
Abstract
Cellular self-organization studies have been mainly focused on models such as Volvox, the slime mold Dictyostelium discoideum, and animal (metazoan) embryos. Moreover, animal tissues undergoing regeneration also exhibit properties of embryonic systems such as the self-organization process that rebuilds tissue complexity and function. We speculated that the recreation in vitro of the biological, biophysical, and biomechanical conditions similar to those of a regenerative milieu could elicit the intrinsic capacity of differentiated cells to proceed to the development of a tissue-like structure. Here we show that, when primary mouse embryonic fibroblasts are cultured in a soft nanofiber scaffold, they establish a cellular network that causes an organized cell contraction, proliferation, and migration that ends in the formation of a symmetrically bilateral structure with a distinct central axis. A subset of mesodermal genes (brachyury, Sox9, Runx2) is upregulated during this morphogenetic process. The expression of brachyury was localized first at the central axis, extending then to both sides of the structure. The spontaneous formation of cartilage-like tissue mainly at the paraxial zone followed expression of Sox9 and Runx2. Because cellular self-organization is an intrinsic property of the tissues undergoing development, this model could lead to new ways to consider tissue engineering and regenerative medicine.
Introduction
The self-assembling peptide RAD16-I, which forms promising three-dimensional (3D) scaffolds for tissue-engineering applications, has been used to promote growth and proliferation of multiple cell types, including chondrocytes, hepatocytes, endothelial cells, osteoblasts, and neuronal cells, as well as embryonic and somatic stem cells.11–18 In a recent work, we demonstrated that, after culturing primary mouse embryonic fibroblasts (MEFs) into a th3D self-assembling peptide scaffold for several days, they up-regulated osteopontin (OPN) and two metalloproteinases (MMP-2 and MMP-9), known as type IV and V collagenases or the 72-kDa gelatinase A and 92-kDa gelatinase B, respectively, characteristic of an embryonic regenerative system.19,20 Moreover, osteoinduced MEF 3D cultures were able to develop mineralized matrix according to von Kossa staining but not 2D cultures or 3D culture controls. Alkaline phosphatase activity (ALP) was highly expressed in all 3D cultures, indicating that the 3D environment promotes ALP activity independently of the osteogenic conditions. Because the RAD16-I peptide scaffold does not contain any specific peptide-signaling motif, we define this environment as “non-instructive” from the point of view of cell receptor recognition and activation (integrin- or growth factor–like receptor), suggesting that the 3D environment per se promotes cellular responses. Finally, the system produced collagen type I and up-regulated the expression of runt-related transcription factor 2 (Runx2), suggesting that the cells acquire an osteoblast-like phenotype. 21 Additionally, other groups have shown previously that mesenchymal cells from bone marrow origin, mouse embryonic stem cells, and mouse embryonic fibroblasts can be differentiated into cartilage-, fat- and bone-like tissue but under specific inductive medium conditions.22–30
The in vitro cellular system we describe here undergoes a process that resembles many aspects of animal development, including cell aggregation, proliferation, migration, and tissue specification and, most importantly, morphogenesis and pattern formation.
Materials and Methods
Culture of MEFs
MEFs isolated from C57BL/6 embryos at day 14 were purchased from the American Type Culture Collection (scrc-1008; ATCC, Manassas, VA). These fibroblasts (<9th passage) were cultured in 75- or 175-cm2 flasks in fibroblast medium (FM), which contains high-glucose Dulbecco's modified Eagle medium (DMEM; 15-013-CV; Mediatech, Manassas, VA) supplemented with 15% (v/v) fetal bovine serum (FBS; 14-501F, Lonza, Allendale, NJ), 4 mM L-glutamine (82007-322, VWR, West Chester, PA), 100 μM minimum essential medium non-essential amino acids (11140-050, Invitrogen, Carlsbad, CA), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (20001, JR Scientific, Woodland, CA). In some cases, the medium was supplemented with 10 to 20 nM of insulin-like growth factor (IGF)-I (I8779 Sigma-Aldrich, St Louis, MO) and 1 to 20 nM of platelet-derived growth factor (PDGF)-BB (recombinant protein from rat, P4056 Sigma-Aldrich).
3D culture technique
For experiments with RAD16-I, MEFs (<9th passage) were trypsinized from the culture flasks, suspended in 20% sucrose (S0389, Sigma-Aldrich). Commercial 1% RAD16-I (Puramatrix, Becton & Dickinson, Franklin Lakes, NJ) was diluted with water to obtain a solution of 0.5% RAD16-I. Then equal volumes of the cell suspension in 20% sucrose and RAD16-I (1% or 0.5%) were mixed to render a final cell suspension containing 2 × 106 MEFs/mL, 10% sucrose, and either 0.25% or 0.5% of RAD16-I. Alternatively, freshly isolated MEFs were suspended in 10% sucrose and mixed with 0.5% RAD16-I in 10% sucrose, to obtain a final MEF concentration of 2 × 106cells/mL, 10% sucrose, and 0.25% RAD16-I. This suspension (80 μL) containing cells and RAD16-I was loaded into 9-mm-diameter cell culture inserts (PICM01250, Millipore, Billerica, MA) previously placed inside 6-well culture plates (Nunc, Rochester, NY). Then 500 μL of FM was added to the wells, outside the inserts. The medium penetrates the insert from the bottom membrane, inducing a bottom-to-top self-assembly of RAD16-I and thus the formation of a gel with MEFs suspended inside the scaffold. After the gel was formed, 500 μL of FM was added on top of the hydrogel. The FM flows trough the gel, washing out the remaining sucrose (Fig. 1A). This step was repeated two more times. Finally, 500 μL of FM was loaded inside the insert and 2.5 mL of FM in the well outside the insert. Incubation was performed at 37°C with 5% carbon dioxide, and medium was changed every 2 days.
