
Abstract
Tissue engineering of vascularized constructs has great utility in reconstructive surgery. While we have been successful in generating vascularized granulation-like tissue and adipose tissue in an in vivo tissue engineering chamber, production of other differentiated tissues in a stable construct remains a challenge. One approach is to utilize potent differentiation factors, which can influence the base tissue. Endothelial precursor cells (EPCs) have the ability to both carry differentiation factors and home to developing vasculature. In this study, proof-of-principle experiments demonstrate that such cells can be recruited from the circulation into an in vivo tissue engineering chamber. CXC chemokine ligand 12 (CXCL12)/stromal cell–derived factor 1 was infused into the chamber through Alzet osmotic pumps and chamber cannulation between days 0 and 7, and facilitated recruitment of systemically inoculated exogenous human EPCs injected on day 6. CXCL12 infusion resulted in an eightfold increase in EPC recruitment, 2 (p = 0.03) and 7 days postinfusion (p = 0.008). Delivery of chemotactic/proliferation and/or differentiation factors and appropriately timed introduction of effective cells may allow us to better exploit the regenerative potential of the established chamber construct.
Introduction
To enhance the utility of VTE, the growing tissue may be manipulated by either the introduction of additional cell types and/or chemotactic, growth, or differentiation factors. One such chemotactic factor is CXC chemokine ligand 12 (CXCL12), an angiogenic chemotactic cytokine previously called stromal cell–derived factor-1 (SDF-1). 6
As an angiogenic chemokine, CXCL12 has important effects on new blood vessel development. These are both on endogenous angiogenesis (directly and via secondary cytokines such as vascular endothelial growth factor-A [VEGF-A]7,8) and vasculogenesis—the formation of blood vessels from endothelial precursor cells (EPCs).9,10 EPCs have been shown to move from the bone marrow, circulate in the bloodstream, and differentiate into mature endothelial cells in situ to contribute to neovascularization.
EPCs homing to areas of neovascularization require expression of CXCR4 on their cell surface and the presentation of CXCL12 in the target tissue.11–16 CXCL12 may enhance VTE endpoints by increasing blood vessel growth (by angiogenesis and vasculogenesis) and by enhancing the homing of EPCs to the growing tissue.
The aim of this study was to analyze the effects of CXCL12 on a vascularized tissue construct, particularly the homing of EPCs. We assessed the tissue effects by histomorphometric analysis to quantify any differences seen. Immunohistochemistry was utilized to delineate the endothelial-lined blood vessels and leucocyte populations within the tissue. The effects of CXCL12 at a molecular level were analyzed by quantitative RNA determination of the inflammatory chemokine CXCL1, CXCL12 itself, and the angiogenic growth factor VEGF. Finally, fluorescently labeled EPCs were tracked to the tissue construct to determine the effect of CXCL12 on the incorporation of EPCs as this tissue grows.
Materials and Methods
Animal experiments
Nude (CBH/Rnu/nu) rats weighing 200–250 g (Animal Resources Centre, Murdoch University, WA) were housed in microisolator cages. Animal care was in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (NH&MRC, sixth edition, 1997), and experiments were approved by the Animal Ethics Committee, St. Vincent's Hospital, Melbourne.
The VTE model used was essentially as described previously.1,2,5 Under general anesthesia (intraperitoneal ketamine [75 mg/kg; Parnell Labs Pty, NSW, Australia] and xylazine [10 mg/kg; Troy Labs Pty, NSW, Australia]) an AVL was formed from a length of left femoral vein microsurgically anastomosed between the right femoral vessels and placed within a 0.5 mL volume polycarbonate chamber (made by the Department of Chemical and Biomolecular Engineering, University of Melbourne) subcutaneously in the right groin. The opening of the chamber was placed superiorly and attached to the inguinal ligament, and the femoral vessels passed through this opening allowing the whole of the AVL to lie within the chamber.
