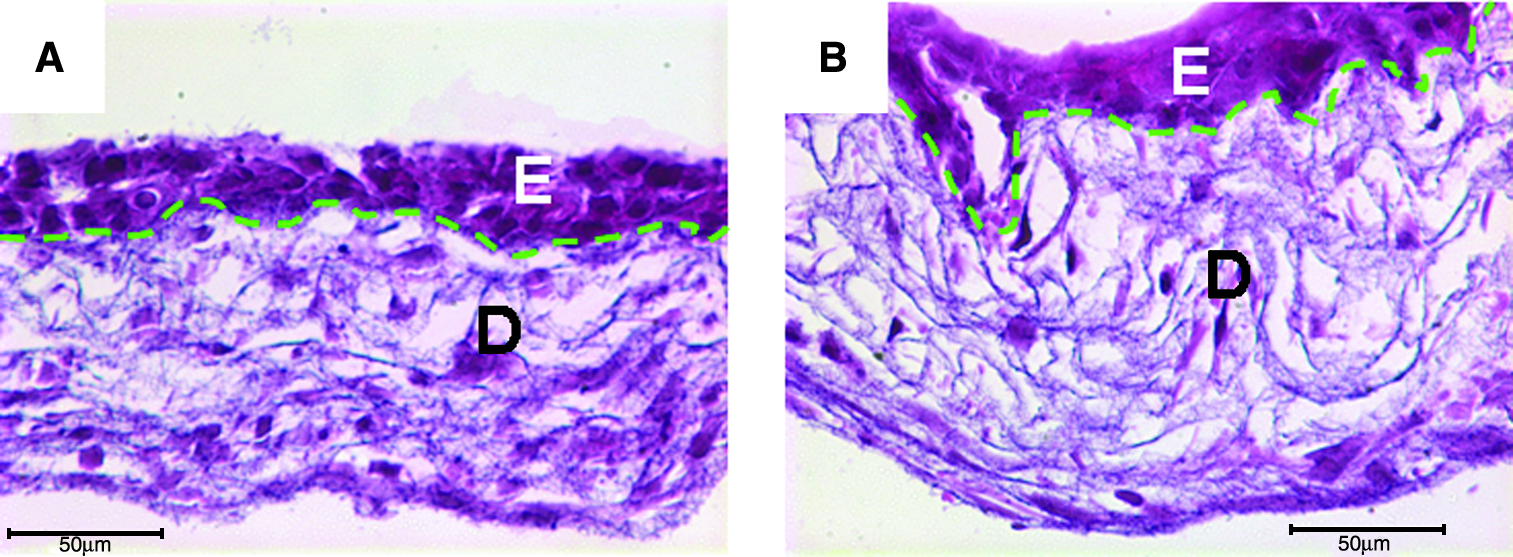
H&E-stained cross sections of bilayer skin tissues composed of epidermal (E) and dermal (D) layers and formed by culturing L-b-L cell-assembled constructs (see “Materials and Methods” section) for 3 days (
Discussion
PCL/collagen nanofibers favorable to skin cells
The advantage of electrospun nanofibers in promoting cell adhesion and maintaining cell phenotype, as a result of its dimensional similarities to ECM fibrils, has been reported.18,33 As a temporary substrate to support cell growth and retain the tissue shape in tissue engineering, the nanofibrous scaffold should have both bioactivity and good mechanical property. In this study, electrospun nanofibers of PCL blended with collagen were considered superior to either pure PCL or collagen nanofibers, having both biological activity and mechanical stability. The biological superiority was demonstrated by the high cell adhesion (88.2%) and rapid cell spreading with spindle-like morphology on its surface (Fig. 2). The promotion of cell adhesion onto PCL/collagen fibers mainly results from the presence of collagen that can interact with cells through cell membrane integrins such as α2β1.34,35 Pure collagen can be electrospun into nanofibers;25,35 however, its rapid degradation often requires a crosslinking step to maintain its structural integrity. In comparison to PCL only, PCL/collagen nanofibers represent a better mimicking of the native ECM in both topology and composition. The advantages of PCL/collagen hybrid fibers have also been reported in the recent studies on the support of glial cells and fibroblasts.36,37
L-b-L cell assembly into 3D constructs with a uniform cell distribution
Despite the advantage of PCL/collagen nanofibers in favoring cell adhesion and proliferation,36,37 how to infiltrate cells into the nanofibrous meshes remains a major challenge. As measured in this study, the interfiber distance (i.e., pore size) of PCL/collagen meshes is less than 5 μm, smaller than the diameter of detached cells, and this can constrain the cells penetrating into the meshes (Fig. 2C) and most likely lead to tissue formation only in periphery.38,39 Some attempts have been made to improve the cell infiltration by using enzyme-degradable natural polymers, 25 fabricating meshes with pore size large enough for cells to migrate in, 40 or coelectrospraying with cells. 41 Due to the intrinsic existence of nutrition gradient from exterior to interior, 42 the improvement by scaffold design is still limited. Coelectrospraying shows promises in bringing cells inside of the fibers, yet with some drawbacks, including less control of cell distribution as well as using high voltage for cell electrospraying. 41 We find that L-b-L cell assembly technique presented in this study represents another promising solution to this hurdle. Following the procedures shown in Figure 1, L-b-L alternation of cell seeding and nanofiber deposition has resulted in a 3D cell–nanofiber construct with homogeneous cell distribution in between the nanofibers (Fig. 5A, B), which is similar to in vivo tissues where cells are embedded in ECM fibers. In addition, almost all the seeded cells were retained in the fibrous meshes, which is highly beneficial