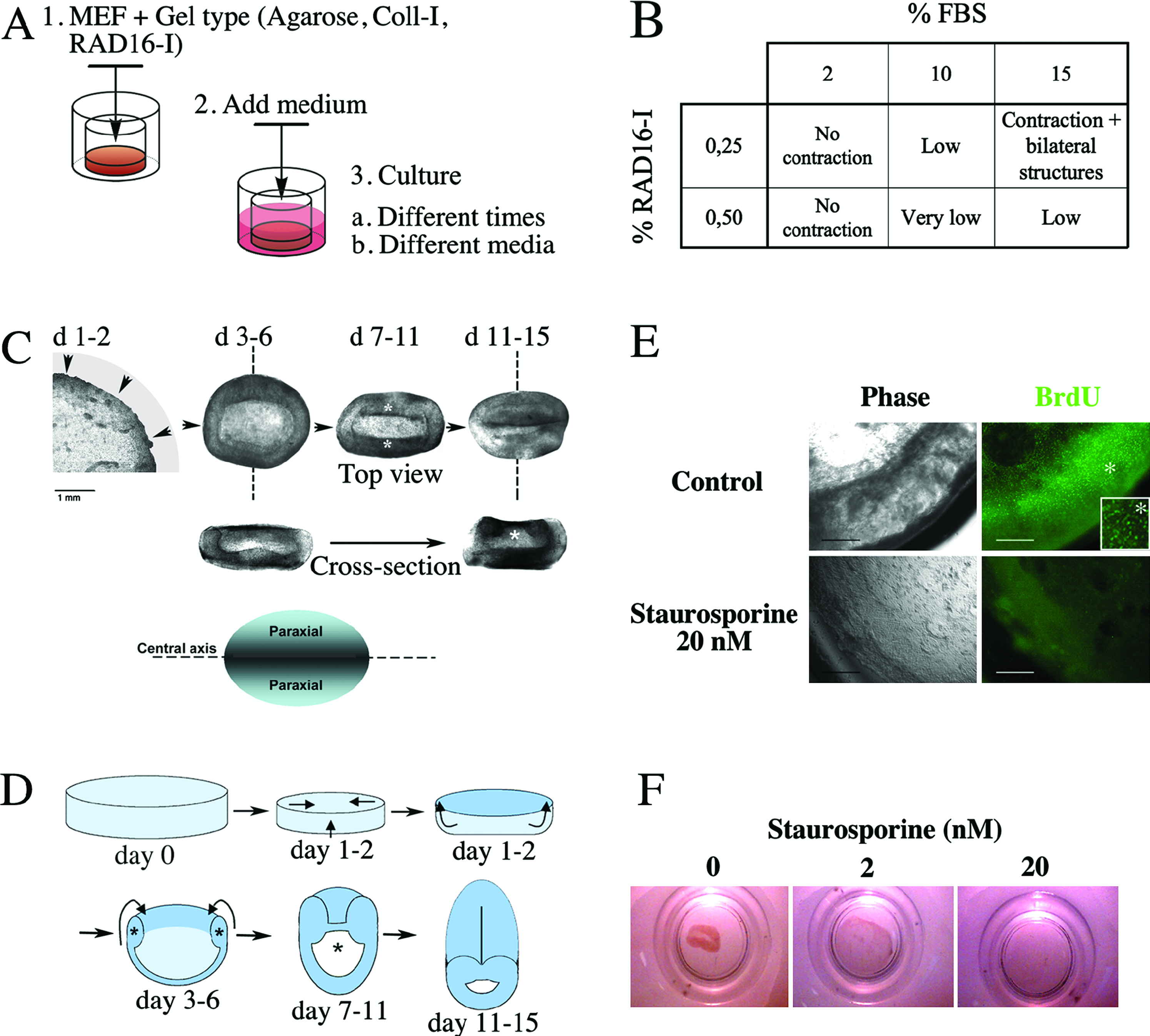
Morphogenetic changes of mouse embryonic fibroblasts (MEFs) in three-dimensional (3D) cultures. (
For the experiments with agarose, the same cells were suspended in FM. This cell suspension was mixed with an equal volume of 0.5% Omnipur agarose (EM Science, Gibbstown, NJ) in 10% sucrose (S0389, Sigma-Aldrich) to obtain a final cell density of 2 × 106 cells/mL in 0.25% agarose. The cell suspension was briefly cooled until the agarose rendered the gels, which were then equilibrated in FM and incubated as described above.
For the experiments with collagen type I, the same cells were suspended in FM. Isotonic collagen 0.4% was prepared by diluting commercial collagen type I (354236, Becton & Dickinson) with filtered 10× phosphate buffered saline (PBS; 14040, Invitrogen) and sterile water (Mediatech). This solution was neutralized with sterile 1 M sodium hydroxide (Sigma-Aldrich) using phenol red (P4633, Sigma-Aldrich) as an indicator. Imediately after neutralization, equal volumes of the freshly prepared collagen solution and the cell suspension were mixed, to obtain a final cell density of 2 × 106 cells/mL in 0.2% collagen, and poured inside cell culture inserts. The suspension was heated at 37°C so that the collagen fibers formed a gel inside the inserts. Gels were cultured in FM and incubated as described above.
Differentiation assays
MEFs were cultured in the 3D hydrogels with FM for 12 days, and after that, medium was changed to differentiation medium. MEFs were maintained in differentiation medium for 21 days. As controls, MEFs were cultured for 33 days in regular fibroblast medium. For osteogenic differentiation, the medium was high-glucose DMEM with 10% FBS, 1% (v/v) penicillin streptomycin, and 4 mM L-glutamine supplemented with Lonza's osteogenic factors: 0.05 mM ascorbate, 10 mM β-glycerophosphate, 0.1 μM dexamethasone, and 20 nM 1α,25-(OH)2 vitamin D3 (PT-3002, Lonza).
The medium to induce chondrogenic differentiation was provided by Cambrex (Charles City, IA) and contained basal medium supplemented with R3-IGF, transforming growth factor beta (TGF)-β, insulin, transferrin, FBS, and gentamycin/amphotericin. The concentrations of the supplements were not available from the manufacturer.
For the adipogenic differentiation, the medium was DMEM with 10% FBS, 8 μg/mL biotin, 4 μg/mL pantothenate (C8731, Sigma-Aldrich), 0.5 mM 3-isobutyl-1-methylxanthine (I7018, Sigma-Aldrich), 1 μM dexamethasone (PT-3002, Lonza), and 10 μg/mL insulin (CC-4182, Lonza).