A CXCL12-containing pump was implanted in the subcutaneous right flank with a catheter passing immediately superficial to the femoral vessels, through the opening of the polycarbonate chamber and terminating at the apex of the AVL within the chamber (Fig. 1a). Recombinant human stromal cell–derived factor-1α (SDF-1α/CXCL12, #557912; BD Biosciences, San Diego, CA) was diluted in 0.1% bovine serum albumin (BSA; CSL, Parkville, Victoria, Australia) to a concentration of 10 μg/mL. This was loaded into an Alzet® microosmotic pump (Model 1007D; Durect, Cupertino, CA) according to the manufacturer's instructions. The specifications of the pumps were provided by the manufacturer to be a mean pumping rate of 0.51 μL/h (SD = 0.03 μL/h) and a mean fill volume of 96 μL (SD = 2 μL). The dimensions of the pump were approximately a 1.5-cm-long, 5-mm-wide cylinder with rounded corners, and a catheter approximately 5 cm long (Fig. 1c). The vinyl catheter tubing contained a volume of 14.9 μL. Therefore, taking into account the overnight presoaking and the residual volume of the catheter, the pump would release a total of 1 mg of CXCL12 protein over a period of 7 days postoperatively. Human EPCs were injected intracardially on day 6. At the conclusion of animal experiments using pumps, the pumps were removed from the animal and checked to ascertain that they were working. All pumps were working at the conclusion of experiments.
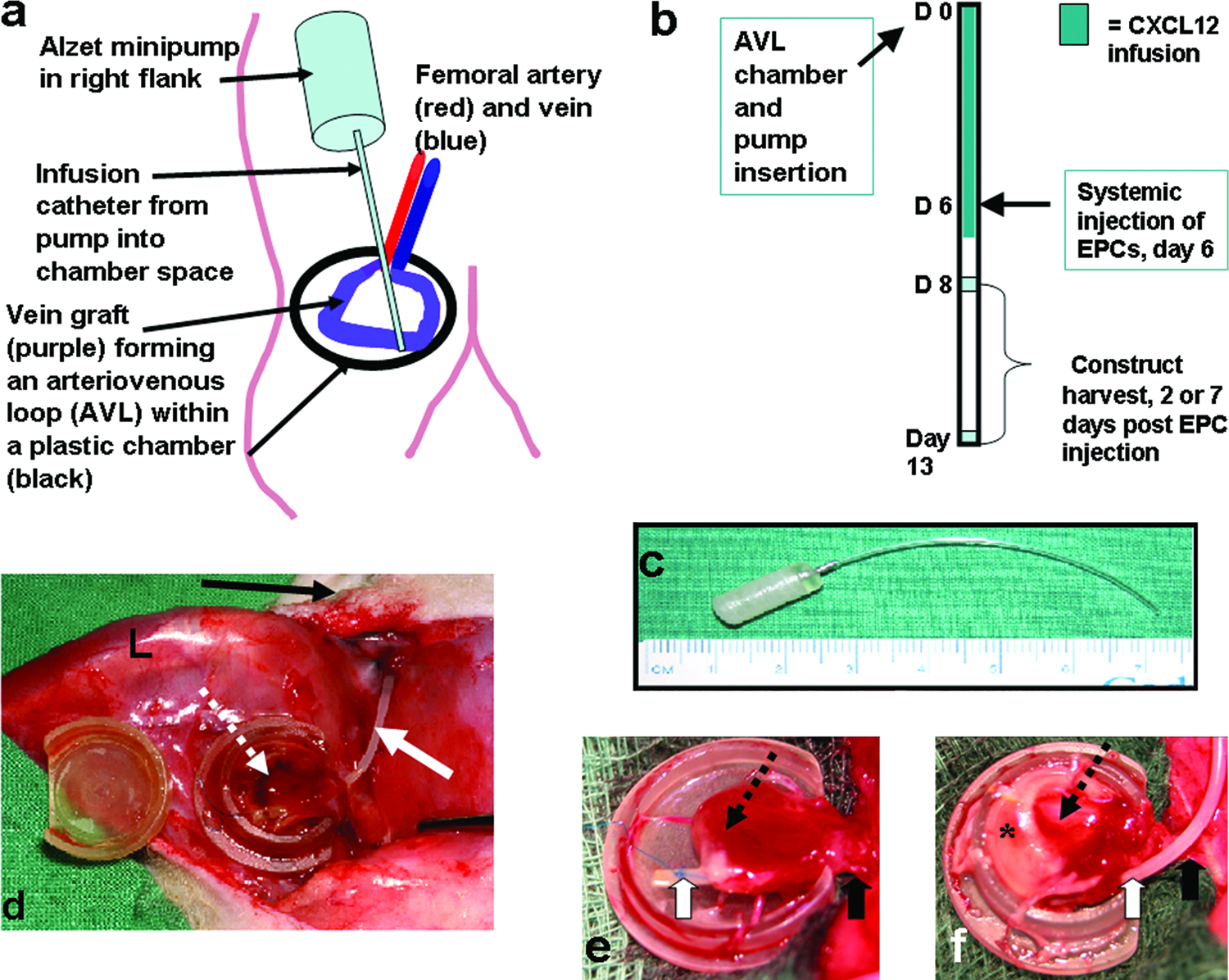
(
Animals were divided into 7 groups and each group comprised four animals. Four groups (as in Table 1) were harvested 2 days following injection of EPCs, and 3 groups (as Groups 2, 3 and 4 in Table 1) were harvested at 7 days following EPC injection. Tissue harvest was therefore 8 or 13 days after creation of the AVL chamber model (Fig. 1b). The contents of the chamber were measured for weight and volume displacement. 17 The tissue was sliced and one slice immediately immersed in liquid nitrogen (for RNA anaylis). The remaining slices were placed in fresh 4% paraformaldehyde fixative for 8-12 h before histological processing.
