
Abstract
Articular chondrocytes (AC) expanded in vitro for tissue engineering rapidly turn off collagen type II (COL2) synthesis. We wanted to inhibit this process sufficiently to obtain therapeutically useful numbers of AC without losing COL2 synthesis. To this end, AC were expanded on their own extracellular matrix (ECM) in structures designated chondrocytes in autologous ECM (CA-ECM). Here, AC maintained a rounded shape and proliferated rapidly. After 13–15 days in culture, 40 × 106 cells (median) could be obtained from a cartilage biopsy. Real-time RT-PCR showed a reduced, but persistent, production of COL2A1 mRNA at this time. Flow cytometry showed high levels of intracellular COL2, and immunogold electron microscopy showed high density of well-organized COL2 fibrils in newly synthesized ECM. Interestingly, high levels of COL1A1 mRNA and intracellular protein were detected, but no COL1 was found in the ECM. The slow loss of COL2A1 mRNA was paralleled by a loss of the COL2 regulating transcription factor SOX9 mRNA. Chromatin immunoprecipitation assays could not identify epigenetic histone modifications that would explain the observed changes in COL2 synthesis. Thus, the CA-ECM strategy allows AC to proliferate to clinically useful numbers while maintaining COL2 synthesis and secretion. This strategy may improve tissue engineering of joint surfaces.
Introduction
Human AC could be a useful cell source, as they are the origin of hyaline extracellular matrix (ECM) in vivo. Such cells may be isolated from a biopsy obtained from an area of lesser weight bearing of the femoral condyle of the patient, but in numbers they are greatly insufficient for therapeutic purposes. To reconstitute the approximately 1.5 × 106 chondrons found per cm2 of hyaline cartilage in the human knee, 7 a large number of AC is required. This can only be achieved following in vitro expansion of the cells. However, in monolayer cultures established for rapid expansion of AC, the synthesis of collagen rapidly changes from type II (COL2) to type I (COL1). This change has been regarded as a dedifferentiation process, 8 leading to the generation of cells that, upon transplantation into a focal lesion of the knee joint, form tissue with fibrous and fibrocartilaginous features. 9
For AC established in culture in vitro, adherence to the tissue culture polystyrene plastic surface seems to be a prerequisite for rapid proliferation. Plastic adherence and rapid proliferation, in turn, lead to dedifferentiation. Attempts to interfere with this sequence of events have been made by culturing the AC on surfaces coated with specific substances, 10 on nonadherent surfaces, 11 in agarose, 12 or in alginate gels. 13 In monolayer cultures, expansion on COL2 was found to increase COL2A1 mRNA concentration upon redifferentiation. 10 In three-dimensional cultures, while the cells retained some of their capacity to produce the constituents of ECM of hyaline cartilage, the proliferation rates were greatly impaired. 2 In other experiments, reduced oxygen tension in the incubator air has been investigated as a means to counteract the process of dedifferentiation. Under these conditions, a small but insufficient increase in COL2A1 mRNA concentration was observed, and the problems of insufficient numbers of cells were not resolved.14,15
Recently, it was shown that AC cultured in the presence of their pericellular matrix (PCM) produced ECM of greater volume and better quality than AC cultured in the absence of PCM. 16 In fact, the sensitivity of AC to their immediate surroundings is such that contact with COL1, but not COL2 fibrils, may lead to destabilization of the cartilage phenotype. 17 Using this information, we hypothesized that AC cultured in vitro in close proximity to their native matrix, but released from the restraint to proliferation exerted by intact ECM, might proliferate and, at the same time, produce ECM resembling hyaline cartilage.
