
Abstract
Dental caries remains one of the most prevalent infectious diseases in the world. So far, available treatment methods rely on the replacement of decayed soft and mineralized tissue with inert biomaterials alone. As an approach to develop novel regenerative strategies and engineer dental tissues, two dental stem cell lines were combined with peptide-amphiphile (PA) hydrogel scaffolds. PAs self-assemble into three-dimensional networks of nanofibers, and living cells can be encapsulated. Cell–matrix interactions were tailored by incorporation of the cell adhesion sequence RGD and an enzyme-cleavable site. SHED (stem cells from human exfoliated deciduous teeth) and DPSC (dental pulp stem cells) were cultured in PA hydrogels for 4 weeks using different osteogenic supplements. Both cell lines proliferate and differentiate within the hydrogels. Histologic analysis shows degradation of the gels and extracellular matrix production. However, distinct differences between the two cell lines can be observed. SHED show a spindle-shaped morphology, high proliferation rates, and collagen production, resulting in soft tissue formation. In contrast, DPSC reduce proliferation, but exhibit an osteoblast-like phenotype, express osteoblast marker genes, and deposit mineral. Since the hydrogels are easy to handle and can be introduced into small defects, this novel system might be suitable for engineering both soft and mineralized matrices for dental tissue regeneration.
Introduction
Dental tissues harboring stem cells are easily accessible and most often discarded as a byproduct of routine surgical treatment. Stem cell characteristics can be detected in cells isolated from the pulp of deciduous 1 as well as permanent teeth,2,3 periodontal ligament,4,5 or periapical follicle. 5 Using stem cell markers such as STRO-1 or CD 146, mesenchymal stem cells, which represent < 4% of the total cell population, can be tagged and sorted by fluorescence-activated cell-sorting analysis. 1 Tooth-derived stem cells are capable of differentiating into adipocytes, neurons, and odontoblast-like cells.1,6 They form mineralized nodules in vitro 5 and create bone or dentin–pulp-like complexes after transplantation into immunocompromised mice.1,3
As new sources of stem cells are explored and optimal permissive conditions for their differentiation are investigated, there is a strong need for advanced biomimetic scaffolding materials, which are versatile enough to be targeted for tooth-specific applications. These scaffolds have to provide a suitable three-dimensional (3D) network to accommodate cells and guide their growth, organization, and subsequent differentiation. In vivo, these features are carried out by the extracellular matrix (ECM). Fibrillar proteins account for most of the ECM network; they self-assemble and form a well-organized structure. It surrounds cells and offers physical support and specific ligands for cell adhesion and migration. ECM also regulates cell proliferation and dynamic characteristics through various growth factors and signaling molecules. Recently, a novel class of hydrogel scaffolds has been developed, which offers several properties of natural ECM.7–13 These peptide-amphiphile (PA) molecules consist of a peptide segment coupled to a fatty acid chain; they assemble into 3D nanofiber networks to form self-supporting gels. The process is driven by formation of a hydrophobic core composed of closely packed alkyl tails, whereby fibrous strands can build because of hydrogen bond formation between the amino acids of adjacent PA molecules. Long cylindrical structures that are nanometers in diameter and microns in length create a gel by trapping water. Whereas the PAs remain amorphous aggregates at neutral pH due to the repulsive negative charge, addition of polyvalent ions eliminates the charge and allows self-assembly into cylindrical micelles, which undergo physical crosslinking to provide the gelled macrostructure. Self-assembly can be triggered upon mixture of PA solutions with cell culture media or other physiological fluids that contain polyvalent metal ions. When cells are suspended in the fluid, these can be encapsulated in the nanofibrillar matrix. It has been shown that cells can move, proliferate, and differentiate within the hydrogel.10,12–15 Several modifications of PAs have been described in the literature, allowing for mineral deposition, 16 optimized cell adhesion,11,17 selective cell differentiation into neurons, 14 or ectopic bone formation. 15
A modification of a PA system that has previously been described was used in this study. 10 The structure of these molecules can be divided into four regions of function (Fig. 1). The peptide sequence contains an enzyme-cleavable site composed of GTAGLIGQ; a glutamic acid to assist in calcium binding; and RGDS, a cell-adhesion sequence that was first described in 1987, 18 which is present in natural ECM and has been integrated into various bioengineering scaffolds. The GTAGLIGQ is expected to be cleaved between glycine and leucine residues by matrix metalloproteinase-2 (MMP-2). Members of the MMP family are able to hydrolyze most of the proteins found in ECM; and MMP-2 is the major matrix metalloproteinase expressed by human pulp cells to remodel their environment. 19 Incorporation of this specific cleavage site is expected to result in cell-mediated proteolytic degradation of the network, enabling cell migration and remodeling of the matrix with natural ECM. The fourth region of functionality is added after peptide synthesis by N-acylation with palmitic acid, which provides the driving force for self-assembly. These modifications of PA hydrogels are first steps to optimize tissue-engineering scaffolds for a specific cell type.