Antibodies
Anti-digoxigenin (DIG) sheep polyclonal antibody (11333089001) was purchased from Roche Diagnostics (Indianapolis, IN). All other antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-collagen type II (sc7764), anti-brachyury (sc17743), anti-nestin (sc21248), and anti-actin (sc1615) goat polyclonal antibodies and anti-PEBP2αA (sc10758; also known as anti-Runx2) and anti-Sox9 rabbit polyclonal antibodies were used as primary antibodies. Anti-sheep (sc2473), anti-goat (sc2020), and anti-rabbit (sc2317) donkey antibodies horseradish peroxidase (HRP)-conjugated and fluorescein isothiocyanate (FITC)-labeled chicken anti-goat (sc2988) antibodies were used as secondary antibodies.
Cell-cycle arrest assays with staurosporine
Cultures of MEFs were incubated in FM with 20 nM staurosporine (569397, Calbiochem, Gibbstown, NJ) for 7 days while controls were incubated in FM. At day 6, a pulse of bromodeoxyuridine (BrdU) was applied overnight (16 h). MEFs were fixed at day 7 and stained with an anti-BrdU FITC-conjugated mouse monoclonal antibody immunoglobulin G 1 (Becton, Dickinson and Company). To assess the dose-response effect of staurosporine on MEFs, these were incubated with different concentrations of staurosporine (0, 2, 20 nM) for 14 days. The cell mass contraction was visually inspected during these 14 days.
Phenotype assignment
To assign osteoblastic, chondrogenic, or adipogenic phenotypes, we stained for specific elements of each phenotype. MEFs underwent the previously described differentiation protocols and were stained for calcium deposition (Von Kossa), for glycosaminoglycan (GAG) synthesis (Toluidine blue), and for lipidic accumulation (Nile Red) to assess osteogenic, chondrogenic, and adipogenic commitments, respectively. Stainings were performed as previously described. 21,31,32
Western blot analysis
MEFs cultured in different conditions were subjected to lysis in radioimmunoprecipitation assay buffer (BP-115D, Boston Bioproducts, Worcester, MA) containing protease inhibitors (Complete-Mini, Roche Diagnostics). Two-dimensional cultured MEF lysates were used as control protein samples in osteogenic differentiation assays. Cell lysates from these cultures were subjected to NuPAGE (NP0321, Invitrogen), transferred onto a polyvinylidene fluoride membrane (Invitrolon, Invitrogen) and blocked using tris buffered saline/Tween with 5% dry milk. PEBP2αA, collagen type II was blotted with the indicated antibodies. Actin was used as an internal protein standard.
Real-time reverse transcriptase polymerase chain reaction
mRNA was isolated from MEFs using Trizol (15596-026, Invitrogen), cultured in RAD16-I, and cultured in two dimensions (controls) at days 3, 7, 11, and 15. After degradation of cellular DNA, first-strand cDNA was synthesized from the mRNA using Taqman Reverse Transcriptase (N808-0234, Applied Biosystems, Foster City, CA). Real-time reverse transcriptase polymerase chain reaction (RT-PCR) reactions were performed in SYBR green PCR master mix (204143) using commercial primers of Sox9 (QT00163765), Runx2 (QT00102193), Col2a1 (QT01055523), Col1a1 (QT00162204) and Col1a2 (QT01055572) (all from Qiagen, Valencia, CA). Relative gene fold variations were all determined according to the 2−ΔΔCt method using the ribosomal unit 18S (QT01036875, Qiagen) as a housekeeping gene.
Reverse transcriptase polymerase chain reaction
mRNA was isolated from MEFs using Trizol (15596-026, Invitrogen), cultured in RAD16-I, and cultured in two dimensions (controls), at days 3, 7, 11, and 15. After degradation of cellular DNA, 0.2 μg of mRNA was retrotranscribed with Omniscript (205111, Qiagen). The cDNA obtained was amplified using PCR (95°C 2 min, 35 cycles of 95°C 20 s, 58°C 30 s, and 68°C 30 s) using Accuprime Pfx (12344-024, Invitrogen). The primers were kindly provided by Dr David Shaywitz, Harvard University (forward: 5’-CATGTACTCTTTCTTGCTGG-3’; reverse: 5’-GGTCTCG GGAAAGCAGTGGC-3’).
GAG quantification
GAGs were quantified using 1,9-dimethyl-dimethylene blue (DMMB; 23481-50-7, Polysciences, Warrington, PA). The samples were treated overnight with pronase at 60ºC and centrifuged at 14,000 g. The supernatant (40 μL) was incubated with 360 μL of a solution of DMMB (0.16% DMMB in 0.2% formic acid [251364, Sigma-Aldrich] with 2.5 mg/mL sodium formate [71541, Sigma-Aldrich]; pH 3.5) and read in a spectrophotometer with visible light (λ = 535 nm). 33 Standards for chondroitin 6 sulfate from shark cartilage (27043, Sigma-Aldrich) were used to prepare a calibration curve with values between 0 and 100 μg/mL.
Immunostaining of MEFs cultured in RAD16-I
MEFs cultured in RAD16-I for 7 to 15 days with FM were fixed with 2% PFA and washed twice with PBS 1 × prepared from 10 × PBS (EM-6508, VWR). Endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in PBS Tween 20 at room temperature for 30 min. Samples were blocked with 20% (v/v) FBS in PBS with 0.1% (v/v) Triton X-100 (T-9284, Sigma-Aldrich) and 1% (v/v) dimethyl sulfoxide (O81-1, Honeywell Burdick and Jackson, Morristown, NJ). Incubation with the indicated primary antibodies was performed overnight at 4°C. After a 24-h washing step, samples were incubated overnight at 4°C with the secondary HRP-conjugated antibodies. Localization of proteins was finally revealed by reaction with diaminobenzidene (DAB) substrate (1718906, Roche Diagnostics).