EPC preparation, labeling, and injection
Human EPCs were provided by the laboratory of Prof. Silviu Itescu, University of Melbourne, Department of Medicine, St. Vincent's Hospital, Melbourne. The cells were retrieved by leucophoresis of patients following up to 3 days of granulocyte colony-stimulating factor stimulation of the bone marrow by daily subcutaneous injection. Cell sorting was by magnetic affinity cell sorting using the CD34 Progenitor Cell Isolation Kit (#467-02; Miltenyi Biotec, Auburn, CA) resulting in a >96% CD34+ purity. 18 The cells were stored frozen until required for experimental use, at which time the viable cells were counted by the Typan Blue exclusion method. Cells from single individual patients were injected into individual rats; that is, EPC samples were not pooled for intracardiac injection.
Cell labeling immediately prior to injection was performed with 3 μg of the fluorescent cell trackers CM-DiI (#C-7000; Molecular Probes, Eugene, OR) for every 1 × 106 cells of cell suspension at a concentration of 1 × 106 cells/mL of phosphate-buffered saline (PBS) for 5 min at 37°C followed by a further 15 min on ice. The cells were then washed three times with PBS (centrifuged at 750 g for 10 min). The cell pellet was resuspended at a concentration of 1 × 106 cells/0.1 mL in saline for injection.
Uniform staining of cells was confirmed on a Zeiss Axioskop 2 microscope with a rhodamine band-pass filter set using an excitation filter wavelength of 546 nm and emission filter wavelength of 590 nm (Zeiss block 15). Membrane-bound CM-DiI has an absorption maximum of 553 nm, an excitation coefficient of 134, and an emission maximum of 570 nm.
Six days following the formation of the AVL, CM-DiI–labeled EPCs (2 × 106 cells in 0.2 mL sterile saline) were injected into the systemic circulation of the rats via an intracardiac injection. 19 The animals were anesthetized with an induction dose of 4% halothane (AstraZeneca, North Ryde, NSW, Australia) in 4 L/min O2.