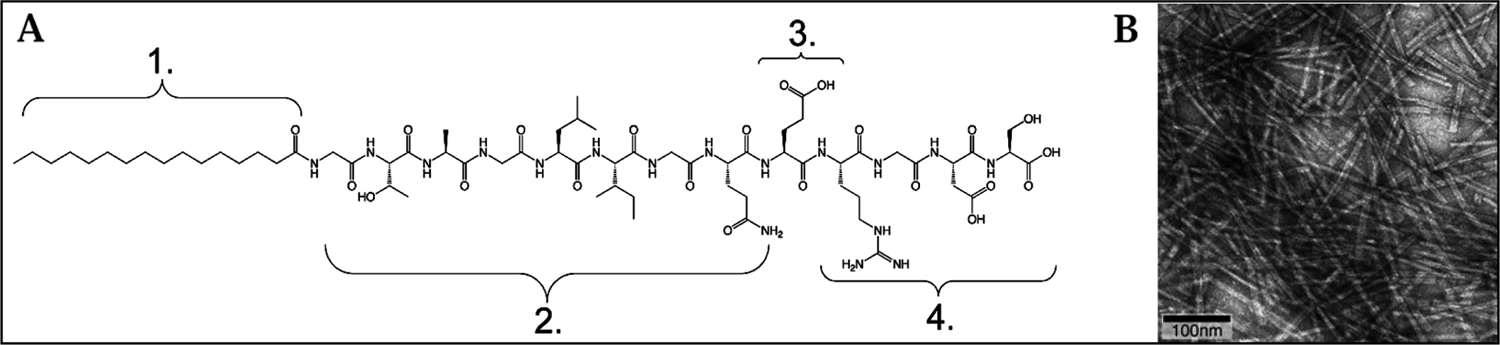
(
In this study, we tested the use of the PA hydrogel scaffold with the following two well-characterized postnatal stem cell lines: SHED, which were isolated from human deciduous incisors, 1 and DPSC from impacted wisdom teeth.2,3 Our goal was to explore the compatibility of PA nanofibers with these two cell lines and to assess their potential as a suitable scaffold for cell proliferation and differentiation. Combinations of dexamethasone with inorganic phosphate (β-glycerophosphate or potassium phosphate [KH2PO4]) have previously been used as osteogenic supplements, and induced calcium accumulation in SHED and DPSC.1,3,20,21 Cell differentiation was monitored by alkaline phosphatase (ALP) assay, histologic analysis, and quantitative real-time PCR analysis, which included marker genes for ECM synthesis and for differentiated osteoblasts and odontoblasts (collagen types I and III, ALP, bone sialoprotein [Bsp], osteocalcin [Oc], Runx2, and dentin sialophosphoprotein [Dspp]).
The results of our study show compatibility of both dental stem cell lines with the PA nanofibers. Cells spread, proliferate, and differentiate within the hydrogels. However, we provide evidence that there are distinct differences between the two cell lines and their reaction to specific osteogenic supplements. SHED appear more adept for soft tissue regeneration, which is enhanced by β-glycerophosphate, while DPSC have a greater potential for terminal differentiation and subsequent mineralization, especially in combination with KH2PO4. The results of this study provide the basis for further optimization of PA nanofibers as a scaffold for dental stem cells and for future tissue-engineering strategies in order to more effectively regenerate dentin after exposure of the dental pulp and to conserve a functional dentin–pulp complex.
Materials and Methods
Preparation of peptide amphiphiles
PA molecules were synthesized as previously described as a 13-amino-acid peptide (GTAGLIGQERGDS) by standard solid phase chemistry on an Advanced Chemtech Apex 396 peptide synthesizer. 10 Preparation of the peptide portion was followed by acylation of the N-terminus. One hundred and fifty milligrams of crude peptide was dissolved in 50 mL DI water at pH 7.0. For purification purposes, peptides were precipitated at pH 3, centrifuged (4000 rad/min, 5 min), the supernatant was removed, and peptides were freeze-dried for 24 h. PA stock solution of 2% by weight was prepared in DI water by adjusting the pH with NaOH to 7.0. PAs were characterized by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry and were found to have the expected molecular weight. Stock solutions were kept under UV light overnight for sterilization purposes.