Materials and Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Experimental design
Human articular cartilage was obtained as 200–300 mg biopsies. For patients undergoing autologous chondrocyte implantation (ACI) therapy, the biopsies were harvested from a low-load-bearing area on the proximal part of the lateral femoral condyle of the injured knee. An initial series in 2003–2004 of 18 patients (median age 41 years, range 20–54) received AC cultured as filtered, single cells. A subsequent series in 2004–2006 of 19 patients (median age 37 years, range 19–52) received AC cultured as chondrocytes in autologous ECM (CA-ECM). Based on the estimate that a minimum cell number of 1.5 × 106 per cm2 lesion area, 7 but preferably >20 × 106 cells, was required for repair, all of these cultures were maintained until the minimum target was reached. However, several cultures were continued beyond this, until the planned day of implantation. This day was established based on our knowledge of the time required to obtain >20 × 106 cells from the two different culture procedures. For in vitro studies, the biopsies were obtained as leftover material from the lateral femoral notch of patients undergoing anterior cruciate ligament (ACL) surgery. As part of the surgical procedure, a small amount of cartilage and bone was removed from the lateral side of the intercondylar notch to create sufficient space for the ACL graft (“notch-plasty”). The bone fragments were removed before further processing of the biopsy. From such biopsies, AC from three donors were established as filtered, single cell cultures while cells from two donors were established as CA-ECM. In all cases informed consent was obtained, and the study was approved by the Regional Committee for Ethics in Medical Research.
In vitro culture of AC
The biopsies were digested in two different ways: for the single-cell procedure, biopsies were washed and minced in physiological saline (B. Braun Meslungen AG, Meslungen, Germany) supplemented with antibiotics and amphotericin B. The minced biopsies were digested in DMEM/F12 supplemented with antibiotics and amphotericin B (culture medium) containing collagenase type XI (1.2 μg/mL) and deoxyribonuclease I (0.1 μg/mL) at 37°C in humidified room air with 5% carbon dioxide (CO2) for 3–5 h. The digested biopsies were then filtered through 70-μm cell strainers (BD Biosciences, Franklin Lakes, NJ) and washed. The resulting single cells were resuspended in culture medium supplemented with ascorbic acid (50 μg/mL) and 20% autologous serum (AS), and seeded as regular monolayer cultures. AS was prepared from 120 to 150 mL of whole blood as described previously. 