especially in autologous tissue engineering to minimize the biopsy size needed for cell isolation. The well control of fiber composition for each layer (Fig. 4B) and the thickness of fiber layer (Fig. 5C, D), as well as the free change of cell types for each cell layer have implied its promising applications in generating layered tissues such as skin and vessels or creating cell-specific microenvironment for engineering hierarchical tissues. Necessary to mention, layer separation was observed in the prepared cell–fiber 3D constructs (Fig. 5A), but this became quickly invisible over the time of culture, and integrated tissues were formed by day 7 in our study (Fig. 7C). This is ascribed to the continuous increase of cells and newly formed ECM (Fig. 8), both act as “glue” and “space filler” to bridge the void space between layers. In addition, electrospinning of nanofibers onto medium surface specifically developed for our 3D cell assembly not only guarantees the seeded cells to be hydrated and viable on the fibers throughout the entire process, but also allows a rapid and uniform cell attachment.
Creation of 3D microenvironment by L-b-L approach
Culture of cells in a 3D environment is evidently favorable to maintain cell phenotypic fate. 43 The in vivo microenvironment for each cell type varies from tissue to tissue and from site to site, and this variation provides and conveys specific cues to cells for specific function. 2 Our L-b-L cell assembly approach provides the cells with a 3D microenvironment right at the moment of cell seeding (Fig. 4). In particular, the use of nanofibers expectedly leads to a 3D cell anchorage. 44 Although there is less control of the exact intercellular distance in the same cell layer (X–Y), where to some degree it can be manipulated by varying the cell seeding density, this approach offers precise control of the distance between neighboring cell layers by controlling the fiber-collecting time for each fiber layer. As shown in Figure 5C and D, at a constant flow rate, when doubling the electrospinning time from 30 s to 1 min for each nanofiber layer, the thickness of nanofiber layers was also correspondently doubled. Thus, spatial arrangement of cells in a 3D environment can be achieved by this L-b-L cell assembly.
The homogeneous inclusion of FITC–BSA into electrospun fibers (Fig. 2B) also offers the opportunity to create a microenvironment with specific bioactivity by including growth factors in the nanofibers and locally releasing to the attached cells, regardless a rapid release has been reported previously. 45 In addition, it has been reported that scaffolds with sequential composition or porosity can be prepared by nanofibers.46,47 In this study, we have demonstrated that it is flexible to vary cell types for each layer while cell seeding and allow integrating multiple cell types within the same construct (Fig. 9). All these together demonstrate the potentials of the L-b-L cell assembly approach in creation of a cell-specific 3D microenvironment by carefully selecting nanofiber composition and incorporating bioactive molecules. This would be a useful tool to study cell–cell and cell–matrix interaction in a 3D environment similar to that in vivo, but with more defined parameters.