Cell culture of dental stem cells
Two mesenchymal human stem cell lines were used in this study: DPSC derived from adult third molars 3 and SHED from exfoliated deciduous teeth. 1 Cells were cultured with α-MEM supplemented with 15% fetal bovine serum, 50 μg/mL L-ascorbic acid 2-phosphate, 100 U/mL penicillin, and 100 μg/mL streptomycin, and incubated at 37°C with 5% CO2. Subconfluent cells of passage 5 were detached using trypsin EDTA (Invitrogen, Carlsbad, CA), and 1.0 × 105 cells were seeded per gel. Therefore, 50 μL of PA stock solution (2% by weight) was placed in wells of a 96-well plate. Self-assembly into nanofiber networks was triggered by addition of 50 μL of cell suspension (2 × 106 cells/mL) containing 0.1 M CaCl2 (pH 7.4). The solution was gently and briefly mixed, and gel formation was observed after 2–3 s. For each assay to be performed and for each time point and culture condition, triplicates were seeded. After 30 min, 200 μL of media was added to each well. The medium was changed after 24 h, and osteogenic supplements were added according to previous reports.3,20–22 Three different conditions were established: one group of gels was cultured with medium as described above (0, control); for the second group, 10 mM β-glycerophosphate and 10 nM dexamethasone (SIGMA-Aldrich, St. Louis, MO) were added (βGP + dex); for the third group, 10 mM KH2PO4 (SIGMA-Aldrich) and 10 nM dexamethasone were added to the media (KPh + dex), all of the above being final concentrations. Gels were cultured for up to 4 weeks, media were changed every other day, and samples for different assays were collected at several time points.
Measurement of DNA content
After 3, 7, 14, and 28 days, cell samples were harvested after enzymatic digestion as described above, and cell pellets were frozen down at −80°C for further analysis. After completion of sample collection, these were thawed, and assays and measurements were performed on all samples at the same time. The number of cells in all samples was determined by fluorometric quantification of DNA content using CyQuant cell proliferation assay kit (Invitrogen, Molecular Probes, Carlsbad, CA) and a FLUOstar Optima fluorescence plate reader (BMG Laboratories, Durham, NC). Actual cell numbers were calculated based on a standard curve created from suspensions of known cell densities.
Alkaline phosphatase activity
Samples for the detection of ALP activity were prepared as described for the DNA assay. Cell pellets were resuspended in 60 μL of PBS. After addition of 60 μL of alkaline buffer and 100 μL alkaline substrate solution (SIGMA-Aldrich), samples were incubated at 37°C for 30 min and the liberated p-nitrophenol was measured spectrophotometrically at 410 nm. Samples were compared to a dilution series of p-nitrophenol standard (SIGMA-Aldrich), and ALP activity was normalized to the corresponding cell numbers obtained from the proliferation assay.
Quantitative real-time PCR
To assess the effect of osteogenic induction on the expression of genes involved in differentiation, matrix formation, and mineralization, real-time PCR was performed on samples after 28 days of culture. RNA was extracted using RNA Stat 60 (Tel-Test, Friendswood, TX) from six gels per group at a time to get sufficient amounts for reverse transcription, which was performed according to standard protocols for cDNA synthesis using an oligo-dT primer. One hundred nanograms of RNA was used for one reaction of reverse transcription, which provided cDNA for 10 real-time PCR reactions. Primer sets for marker genes of osteoblast and odontoblast differentiation were designed from mRNA sequences published in GenBank using primer 3 software (http://biotools.umassmed.edu/bioapps/primer3_www.cgi) and synthesized as follows: collagen α(I)I (Col I), collagen III (Col III), ALP, Bsp, Oc, Runx2, Dspp, MMP-2, and glyceraldehyde 3 phosphate dehydrogenase (GAPDH) as an internal control. Identification numbers and primer sequences are shown in Table 1. Primer efficiency was determined prior to quantification. Conditions for real-time PCR were as follows: after a denaturation step at 95°C for 15 min, 60 cycles were run with 95°C (15 s), 58°C (30 s), and 72°C (30 s), with a final dissociation step to generate the dissociation profile of the PCR products. Experiments were run in triplicates (ABI Prism 7900HT), and gene expression was quantified using SYBR green (QuantiTect SYBR green PCR kit; Qiagen, Valencia, CA) and normalized to GAPDH activity in respective samples. Calculations of fold change in gene expression between controls and treated samples were performed according to the Pfaffl method for relative quantification in real-time PCR as described in http://pathmicro.med.sc.edu/pcr/realtime-home.htm.