18 In the CA-ECM procedure, biopsies were washed and minced to very tiny pieces in culture medium (Fig. 1A). The fine-minced biopsies were digested in 10 mL culture medium containing collagenase type XI (1.2 μg/mL) at 37°C in room air with 5% CO2 for 90 min. The CA-ECM structures formed by this procedure (Fig. 1B) were washed and resuspended in culture medium supplemented with ascorbic acid (50 μg/mL) and 20% AS and seeded as CA-ECM cultures. Each culture represented cells from a single biopsy from a single donor. All cultures were performed in 25 and 75 cm2 culture flasks except for those designated for electron microscopy, which were performed in 24-well plates. Culture medium was replaced every 3–7 days. At the first passage or the medium change at 8–10 days of culture, amphotericin B was removed and 10% AS was used instead of 20% in the culture medium. For each passage, the cells were detached from the plastic surface with trypsin-EDTA. The cells were counted in a fluorescence microscope after staining with an acridine orange/ethidium bromide solution.
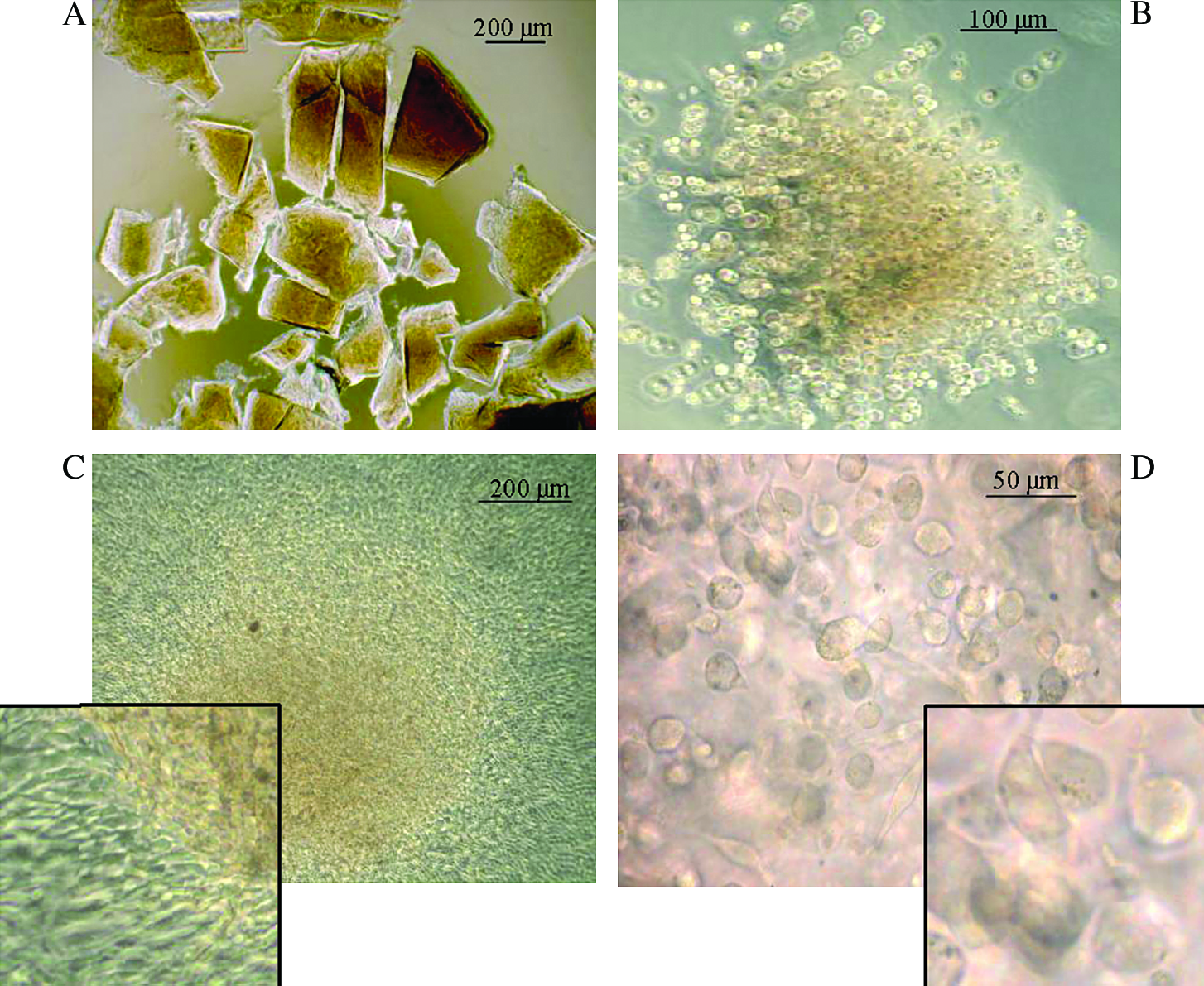
CA-ECM cultures: the cartilage biopsy was cut into tiny pieces (
Real-time RT-PCR
Total RNA was extracted from pellets of AC using Trizol (Invitrogen, Carlsbad, CA). Following treatment with DNase I (Ambion, Huntingdon, UK), reverse transcription (RT) was performed according to the manufacturer's protocol (Invitrogen) with 100 ng total RNA per RT reaction. Assays for COL1, COL2, and aggrecan mRNA were performed as described by Martin et al. (Table 1). 19 Predesigned assay for SOX9 (HS001165814-m1) was purchased from Applied Biosystems (Foster City, CA). Here, the sequences of the primers and probes are not available. All assays were designed to overlay a junction between two exons to avoid hybridization to genomic DNA. 18S (Applied Biosystems) was included as an endogenous normalization control. Quantification of cDNA was performed using the 7300 Real-Time PCR system (Applied Biosystems). From each cDNA, three samples (2.5 μL cDNA, total volume 25 μL) were run in parallel. Cycling parameters were 50°C for 2 min, then 95°C for 2 min followed by 40 cycles of 95°C for 15 s, and 60°C for 1 min, and the assays were performed as single-plexing assays. Gene expression was calculated using the relative standard curve method (User Bulletin 2; Applied Biosystems).