Brachyury in situ hybridization
MEF cultures in RAD16-I were fixed at days 7, 11, and 15. In situ hybridization was performed whole mount and onto 14-μm slices of the tissue-like cell mass obtained after culturing. Slices were obtained by cryosectioning of the cell mass with a Leica CM 3050 S cryostat using optimal cutting temperature compound (Tissue-tek, VWR) as freezing support. The DNA probe used for the in situ hybridization was synthesized with the PCR DIG probe synthesis kit (11636090910, Roche Diagnostics) using primers kindly provided by D. Shaywitz (forward primer: CATGTACTCTTTCTTGCTGG; reverse primer: GGTCTCGGGAAAGCAGTGGC). The in situ hybridization was performed following the company's instructions for embryos and for tissue sections. After hybridization of the DIG-labeled probe, samples were immunostained for DIG using anti-DIG sheep antibody and anti-sheep HRP-conjugated antibody. Reaction with DAB substrate (1718906, Roche Diagnostics) showed the localization of brachyury mRNA.
Results
Fibroblasts suffer a morphogenetic process when cultured in RAD16-I
To understand the influence of the biomechanical and biological environments on the behavior of fibroblasts in the peptidic nanofiber scaffold RAD16-I, we varied the concentration of peptide used to form the scaffold (0.25-0.5%) and the concentration of FBS (2-15%) added to the medium (Fig. 1B). We observed that, when cultured in 0.25% RAD16-I and 15% FBS, fibroblasts engage in a morphogenetic process that renders a compact cell mass with a central axis and two thick parallel structures at each side of the central line (Fig. 1C). During the first 1 to 2 days in culture, the cells contracted the scaffold from a disk shape to a much smaller, flat, dense disk. Four to 6 days after encapsulation, the edge of the disk continued the contraction, compacting its perimeter and turning it into a wheel-like shape with a semicircular cross-section or dome shape. Next, two diametrically opposite zones at the edge of the wheel or dome started actively contracting inward, converging at the center and compressing both sides. As a consequence, the compaction of the cell masses from each side of the dome caused a merging zone that formed a “middle line.” This process seemed to elongate the body along the axis, producing two large, dense paraxial structures, which resulted in a 3D bilateral assembly 11 to 15 days after encapsulation (Fig. 1C). At this point, the cell mass had gone through the main morphological changes. For better understanding, we show a schematic representation suggesting the main morphological process that the system undergoes to develop into a 3D bilateral structure (Fig. 1D). Optical cross-sections at two time points suggested the formation of an internal cavity as a result of this morphogenetic process (Fig. 1C). The final shape presents bilateral symmetry (3D bilateral) and resembles some aspects of a vertebrate embryo undergoing axis formation. The development into a bilateral structure happened in 66% of the cases estimated after performing three independent experiments. In addition, we have found that the quality and origin of the serum affects success. To normalize those differences, we are currently testing the addition of growth factors naturally present in serum, such as PDGF and IGF-1. Preliminary results indicate that the addition of PDGF has a positive effect on the contraction and structure development (not shown). This is consistent with other investigations that show how PDGF and TGF-β enhance fibroblast-mediated collagen gel contraction.34,35 In fact, we eventually observed the contraction of collagen type I gels mediated by MEFs (Supplemental Fig. G, H, available online at www.liebertonline.com/ten), although the formation of a bilateral structure did not accompany these contractions. Moreover, PDGF-α has been shown to promote chondrogenesis at early stages of limb development, 36 which may in some way be related to the effect that PDGF exerts on the system described here.
Proliferation is required to complete the morphogenetic changes
To further understand this phenomenon, fibroblasts, cultured as explained above, were treated with staurosporine (an inhibitor of the protein kinase C, that arrests cells in G1). Staurosporine clearly abrogated the development of the 3D bilateral structure. Figure 1E shows that no evident incorporation of BrdU was detected in staurosporine-treated samples, whereas control samples exhibited high proliferation in the outermost circle of the cell mass. Moreover, a dose-response inhibitory effect on the 3D bilateral structure development can be observed when diluted concentrations of staurosporine are added to the cultures (Fig. 1F). These results indicate that, during this morphogenetic event, the cell density increases not only because of contraction, but also because of proliferation, suggesting that the system needs to reach a critical cell mass for this phenomenon to occur.
Fibroblasts have multipotentiality and default chondrogenic commitment in RAD16-I
Fibroblasts have been shown to have multipotential capacity to differentiate into mesenchymal lineages.21–30 To further assess the multipotentiality of MEFs, we evaluated the osteogenic, adipogenic, and chondrogenic capacities of fibroblasts cultured in the RAD16-I nanofiber scaffold, in agarose (a non-instructive 3D environment), and in collagen type I (the natural extracellular matrix (ECM) of dermal fibroblasts and bone). We questioned whether these three chemically unrelated matrices promoted the acquisition of mesenchymal potentiality, as observed before in RAD16-I. 21 Osteogenic, adipogenic, and chondrogenic phenotypes were assessed according to Von Kossa, Nile red, and toluidine blue staining, respectively. Figure 2H shows that fibroblasts undergo acute osteogenesis in all 3D environments after induction with osteogenic supplements, which the high extracellular deposition of calcium salts indicates (Fig. 2A, D and Supplemental Fig. G–K available online at www.liebertonline.com/ten). Osteogenesis occurred only with the addition of osteogenic supplements, whereas adipogenesis occurred with (not shown) and without adipogenic supplements (Fig. 2C, F, G). Chondrogenesis arose only in the self-assembling peptide scaffolds, with (Supplemental Fig. C) and without chondrogenic supplements (Fig. 2B, E and Supplemental Fig. F), indicating that there is some relationship between chondrogenesis and the biomechanical and non-instructive characteristics of RAD16-I.