Histomorphometry
Tissue from each chamber was sliced coronally into 5–8 slices and processed for paraffin embedding (one slice was reserved for RNA analysis, the remaining slices (4–7) were processed for histology). The chamber entrance (where the recipient artery and vein entered the chamber) was considered the front of the chamber, and vertical (top of construct [adjacent to chamber lid] to base of construct [lying on chamber base]) slices (1–2 mm thick) were cut from the chamber entrance to the opposite side of the construct. Tissue slices for histology were placed cut surface down in a single histology block mold for paraffin embedding. It was this coronal slice surface that was microtome sectioned for 5-μm sections for subsequent counting/histomorphometric assessment. This sampling and sectioning technique was standard for all AVL constructs. Five-micron-thick sections of all tissue slices/construct (i.e., 4–7 entire sections per construct, every construct was examined) were stained with toluidine blue or hematoxylin and eosin (H&E), and morphometric analysis of the H&E stained tissue was carried out using the Computer Assisted Stereology Toolbox (CAST) system (Olympus, Denmark). The system was set to systematically and randomly sample an overall fraction of 12% of the entire chamber section using the ×10 objective with a grid of 9 points. (Observer, J. Simcock, was blinded to tissue identity.) It was determined whether each point fell on the AVL, new fibrovascular tissue, fibrin, or fibrin-containing white blood cells (WBCs).
In calculating volume proportions of tissue components, the entire construct is given a value of 1. The volume proportion for each tissue component of each construct was determined by dividing the total points falling over that particular tissue component in every section counted for that construct, by the total points counted (including all tissue components) for that construct. 20 These data are presented as mean tissue volume proportions ± standard error of the mean (SEM) in Figure 3a for all constructs in each group. In addition, within the fibrin compartment only of the construct, the volume proportion of the WBC (leucocyte) component was determined by counting the number of points falling on the WBCs infibrin in all sections of a construct and dividing this figure by the total number of points falling on total fibrin (with and without WBCs) in all sections of the same construct. These data are presented as mean volume proportion of WBCs in fibrin (±SEM) in Figure 3b.
Fluorescent microscopy
Ten-micron-thick sections from the same paraffin blocks (including 4–7 tissue sections from each construct) were dewaxed and hydrated in sequential graduated ethanol. Nuclear counterstaining was performed with 6-diaminido-2-phenylindole (DAPI) (#D1306; Molecular Probes, Invitrogen, Mount Waverley, Victoria, Australia). When bound to dsDNA, this stain has an excitation maximum of 358 nm and an emission maximum of 461 maxima. For staining, 300 μL of 0.1 μM DAPI diluted in PBS was placed on each hydrated section for 1 min. It was removed with three PBS washes and coverslipped with fluorescent mounting medium (Dako, Carpintaria, CA). These slides were stored at room temperature in the dark.
The tissue was viewed with the ×40 magnification objective in a systematic fashion across all chamber slices; that is, the entire section area from every section of every construct (4–7 sections/construct) was examined in a systematic fashion. The observer (J. Simcock) was blinded to the identity of the sections. Individual CM-DiI–stained cells were identified, and their number was recorded per slice of tissue.
Immunohistochemistry
Visualization of vascular endothelium was achieved by immunohistochemical staining with biotinylated Griffonia simplicifolia lectin (rat lectin, #B1105; Vector Labs, Burlingame, CA) as described previously.5,21
To determine the nature of the mononuclear cell population within the AVL tissue, immunohistochemical staining with ED1 (mouse anti-rat CD68 IgG, #MCA341R; Serotec, Oxford, UK), a marker of activated rat macrophages, 22 was performed as described previously. 5
The entire area of all tissue sections available from each construct (4–7 sections/construct) was viewed for both lectin labeling and ED1 labeling to determine the vascularity and macrophage infiltration into each construct. The observer (J. Simcock) was blinded to the identity of the sections during this assessment.
RNA extraction
RNA was extracted from a slice of AVL tissue taken from each construct using TriReagent™ (Molecular Research Center Inc., Cincinnati, OH) under sterile conditions following manufacturer's instructions. RNA was purified using the NucleoSpin RNA II kit (Macherey-Nagel, Duren, Germany) with mini spin columns according to the manufacturer's instructions. RNA concentration and purity was determined for each sample using the Nanodrop spectrophotometer (ND-1000, V 3.1.0; Nanodrop Technologies, Wilmington, DE).