Histologic analysis
For histological analysis, gels after 28 days in culture were fixed in 4% paraformaldehyde for 3 h, dehydrated through ethanol series, embedded in paraffin, and sectioned at 5-μm thickness. Sections were stained with hematoxylin and eosin (H&E), Masson's trichrome for collagen detection, and von Kossa stain for calcium deposition using standard methods.
Cryo-transmission electron microscopy
To further investigate the mineralization properties, cryo-transmission electron microscopy (cryoTEM) analysis was performed for all groups and samples. Three samples per cell line and condition were cultured as described above. As a cell-free control, additional gels were incubated with osteogenic supplements for 4 weeks. A small quantity of the sample solution (2–3 μL) was applied to a TEM copper grid with holey carbon film purchased from Quantifoil (400-mesh Cu grid, 1.2-μm hole diameter), and blotted with filter paper using a Vitrobot type FP 5350/60 under 100% relative humidity for 2 s to create a thin layer of sample on the surface of the grid. The grid was plunged into liquid ethane and quickly transferred to liquid nitrogen. The sample was then analyzed using JEOL 2010 TEM at an accelerating voltage of 200 kV under low-dose imaging conditions.
Results
Cell proliferation
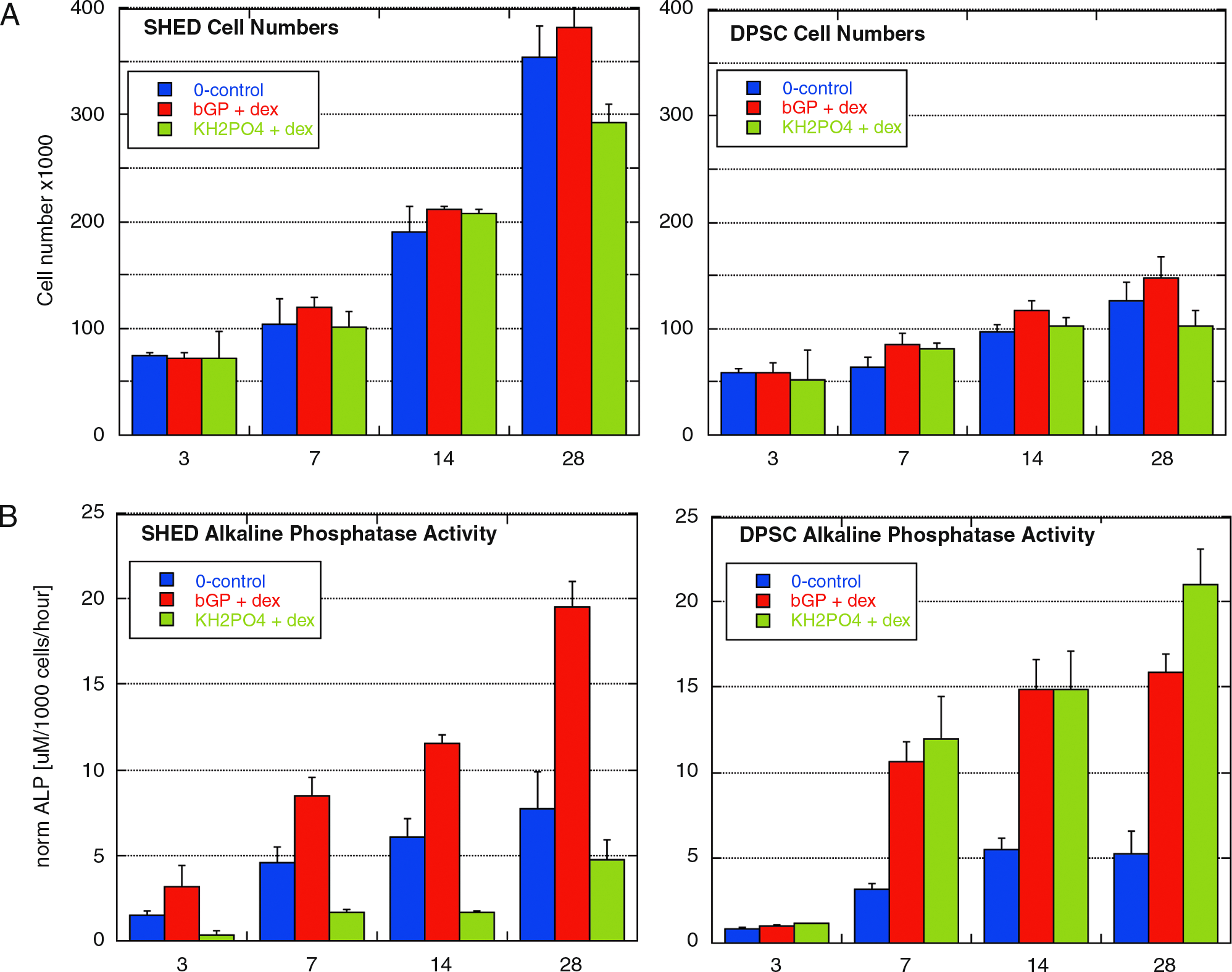
When embedded in 3D PA hydrogels, SHED proliferation was markedly higher as compared to DPSC. Addition of βGP + dex resulted in slightly increased proliferation rates in both cell lines, whereas KPh + dex showed attenuated cell growth. The DNA assay yielded corroborative data. After 28 days, cell numbers of SHED were close to 4 × 105 cells when cultured with βGP + dex, but DPSC did not exceed 1.5 × 105 cells per gel. Growth curves under different culture conditions as well as actual cell numbers for different time points for both cell lines are shown in Figure 2.
(
Differentiation of dental stem cells with different culture conditions
Quantitative measurements of ALP activity showed differences between the two cell lines as well as between groups treated with different osteogenic media. The general trend shows a dramatic increase in both cell lines over time. SHED responded to treatment with βGP + dex with a considerably higher enzyme activity compared to controls, whereas KPh + dex seemed to have opposite effect. DPSC showed a different profile, where KPh + dex evoked slightly higher ALP levels than βGP + dex.