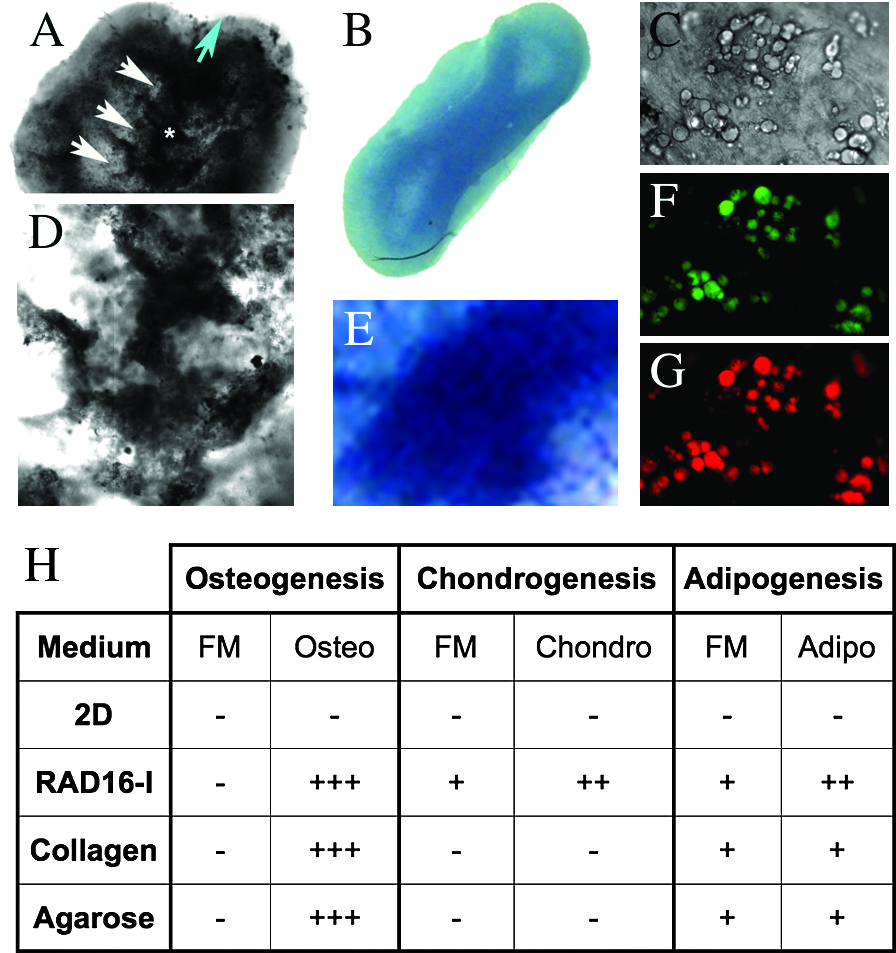
Multipotencial capacity of mouse embryonic fibroblasts (MEFs). (
Western blot analysis revealed that the pre-osteogenic transcription factor Runx2 was more upregulated in MEFs cultured without inductive supplements in the nanofiber scaffold RAD16-I than expression levels of the same cells cultured in 2D (Fig. 3A). Then the addition of differentiating supplements caused an upregulation of the expression of Runx2 in 2D cultures, whereas in 3D cultures, Runx2 remained constant. Runx2 is a transcription factor present in hypertrophic chondrocytes (which mineralize the ECM only when osteochondral ossification begins37,38) and normally during osteogenesis. 39 In our system, mineralization happens in 3D cultures only in the presence of osteogenic supplements. Moreover, the expression of Runx2 is upregulated in 3D cultures with and without osteogenic induction, suggesting that, in our system, fibroblasts develop by default all the way to hypertrophic chondrocytes but not to osteoblasts. This implies that other factors known to induce osteogenesis in hypertrophic chondrocytes are not present in our system (e.g., the vascular cross-talk signaling). two-dimensional cultures of MEFs induced with osteogenic factors upregulated the expression of Runx2 but failed to mineralize the matrix, suggesting that, in the 3D system, in addition to the upregulation of Runx2, there must be other factors that determine the osteogenic commitment.
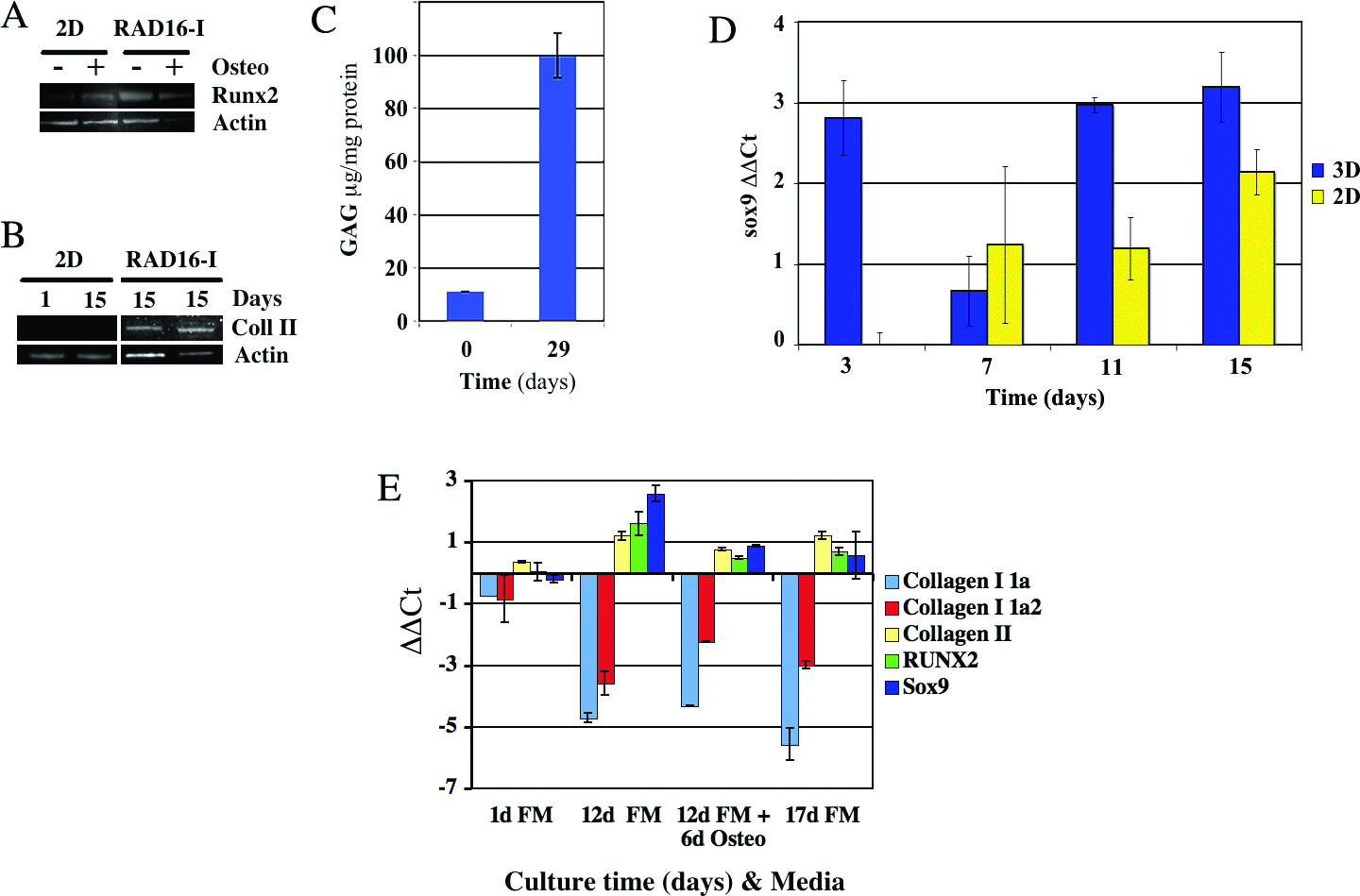
Molecular characterization of the mesodermal induction process. (
Default chondrogenic commitment of MEFs was observed using Western blot analysis of collagen type II (Fig. 3B). Real-time RT-PCR of the transcription factor Sox9 presented a pattern of expression with two peaks of upregulation, one at day 3 and the second at day 11, separated by a downregulation zone at day 7 (Fig. 3D). These changes in expression correlate well with the morphogenic changes previously described. For instance, at day 3, the structure contraction is isotropic, continuing until day 6 (Fig. 1C). At days 7 to 11, the structure starts bilateral contractions, suggesting a dramatic change in the morphogenic process that might affect the expression of Sox9. After several days (11-15 days), the morphogenetic changes finish, and the bilateral structure up-regulates Sox9 again although localized at the paraxial zone (Fig. 4D). In 2D cultures, Sox9 upregulation was less intense, with a gradual increase over time (Fig. 3D). To further understand the gene expression of the 3D system during default chondrogenesis and after osteogenic induction, we evaluated the expression of the transcription factors Runx2 and Sox9, as well as ECM proteins such as collagen type Ia1, collagen type Ia2 and collagen type II using real-time RT-PCR. Results show that, at day 12, when the 3D bilateral structure closes, Sox9, Runx2, and collagen type II are more over-expressed than MEFs cultured in flasks (Fig. 3E). At the same time, the synthesis of collagen type I is strongly down-regulated (Fig. 3E), meaning that MEFs cultured in RAD16-I gradually loose their