Flow cytometry
Cultured AC were examined for intracellular expression of ECM molecules using flow cytometry. AC were fixed in 1% paraformaldehyde for 4 h and incubated overnight in 1% Tween-20 at 4°C, washed and stained with unconjugated antibodies specific for COL1 or COL2 (MP Biomedicals, Aurora, OH) or aggrecan (Biosource, Nivelles, Belgium) proteins for 30 min, washed and incubated with phycoerythrin-conjugated goat anti-mouse IgG1 or IgG2a secondary antibodies (Southern Biotechnology, Birmingham, AL) for 30 min at 4°C. Cells were washed, fixed in 1% paraformaldehyde, and analyzed using an FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). Gates were set so that no more than 1% of the cells were positive using irrelevant antibodies.
Electron microscopy
For electron microscopy, CA-ECMs were cultured in 24-well plates for 16–18 days. Specimens were fixed in a phosphate-buffered mixture of paraformaldehyde and glutaraldehyde and embedded at low temperature in Lowicryl HM23 (Chemische Werke, Waldkraiburg, Germany). 19 Ultrathin sections were subjected to immunogold analysis against COL1 (monoclonal mouse anti-human COL1; MP Biomedicals) at 1:10 and COL2 (polyclonal rabbit anti-human COL2 antibody, Novocastra, Newcastle upon Tyne, UK) at 1:2 using protein A coated with 10-nm colloidal gold for detection. Electron micrographs were sampled randomly with respect to distribution of gold particles, and marker density for each tissue compartment was measured by semiautomatic interactive image analysis. 20
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed essentially as described earlier, 21 in buffers containing 20 mM sodium butyrate with a few modifications. In short, 106 cells were cross-linked with formaldehyde, lysed in lysis buffer/2% sodium dodecyl sulfate (SDS), sedimented, and the pellet was sonicated in lysis buffer/1% SDS to produce chromatin fragments of ∼300 bp. ChIPs were performed using 100 μL sonicated chromatin (corresponding to ∼5000 cells) using antibodies to acetylated H3 lysine 9 (H3K9ac) and trimethylated H3K9 (H3K9m3) and H3K27m3 (Abcam #ab6002). Immunoprecipitated DNA was used as template for quantitative PCR. 21 Primers used are described in Table 1.
Results
Cell proliferation in monolayer culture of single chondrocytes
For a period of time, in vitro culture of single AC in monolayers in room air and 5% CO2 was used in our laboratory to produce cells for ACI. The median culture period for 18 consecutive donors was 21 days (range 14–23 days), and the median number of AC obtained was 28.7 × 106 (range 9.1–68 × 106 cells) with a median viability of 93% (86–98%).
AC expanded in vitro in CA-ECM
The cartilage biopsies were cut into tiny pieces (Fig. 1A), and then exposed briefly to collagenase. A light micrograph of a CA-ECM immediately after collagenase exposure is shown in Figure 1B. The collagenase removed the physical restraint of the ECM on the chondron, allowing the AC to proliferate while still attached to their PCM/ECM. At the end of the first passage, after 8–10 days, most of the AC were still organized as clusters in CA-ECM, while some had spread on the plastic surface to form monolayer culture (Fig. 1C). As long as the CA-ECM persisted, the AC on the CA-ECM remained as small, round cells (Fig. 1D). At the end of P1, after 13–15 days in culture, the CA-ECMs dissolved upon trypsinization, leaving the AC as a single-cell suspension. At this stage, only, cell counts could be performed in these cultures. Cells to be used for ACI were cultured in this way in room air and 5% CO2 from mid-2004 onward in our GMP laboratory. The median period for 19 consecutive ACI cultures performed this way was 13 days (range 13–20 days), and the median cell count was 39.7 × 106 cells (range 8.1–138.9 × 106 cells) with a median viability of 92% (82–99%).