Analysis of marker genes for osteoblast and odontoblast differentiation (Fig. 3) revealed increased expression levels for all the genes investigated after 28 days in culture, with one exception, DPSC ceased to express dentin-specific Dspp after treatment with KPh + dex. In SHED, higher expression levels of genes coding for ECM components (Col I and Col III) could be observed, especially with βGP + dex, and levels of MMP-2 were noticeably elevated. SHED treated with KPh + dex show an increase in osteoblast marker genes, such as Bsp, Oc, and Runx2. The prominent finding for DPSC is that when cultured with KPh + dex, the genes involved in the mineralization process, namely, ALP, Bsp, and Oc, show a substantial increase, which is higher than in SHED. Histologic analysis revealed degradation of the PA gel (Fig. 4A) and replacement with ECM as observed by Masson's trichrome (Fig. 4B, C). Whereas SHED formed clusters of cells (Fig. 4A, B), DPSC were more sparsely distributed within the gel (Fig. 4C). They showed a round, osteoblast-like morphology, and mineral deposition with both osteogenic supplements, but to a higher degree with KPh + dex (Fig. 4D). SHED treated with βGP + dex did not produce mineral deposits, but few deposits were visible for cells treated with KPh + dex after von Kossa stain (data not shown).

Gene expression profile of SHED and DPSC after 4 weeks of osteogenic induction. Data obtained from controls were set to 1 as indicated by the dotted line. The graph shows the fold increase of gene expression compared to control cells cultured in media without osteogenic supplements. Columns show mean values of triplicate samples and error bars the standard deviation. (

(
Vitreous ice cryoTEM was performed on the PA scaffold surrounding the cultured cells. Mineralization of the artificial matrix was observed mirroring the mineralization seen by histology. cryoTEM images revealed little mineral deposition for SHED cultured with βGP + dex (Fig. 5B), but a slightly higher mineralization rate with KPh + dex (Fig. 5C). Mineralization potential in DPSC is much higher; βGP + dex induces deposition mainly of larger crystals (Fig. 5E), whereas KH2PO4 appears to be most conducive for the formation of multiple mineral deposits (Fig. 5F). Control gels with cells cultured with regular media (Fig. 5A, D) or gels incubated with osteogenic supplements for an equivalent period of time without cells (images not shown) did not show mineral deposits.