fibroblastic phenotype (characterized by the synthesis of collagen type I) and become chondrocyte-like cells (expressing collagen type II, Runx2, and high levels of Sox9). Then, 5 to 6 days after osteogenic induction, the expression levels of collagen type II, Runx2, and Sox9 decrease (Fig. 3E). Collagen type II remains constant after 17 days of culture without osteogenic supplements (Fig. 3E), consistent with the Western blots (Fig. 3B) and immunohistochemistry (Fig. 4E). The down-regulation of Sox9 and Runx2 appear to be independent of the presence of osteogenic induction (Fig. 3E). In the presence of osteogenic induction, a slightly greater up-regulation of collagen type I 1a2, but not collagen type I 1a, can be observed than under conditions without differentiating factors (Fig. 3E).
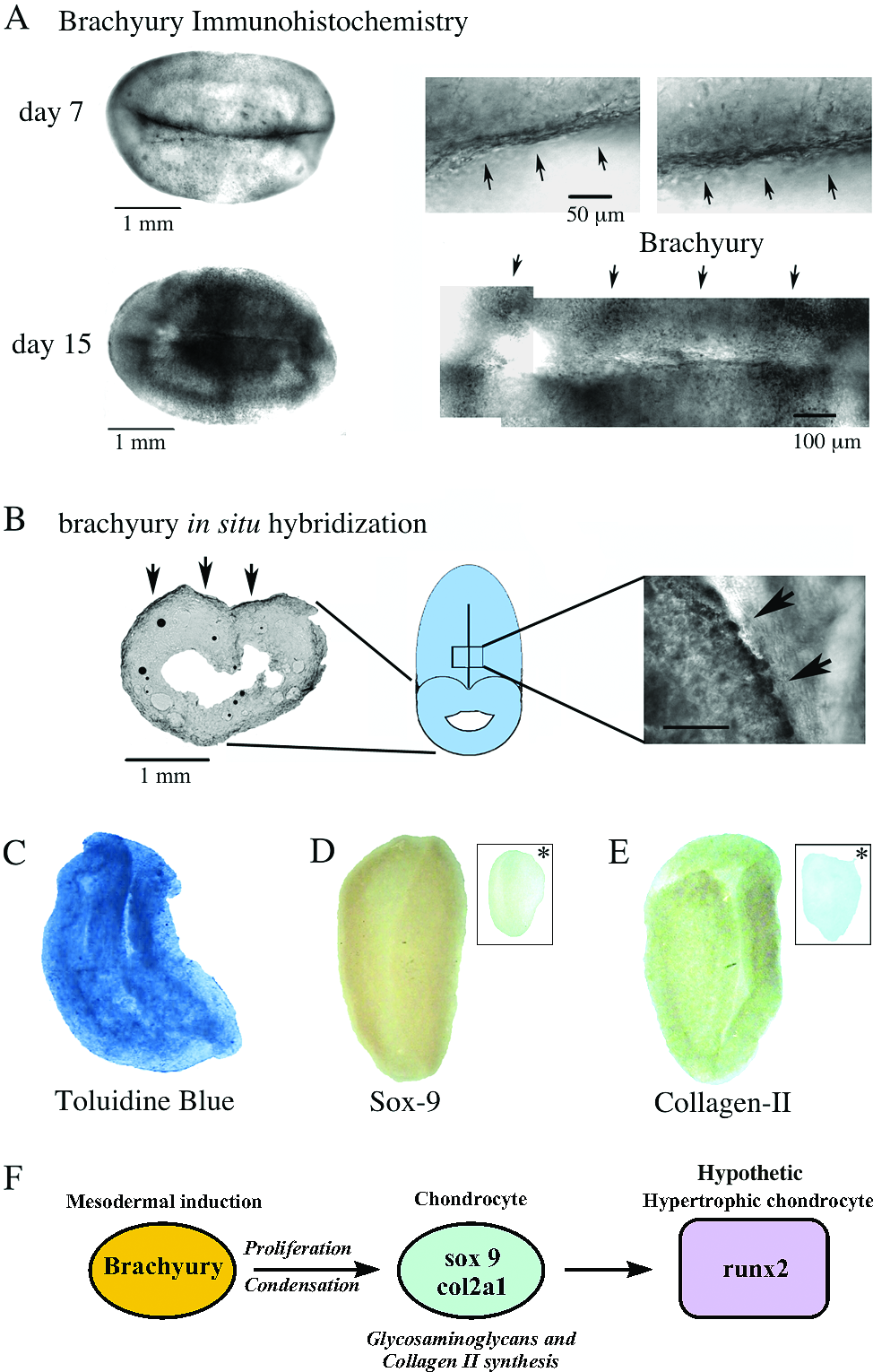
Morphogenesis correlates with pattern formation. (
Finally, MEFs synthesized GAGs when cultured in RAD16-I with FM, confirming the default cartilaginous commitment of these fibroblasts (Fig. 3C). Moreover, when we stained the 3D bilateral structures with Toluidine blue for GAGs, two positive zones parallel to the central axis were detected, suggesting the formation of a paraxial cartilage-like tissue (Fig. 4C). These results confirm the presence of a glycosylated proteoglycan that could be, for example, aggrecan or perlecan or a mix of them. Perlecan is more consistent with the appearance of cartilage during early development. 40
Pattern formation accompanies symmetry appearance
To provide more insight into the possible patterned chondrogenesis, we studied the expression of brachyury, one of the first mesodermal-specific transcription factors that is expressed in the presumptive notochord and in the early mesoderm.41–43 With different culture times, brachyury stained positive according to immunohistochemistry first (day 7) in the central axis and then (day 15) in the entire paraxial zones of the 3D bilateral structure (Fig. 4A). The expression of brachyury was confirmed using regular RT-PCR (see Supplemental Fig., available online at www.liebertonline.com/ten) and its localization was visualized using in situ hybridization. Probes were prepared with the same primers used for regular RT-PCR, and after the staining, we observed that the brachyury mRNA was mainly expressed at the same zone detected using immunohistochemistry, confirming the expression of this early-organizer transcription factor in the 3D bilateral structures (Fig. 4B). Staining of a cross-section of this structure depicts the clear presence of an internal cavity, observed before in Figure 1, and the expression of brachyury at the external paraxial zone (Fig. 4B).
It is also known that the expression of some set of transcription factors, such as Sox9 precedes organized chondrogenic commitment. Thus, we studied the presence and localization of the transcription factor Sox9, a key transcription factor in early chondrogenesis, 44 and immunostaining showed that Sox9 is mainly expressed in the paraxial zones next to the symmetrical axis (Fig. 4D). Finally, the synthesis of the key ECM component of cartilage collagen type II was assessed by immunostaining of the 3D bilateral structures cultured for 15 days in FM. Results showed that collagen type II is also expressed at the paraxial zones next to the central symmetrical axis (Fig. 4E).