Gene expression in AC cultured as CA-ECM
The concentrations of mRNA coding for COL2, COL1, and the transcription factor SOX9 were determined relative to the endogenous control 18S mRNA at the end of P0 and P1 for the CA-ECM, and at P3, after 3 weeks in culture, for the filtered single cells in monolayer. These time points (P1 for CA-ECM and P3 for monolayer cultures) correspond to the times required to obtain clinically useful number of cells in the ACI cultures. The results are shown in Figure 2. For the cells expanded in monolayer cultures, the concentrations of COL2A1 mRNA were too low to give reliable triplicates. For the chondrocytes expanded in CA-ECM, the levels of COL2A1 mRNA concentrations fell from the end of P0 to the end of P1, but at this time, when a clinically useful number of cells could be collected, the COL2A1 mRNA concentrations were still approximately 100-fold higher than for the cells in monolayer cultures (Fig. 2A). The levels of SOX9 mRNA (Fig. 2B) also fell from the end of P0 to the end of P1, similar to the pattern observed for COL2A1 mRNA. When these cells were followed in later passages as monolayer cultures, the SOX9 mRNA levels remained at approximately those observed after P1 (data not shown). The concentration of COL1A1 mRNA was high already at the end of P0, remained high at the end of P1 (Fig. 2C), and remained at this level through several passages as monolayer cultures (data not shown). A pattern similar to that observed for COL1A1 was observed for the mRNA encoding aggrecan (data not shown).
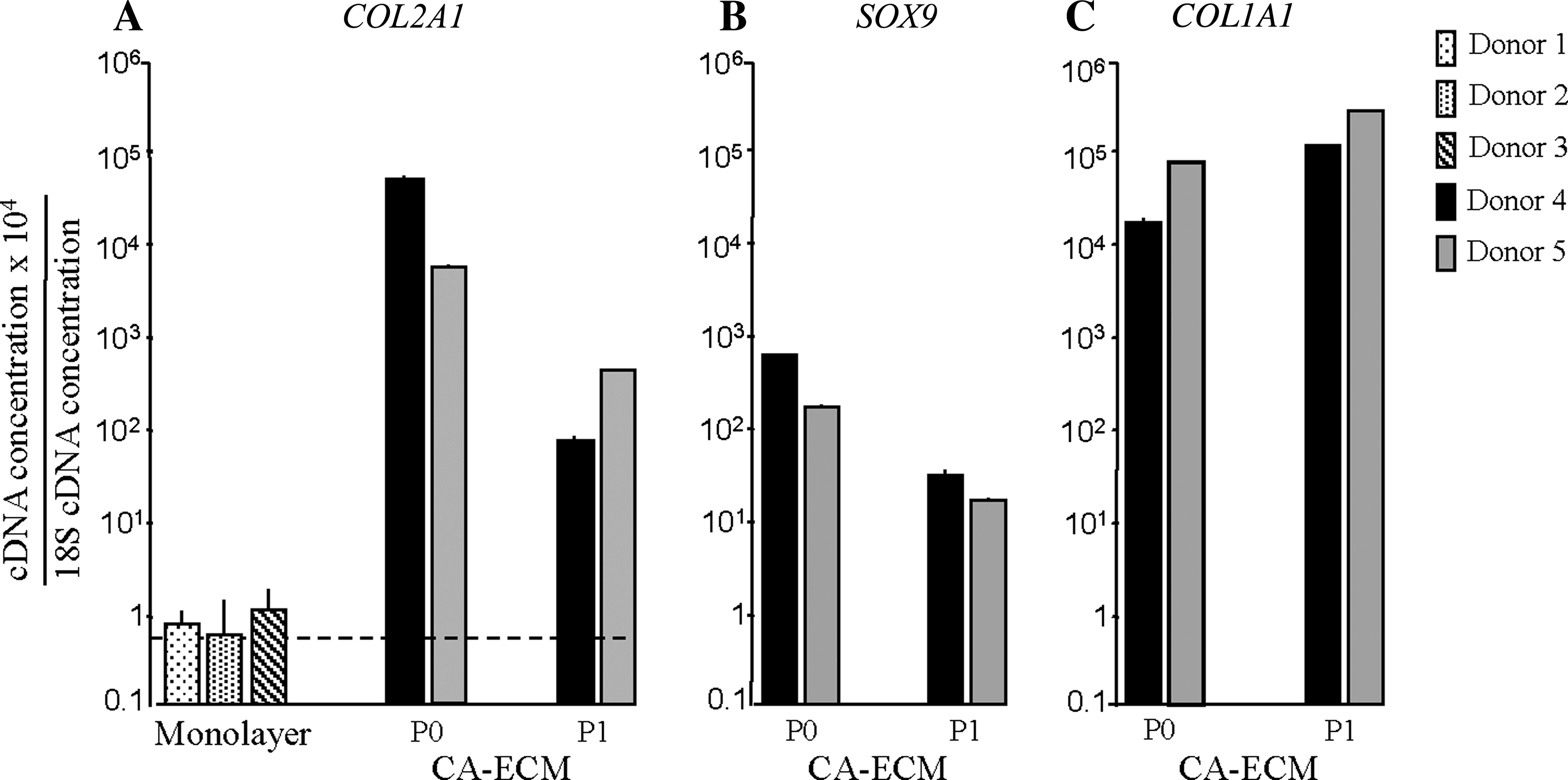
Real-time RT-PCR analysis of mRNA encoding COL2 (
Flow cytometry analysis of intracellular levels of ECM proteins
In monolayer culture, chondrocytes immediately lose COL2 expression and upregulate COL1.2,8 To determine if these changes occurred to the same extent in AC cultured as CA-ECMs, we performed analyses of ECM proteins within the AC and in the ECM of the CA-ECM. Figure 3 shows a flow cytometry analysis of the intracellular concentrations of COL1, COL2, and aggrecan at the end of P1, the first passage at which the cells could be obtained in single-cell suspension, and P3, when the cells were entirely in monolayer culture. Consistent with the reduction in COL2A1 mRNA, the intracellular COL2 concentration was high at the end of