cryoTEM images show mineral deposition only for cells treated with osteogenic supplements after 4 weeks in culture. Whereas SHED produce few deposits, DPSC exhibit a much higher tendency for mineralization. Treatment with KPh + dex enhances mineral deposition significantly. (
Discussion
In this study, we present an in vitro system in which we combined nanofibrous PA hydrogels with two adult tooth–derived mesenchymal stem cell lines. Our data provide evidence that these cells are able to (1) proliferate within the gel, (2) remodel it by enzymatic degradation and deposition of a collagenous matrix, (3) change their morphology and gene expression profile as a sign of differentiation, and (4) deposit calcium and form a mineralized matrix. All these characteristics vary between the two cell lines as well as between groups treated with different osteogenic supplements. SHED maintain a high proliferation rate and a spindle-shaped, fibroblast-like morphology. They produce large amounts of collagen, which goes hand in hand with a higher MMP-2 activity as an indication of matrix degradation and remodeling, and they form coherent soft tissues during a 4-week culture period. β-glycerophosphate with dexamethasone enhances proliferation and induces upregulation of early differentiation markers such as collagen and ALP. In contrast, KH2PO4 and dexamethasone slightly decrease proliferation and affect the expression of markers of terminal differentiation of cells involved in biomineralization. These include Runx2, Bsp, and Oc. DPSC growth is significantly slower in the 3D environment compared to SHED. Histological images and the change in gene expression after 4 weeks suggest that these cells can be driven further toward terminal differentiation, but adopt an osteoblast-like phenotype displaying a round morphology, significant upregulation of osteoblast markers, downregulation of Dspp, and calcium deposition, especially if cultured with KH2PO4. cryoTEM analysis shows that mineral deposition occurs on the synthetic PA matrix in a cell-mediated fashion. The pattern of mineralization observed by TEM on the PA matrix qualitatively mirrors mineralization as seen by histology in deposited natural ECM. Importantly, this mineralization does not occur in the absence of cells.
Our data are in accordance with the established model for terminal differentiation where cells exit the cycle and cease to proliferate. They are also consistent with previous findings, where SHED were described to be distinct from DPSC regarding their higher proliferation rates and differences in differentiation potential in vitro, which led to the conclusion that they might represent a more immature population of stem cells, 1 whereas DPSC seem to be further advanced in lineage commitment toward a mineralizing cell type. Further, our observations reflect the behavior of these cells in vivo and the differences regarding their mineralization potential in deciduous and permanent teeth. Clinical studies have shown that pulp capping after exposure of the soft tissue in primary teeth shows high failure rates. 23 In permanent teeth, reparative dentin formation after application of calcium hydroxide has been reported in numerous studies. 24 However, formation of dentin with its unique tubular structure can only be observed if the odontoblast layer remains intact; destruction of these cells leads, in many cases, to deposition of an osteoid matrix by replacement cells. Whereas bone and dentin are similar in their matrix protein composition, and osteoblasts and odontoblasts are closely related lineages, their organ structure, cell phenotype, and gene expression profile are distinct. However, the detailed mechanisms underlying the initiation and maintenance of an osteoblast- versus an odontoblast-like phenotype remain to be elucidated. The conditions used in this study seem to favor a differentiation pathway toward the osteoblast lineage. Although both stem cell lines have been shown to differentiate into odontoblasts in vivo, the optimum permissive conditions for odontoblast differentiation in vitro have yet to be clarified. Growth and differentiation factors, such as dexamethasone, TGFβ-1, different BMPs, Dmp-1, or a mixture of dentin noncollagenous proteins, have yielded differentiation of cells into odontoblasts in vitro.25–28 In this study, the cell culture medium was supplemented with dexamethasone to induce cell differentiation, and inorganic phosphate to enhance the mineralization process.
Our data lead to the conclusion that PA molecules provide a nanostructured, cell-responsive matrix that is specifically conducive to dental stem cells. The hydrogels are easy to handle, and due to their mechanical properties, they could be inserted into small defects, such as cavities or periodontal pockets, without difficulties. The two cell lines seeded in PA hydrogels show differences in morphology, proliferation, and differentiation behavior. SHED seem to be a suitable tool for soft tissue regeneration, such as dental pulp, whereas DPSC might be useful for engineering mineralized tissues like bone or dentin, which can be enhanced by the choice of osteogenic supplement. This research may provide us with an applicable system for new treatment strategies to regenerate both soft and mineralized dental tissues.