Rapid formation of 3D tissues by L-b-L cell assembly
Rapid tissue formation is always preferred in order to treat the patients in time, especially for those of time-sensitive cases. Since L-b-L cell assembly approach can yield an initial uniform cell distribution, high seeding efficiency, and fibroblast-favorable environment, we, therefore, hypothesize that a rapid skin tissue formation will take place for the L-b-L built constructs. It was found that a uniform dermal tissue was formed by culturing 10-layer fibroblast/nanofiber constructs for 7 days (Fig. 7C), which in morphology was very similar to the tissue that was created by culturing the fibroblasts in a porous poly-(ethylene glycol terephthalate)-poly(butylene terephthalate) (PEGT/PBT) scaffold for 14 days under an optimal condition. 7 The significant reduction of time might be a result of multiple factors involving cell–cell and cell–nanofiber interaction. Necessary to mention, continuous contraction was observed in the cultured dermal constructs (Fig. 8a), and this could result from the continuous deposition of new ECM molecules (GAG) (Fig. 8c) and proliferation of fibroblasts (Fig. 8b), as the fibroblasts can generate contractile force to the attached matrix. 48 In addition, the deposition of other ECM molecules (e.g., collagen 49 ) may also contribute to the contraction of the constructs. In creation of autologous bilayer living skin grafts containing both epidermal and dermal layers, a “two-step” approach is normally employed, that is, separately forming epidermal sheet and dermal layer, and then combing to form the bilayer grafts after additional culture, which requires a prolonged time (about 3 weeks). In the present study, a bilayer skin construct was created by one step—that is, L-b-L cell assembly of dermal fibroblasts with PCL/collagen nanofibers first and then on top of it assembling two layers of keratinocytes. After culture for only 3 days, a clear bilayer skin construct similar to native skin structure (except nonstratified epidermis) was observed with tight binding between both layers (Fig. 9A). The tight connection can come from the active keratinocyte–fibroblast interaction or strong cell–fiber binding. This finding indicates that the time needed to create bilayer skin grafts can be significantly reduced to less than 1 week, implying a great clinical impact on the current burn treatment. With the extension of culture time, no significant change in morphology was observed, except a mild contraction (Fig. 8a) and new tissue formation (Fig. 8b, c). Interestingly, the early contraction observed in the bilayer skin constructs did not continue with the extended culture, suggesting that the presence of epidermal layer could inhibit the contraction. Keratinocytes have been reported to stimulate the contraction of fibroblast-enriched collagen gel, 50 where they still remained as individual cells; however, in this study the seeded keratinocytes formed a continuous epidermal layer as early as day 3. The epidermal sheets have been reported to inhibit the wound contraction in the in vivo study. 51
Conclusion
A new on-site L-b-L cell assembly approach toward 3D tissue formation was proposed and experimentally proven by using PCL/collagen nanofibers and human skin cells in this study. In this approach, multilayered construct with variable compositions and cell types could be assembled in an alternating manner, that is, alternating cell seeding and nanofiber collection on site of electrospinning (Fig. 1). This new approach has the advantages in providing a uniform cell distribution throughout the nanofiber scaffolds, significantly increasing the cell seeding efficiency (100%), freely formulating the composition of each fiber layer, and allowing coculturing multiple cell types with controlled spatial arrangement. A continuous new tissue formation was observed in the L-b-L–assembled dermal or bilayer skin constructs and was time dependent. The time needed to form skin tissues can be significantly reduced by using this approach. To our knowledge, this on-site layering process for tissue formation is reported for the first time and can provide a useful tool for engineering tissues with complex hierarchical architecture and for in vitro studying cell–cell or cell–material interaction.
Footnotes
Acknowledgments
The study was financially supported by the startup funds from Stevens. The authors would like to thank Dr. Arthur Ritter for his scientific review and important discussion on this manuscript, and also thank Dr. Eugenia Kharlampieva and Veronika Kozlovskaya for their technical help in use of confocal microscope, and Dr. Robert Clancy in providing the histological sectioning.