P1 (fluorescence intensity 5.9 and 7.2 relative to the irrelevant controls for donors 4 and 5, respectively), and considerably reduced but still detectable at P3 (1.8 and 4.2). Again consistent with the mRNA analysis, the levels of COL1 and aggrecan were quite high at P1 (COL1: 8.1 and 12.1; aggrecan: 3.8 and 13.3 for donors 4 and 5, respectively), and remained unchanged at P3 (COL1: 9.5 and 15.4; aggrecan: 23.6 and 17.0 for donors 4 and 5, respectively).

Flow cytometric determination of the intracellular concentration of COL2 (blue), COL1 (red), and aggrecan (black) for cells at the end of P1 (top) and P3 (bottom). Staining using an irrelevant control antibody is shown in green. The results shown are for donor 4. Similar results were found also for donor 5.
EM analysis of de novo ECM synthesis and secretion
The release of new ECM molecules into the ECM by AC in the CA-ECM was evaluated by immunoelectron microscopy. Altogether 360 micrographs were analyzed for each of COL1 and COL2. Representative images are shown in Figure 4. It was easy to distinguish the ECM of the original biopsy material from newly made ECM, because in the original ECM, the collagen fibrils had been cut into small pieces by the collagenase (Fig. 4A, C), while in the newly formed ECM, the fibrils were long, and frequently oriented in the same direction (Fig. 4B, D). Immunogold labeling demonstrated COL2 fibrils, both within the original and the newly formed ECM (Fig. 4A, B). In contrast, the COL1 labeling was similar to background labeling, both within the original and the newly formed ECM (Fig. 4C, D). As a control for the COL1 immunogold labeling procedure, COL1 labeling of sections of bone resulted in very high marker density (data not shown).

Electron microscopic examination of CA-ECM. Original ECM (
Histone modifications associated with COL1A1 and COL2A1 regulatory regions
ChIP assays were performed to map histone H3 modifications associated with COL1A1 and COL2A1 regulatory regions. In agreement with sustained expression of COL1A1 in all cultures, we found that the COL1A1 promoter was associated with high levels of acetylated H3 lysine 9 (H3K9ac), a marker of active genes. Conversely, epigenetic markers of silent genes, such as trimethylated H3K3 and H3K27, were essentially absent from the COL1A1 promoter (Fig. 5A).