Discussion
Fibroblasts are known to be responsible for many regenerating events. A significant example is the regeneration of amputated limbs in amphibians, where fibroblasts generate a set of undifferentiated cells called a blastema that is capable of proliferating and rebuilding the new limb.5–9 Previously, fibroblasts cultured in vitro have also shown multipotential capacity21,30 and have reproduced the mesenchymal condensation in micromass cultures.45,46 Nevertheless, it is still not clear what mechanism drives fibroblasts to this multipotential state and which is the exact phenotype of these undifferentiated fibroblats.6,7 We hypothesize that the biomechanical and non-instructive properties of their environment are key for fibroblasts to have the ability to acquire a less differentiated phenotype that allows them to render most of the cell types of the mesodermal germ line.
Our work suggests that any 3D environment, under the culture conditions tested, and regardless of its chemical nature (self-assembling peptide nanofiber, polysaccharide, or protein matrix), is sufficient to promote dedifferentiation of MEFs into a multipotent mesenchymal progenitor-like cell (Fig. 2). This opens the possibility that the cells “sense” the environment, reprogramming them into a multipotent progenitor. Osteogenic commitment of this progenitor cell in 3D environments occurs only after an osteogenic differentiation protocol, which is evidenced by the deposition of Ca2+ (Fig. 2A, D) and the expression of Runx2 (Fig. 3A) (also known as core binding factor, Cbfa1 and Osf2), positively regulates the expression of specific genes during early osteoinduction in hypertrophyc chondrocytes, suggesting that these cells have acquired such a phenotype. Nevertheless, the specific characteristics of the 3D environment, such as the biomechanical and non-instructive properties, as well as the permissiveness toward cell-to-cell interactions, will provide cells with more or less multipotential capacity. This is clear in the case of RAD16-I, where cells acquired chondrogenic potential by default, as well as the unexpected morphogenetic process here described, suggesting that. in this case. the unique conditions obtained allowed the cells to proceed into a more-organized or emerging state: cellular self-organization. We cannot exclude that diffusion and oxygen gradients may play an important role in the morphogenic process. It is not casual that the final wall thickness of the 3D bilateral structure does not exceed approximately 500 μm, which is the diffusion limit of oxygen in non-vascularized tissues. Moreover, we also speculate that the low oxygen concentration at the inner-most part of the structure could favor the development of paraxial cartilage-like tissue. Notwithstanding this, the gene and protein expression profiles that accompany this morphogenetic event strongly suggest that there is a certain patterning involved.