Histone modifications on the COL2 and COL1 regulatory regions in CA-ECM–cultured human AC. (
Histone modification patterns on the COL2A1 promoter (P) and 3′ enhancer regions (E) are shown in Figure 5B. The level of H3K9ac was low at both loci both at P1 and P3. At the same time, the H3K9m3, considered to be associated with constitutive transcriptional repression, was also low for both loci at both time points. Surprisingly, however, we detected high levels of H3K27m3 on the COL2A1 promoter, even at P1 when the gene was highly expressed (Fig. 5B; P), suggestive of the establishment of a facultative silencing histone mark on this locus. The enhancer also harbored H3K27m3, albeit at a much lower level than the promoter. Lastly, the housekeeping GAPDH gene promoter was highly acetylated on H3K9, while displaying no trimethylation on H3K9 and H3K27 (Fig. 5C) as expected from its constitutive expression.
Discussion
Treatment of focal lesions of the hyaline cartilage of weight-bearing joints using ACI has been an alternative to other surgical repair procedures for several years. 1 The short- and long-term clinical outcomes of the procedure have been reported to be good. 22 In a study comparing the results at 5 years of follow-up between ACI and microfracture repair, neither could be shown to be superior. Approximately 70% of the patients with single grade IV cartilage injuries >2 cm2 on the femoral condyles were found to have good results, although their histology did not show normal hyaline cartilage. 23 Microscopically, ACI yields repair tissue mostly consisting of fibrocartilage. 24 Given that 30% in both groups reported only poor or fair results, that the histological analysis may suggest that there may be clinical problems in the long term, and that improvement of the microfracture technique is difficult to envisage, there is clearly a need for improvement of the current ACI protocol.
The primary ambition of the present study was to expand AC in vitro to clinically useful cell numbers without losing COL2 synthesis and secretion. To this end, the AC were cultured attached to their PCM/ECM in loose structures termed CA-ECM. With the ECM disintegrated sufficiently to allow the AC to escape from the lacunae, the AC proliferated better than in monolayer cultures attached to the plastic surface. This is, to the best of our knowledge, the first report of rapid proliferation of AC established in a three-dimensional structure.
Loss of COL2 expression is a rapid and invariable result of AC proliferation in monolayer cultures.8,25 In the CA-ECM cultures, on the other hand, COL2A1 mRNA levels were quite well preserved after 13–15 days of culture, and the cell counts were increased, compared with monolayer culture under standard incubator gas conditions. Additionally, our CA-ECM COL2A1 mRNA estimations may be falsely low, as some of the cells had attached to the plastic surface at the end of P1 to form monolayer cultures, which presumably led to rapid reduction in COL2A1 mRNA levels in these cells. Preclinical and clinical studies will have to be performed to determine if the maintenance of the COL2A1 mRNA levels by this method is sufficiently well preserved to promote the production of hyaline cartilage in lesions better than that seen following the transplantation of dedifferentiated AC. 24
The molecular mechanisms driving the relationship between cell attachment, cell cycle, and COL2 synthesis have not yet been identified. Recently, the interaction between cell surface integrins and the common integrin binding motif arginine-glycine-aspartic acid was shown to inhibit COL2 expression in MSC induced toward chondrogenesis. 26 In vitro, integrins account for the attachment of AC to plastic surfaces, in which case this mechanism might account also for the loss of COL2 expression in AC in monolayer cultures. Similarly, chondrocytes cultured in gels of fibrils from COL1, but not COL2, COL9, and COL11, synthesized COL1, and not hyaline cartilage collagens. 17 Interestingly, the cells attached to COL1 acquired a fibroblast-like shape. This introduces cell shape as a factor, with small, round AC producing COL2, and fibroblast-like cells producing COL1. 12 In the present study, the AC kept their small size, rounded shape, and attachment to their own PCM/ECM while maintaining COL2 production, further suggesting that both molecules involved in attachment as well as the organization of the cytoskeleton may regulate the dedifferentiation process.