For instance, these fibroblasts undergo spontaneous adipogenesis in three dimensions (Fig. 2C, F, G), but only in the self-assembling peptide do MEFs begin a default chondrogenic differentiation process, presumably by creating a special cell microenvironment (Fig. 2B, E, 4C). This could be due to the chemical differences between these three scaffolds. The polysacharide (agarose) has shown low interaction with fibroblasts, as well as poor contracting capacity; cells adopt a spherical shape and have little movement inside the scaffold (Supplemental Fig.). Instead, collagen type I is the natural ECM component of the dermis and bone, ensuring high interaction between the cells and the matrix. Furthermore, this material could be instructive in guiding embryonic fibroblasts into these lineages, preventing them from spontaneous differentiation into a chondrogenic lineage (Fig. 2H). Finally, viscoelastic studies with RAD16-I (at different concentrations) over the years have shown that RAD16-I peptide concentrations between 1.7 and 2.8 mg/mL have a G’ (elastic modulus) of 120 and 400 Pa, respectively. In our system, we used final peptide concentrations of approximately 2.5 mg/mL, with a G’ close to 370 Pa, a similar G’ to that of extracellular collagen type I matrices, suggesting that the microenvironment in RAD16-I gels is mechanically similar to the natural ECM of fibroblasts. Therefore, because self-assembling peptide scaffolds lack inherent instructive capacity and have the same biomechanical properties as natural fibroblast ECMs, the default chondrogenesis here described could mean that mouse embryonic fibroblasts can naturally undergo chondrogenic lineage differentiation under the mentioned culture conditions (Fig. 2H). Not only does the self-assembling peptide allow spontaneous chondrogenesis, but it also promotes, under certain conditions, a unique in vitro cellular self-organization that resembles early embryonic stages (Fig. 1C). Furthermore, similar to what has been observed in animal development, the progression of this morphogenesis is dependent on proliferation, as evidenced by the effects of staurosporine on system contraction (Fig. 1E, F). It was also noticed that less initial concentration of MEFs (5 × 105 cells/mL) within the RAD16-I resulted in three fold the contraction time of the cell mass and failed to render bilaterality (results not shown). These results indicate that proliferation seems to have a close relationship with the spontaneous chondrogenic differentiation of MEFs, similar to the relationship between embryonic mesenchymal condensation and chondrogenesis within the pre-skeletal tissue. Altogether, as a consequence of the biomechanical and non-instructive properties of RAD16-I scaffolds, we can speculate that the contraction produced during the morphogenetic process could reproduce a mesenchymal condensation phenomenon that explains the default chondrogenic pathway. Hence, the localized natural chondrogenic induction could be the result of mesenchymal condensation and differentiation under the control of an organized process that directs tissue-like patterning. This is a significant difference from the mesenchymal potential of fibroblasts in high-density micromass cultures, because in these cultures, although fibroblasts reproduce mesenchymal condensation and show default chondrogenic commitment, no self-organization is observed.45,46
We have also showed, using immunohistochemistry and in situ hybridization, that the expression of brachyury, and its localization in this system, is highly related to the formation of the 3D bilaterality (Fig. 4A, B). First, brachyury is expressed in a mid-line zone of the structure, to further spread to both sides of the central axis, suggesting that mesodermal commitment continues advancing toward both sides of the cell mass.
The expression of Sox9 and the synthesis of collagen type II and GAGs also indicated tpontaneous chondrogenesis during this morphogenic development (Fig. 3C, D and 4C–E). Sox9, Runx2, and collagen type II, like brachyury, showed a profile of expression strongly related to morphogenic development, although it remains unknown how the morphological changes proceeded as function of the expression of these genes. Loss-of-function experiments would elucidate the exact relationship between gene expression and the morphogenic process. Sox9 was strongly over-expressed at two time-points during the morphogenetic event related to physical changes: strong isotropic contraction (day 3) and closure of the 3D bilaterality (day 11). The over-expression of Sox9 could be explained as a requirement for the cells to condense, as happens in mesenchymal condensation. Then, after closure of the cellular structure, the expression of Sox9 is upregulated again and correlates with the synthesis of collagen type II, meaning that, at this moment, chondrogenesis is taking place. Runx2 and Collagen type II expression levels are upregulated after closure of the structure and are maintained thereafter.
From the mechanical point of view, from the experiments with different concentrations of RAD16-I (Fig. 1B), it seems clear that the biomechanics in early stages govern the process, at least in the first days of isotropic contraction, when cells form a network and contract the system. It is likely that, at early stages of this phenomenon, some genetic programs start taking control of the evolution of the system. At the time when the lateral contractions start to happen, some genetic programs are already “ruling” the process. We are now focusing on early organizer genes to see whether they are co-localized where and when bilateral contractions take place.
Altogether, we observed that the system undergoes organized contraction, developing into a bilateral shape structure with a central axis, called 3D bilateral structure, resembling an early vertebrate embryonic stage. These remarkable morphological changes suggest that the system might be engaging in a cellular self-organization process. We speculate that the initial conditions we provide to the cells (3D culture of MEFs in self-assembling peptide scaffolds) recapitulates cellular and morphological changes of tissues undergoing development (proliferation, migration, and condensation) and the expression pattern of early embryonic genes (organizers and tissue-specific genes) (Fig. 4F). Moreover, the process ends in the production of specific tissues, such as patterned paraxial cartilage-like tissue. Most importantly, a group of differentiated cells under special “environmental conditions” can proceed to a self-organization process, characteristic of systems undergoing development.
In conclusion, the system we described here can be used to culture cells carrying specific mutations or transgenic genes and to obtain 3D bilateral structures that recreate certain morphogenetic processes without the use of pregnant females and their embryos. In future work, other cell sources such as dermal fibroblasts from amphibians and other mammals, including adult humans, will be tested to see whether this particular self-organization phenomenon is common in other metazoans.
Footnotes
Acknowledgments
We gratefully acknowledge our colleagues Kelly Blackburn, Alice Chen, Roger Kamm, Alan Grodzinsky, Hayman Hartman, Hans Jörg Meisel, Jaime Rivera, and Angelo Cardoso for helpful discussions and suggestions during the development and writing of this work. We are indebted to David Shaywitz and the Department of Cellular Molecular Biology at Harvard University for providing brachyury primers. We thank Nicky Watson and the microscopy facilities in the Whitehead Institute for the excellent technical assistance in sectioning. We thank the Catalan Government for the pre-doctoral fellowships 2005 BE 00178 and 2005 FI 00864 to L.Q., and to IQS for the pre-doctoral fellowship to E.G. This work was supported by Grant NIH 1-ROI-EB003805-01A1 and award 1098SF from TRM, Universität Leipzig, Germany, to C.E.S.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.