In the CA-ECMs, we found a logical relationship between COL2A1 mRNA levels, intracellular COL2 protein expression, and ECM structure. A lag phase could be seen between the loss of COL2A1 mRNA levels and loss of intracellular COL2 protein, which presumably reflected the time taken for the secretory granules to release the last COL2 molecules translated from the remainder of the COL2A1 mRNA into the ECM. Here, COL2 fibrils were formed and subsequently organized into bundles, as demonstrated by the EM images. For COL1, however, the situation seemed to be different. True, persistently high COL1A1 mRNA levels were associated with high levels of intracellular COL1 protein. However, no COL1 could be demonstrated in the ECM. Previously, moderate levels of COL1A1 mRNA have been observed in freshly isolated, uncultured AC. 27 The level was found to increase 1000-fold in the course of 2 weeks in monolayer culture, and then to remain relatively unchanged.28,19 Expression of intracellular COL1 protein has, to the best of our knowledge, not been demonstrated in uncultured AC. 28 However, COL1 could be observed at day 21 of monolayer culture. 28 In the CA-ECM published here, the COL1A1 mRNA levels and intracellular COL1 protein level were high at the end of P1, after 13–15 days in culture, but the release of COL1 into the ECM seemed to be blocked. Thus, while further studies have to be performed in order to reveal the true relationship between transcription, translation, and secretion of COL1 and their relationship to in vitro dedifferentiation and disease processes in vivo, the failure of CA-ECM to secrete COL1 suggests that these cells will initially produce a predominantly hyaline-type cartilage following ACI.
As the process of dedifferentiation took place quite slowly in the CA-ECM cultures, this allowed us to investigate some aspects of the mechanisms potentially involved in the regulation of this process. The mRNA encoding the transcription factor SOX9, which has been shown to regulate COL2A1 mRNA expression in dedifferentiated AC in vitro,11,29 was reduced in a way that paralleled the reduction in COL2A1 mRNA levels. This indicates that, also in the CA-ECM cultures, COL2 synthesis is predominantly regulated at the level of transcription factors. Interestingly, both integrin attachment and cytoskeleton rearrangement mechanisms act via the p38 signaling pathway.26,30
However, epigenetic mechanisms could also be involved in the changes observed in COL2 synthesis. To start unraveling the possible role played by epigenetic mechanisms in the regulation of COL2A1 gene expression, we carried out ChIP assays to map histone H3 modifications associated with regions of the COL2A1 enhancer and promoter regions previously shown to be involved in the expression of the human COL2A1 gene.31,32 The H3 modifications on COL2A1 suggest that the gene is accessible to transcription factors (low levels of H3K9m3, albeit relatively low levels of acetylation at K9), supporting the idea that regulation of transcription factors may be the predominant mechanism involved in the regulation of COL2A1 expression. However, high levels of H3K27m3 in the promoter region are consistent with an epigenetic mechanism of transient transcriptional repression. This trimethylation may have been induced early in the CA-ECM cultures, in which case it may indeed be involved in the dedifferentiation process, or it may be a constitutive aspect of the COL2A1 promoter, and not involved in COL2A1 expression in these cultures. Finally, it should be emphasized that the regions examined in the present study are only some of many sequences potentially involved in the regulation of COL2A1 expression. Thus, epigenetic mechanisms may play a more important part in the regulation of COL2A1 expression than suggested by our results.
In conclusion, we have shown that AC expanded in vitro attached to their own PCM/ECM in CA-ECM proliferate rapidly to yield clinically useful number of cells still secreting COL2 and no COL1 at the end of 2 weeks in culture. Further studies are required to determine if the maintenance of COL2 secretion will translate into clinical improvement following ACI.
Footnotes
Acknowledgments
We are thankful to Janke B. Eriksen for help with the cell cultures, Leif Lindeman for assistance with ChIP experiments, and Aileen M. Larsen for assistance with the EM. This work was supported by Storforsk and Stamcelle grants from the Research Council of Norway and Gidske og Peter Jacob Sørensens Foundation for the Promotion of Science. Support was also received from the Oslo Sports Trauma Research Center. The Oslo Sports Trauma Research Center has been established at the Norwegian University Sport and Physical Education through a generous grant from the Eastern Norway Regional Health Authority, the Royal Norwegian Ministry of Culture and Church Affairs, and the Norwegian Olympic Committee and Confederation of Sports.
Part of this material was presented as an oral presentation and an abstract (9.7) at the 7th World Congress of the International Cartilage Repair Society in Warzaw, Poland, in September 2007.