Quantitative RT-PCR
Quantitative RT-PCR was performed to determine the RNA levels of CXCL1, CXCL12, and VEGF. RNA (100 ng) was reverse transcribed to cDNA using the Thermoscript™ RT-PCR system (Invitrogen) and the GeneAmp PCR system (Model 2400; Perkin Elmer, Wellesley, MA) using gene-specific priming (reverse primers shown in Table 2). The product was made up to 100 μL with diethylpyrocarbonate (DEPC)-treated water to give a cDNA concentration of approximately 1 ng/μL.
Quantitative RT-PCR was performed on an ABI Prism 5700 Sequence Detection System (Perkin-Elmer Applied Biosystems, Rowville, Victoria, Australia) on cDNA generated from the equivalent of 2 ng of RNA in 10 mM Tris-HCl (pH 8.0), 2.5 mM Mg(OAc)2, 50 mM KCl, 200 μM dNTPs, 1/40,000 dilution of SYBR Green I (Molecular Probes, Invitrogen), 1 ng/mL 6-carboxy-X-rhodamine (6-ROX) (Molecular Probes, Invitrogen), 8% DMSO, 200 nM primers, and 0.625 U AmpliTaq Gold polymerase (Applied Biosystems, Scoresby, Victoria, Australia) per 25 μL reaction. Reaction conditions were 95°C for 10 min followed by 50 cycles of 95°C for 15 s and 60°C for 1 min. Melt curve analysis was performed at the end of each run from 60°C to 95°C to ensure a single PCR product.
Statistical analysis
Data were analyzed using analysis of variance (ANOVA) on SPSS for Windows v15.0 (SPSS, Chicago, IL). Where possible, two-way ANOVA was used with a Tukey's post hoc test where appropriate. Alpha was set at 0.05. For counts of EPC per tissue slice where numbers were small, an exact Kruskal–Wallis test was used (StatXact v4.0.1, Cytel Software, Cambridge, MA) for an overall p-value with an exact Wilcoxon–Mann–Whitney test for direct comparison between groups.
Results
There was consistency within individual groups regarding macroscopic appearance, general histological architecture, macrophage infiltration, vascularity, and DiI-labeled EPC occurrence in constructs receiving the same treatment. The data relating to each of these assessments for each group appear in the following Result's sections.
Gross appearance and AVL patency
The AVL of all constructs was patent at harvest, and the chamber contents were healthy and showed no significant difference in chamber weight or volume displacement between those treated with CXCL12 or not. Mean construct weights for all groups were between 0.250 ± 0.019 g and 0.305 ± 0.023 g (mean ± SEM), with the CXCL12 infused groups the heaviest (over 0.3 g in each case) at both time points. Mean construct volume varied between 0.228 ±0.016 mL and 0.282 ± 0.02 mL. Constructs that received CXCL12 infusions demonstrated macroscopically the presence of a white infiltrate in the external cuff of fibrin that was not apparent in chambers not infused with CXCL12 (Fig. 1e,f).
Morphology/morphometry
New fibrovascular tissue growth
All groups showed rapid growth of healthy fibrovascular tissue from the AVL into the large fibrin clot that initially surrounds the AVL. At 8 and 13 days all constructs demonstrated a cuff of new fibrovascular tissue (Fig. 2a–d, zone I) around the AVL. The most mature tissue was adjacent to the AVL adventitia, whilst an immature proliferative zone (Fig. 2a, arrow) in which new capillaries were forming was observed adjacent to and radiating into the more peripheral fibrin clot (Fig. 2a, d). In all constructs, outside the new tissue was a hemorrhagic zone (Fig. 2a–d, zone II), and most peripheral was the fibrin clot (Fig. 2a–c, zone III). In all CXCL12-treated constructs an additional zone of leucocyte infiltration was evident between zones II and III (Fig. 2c, d).
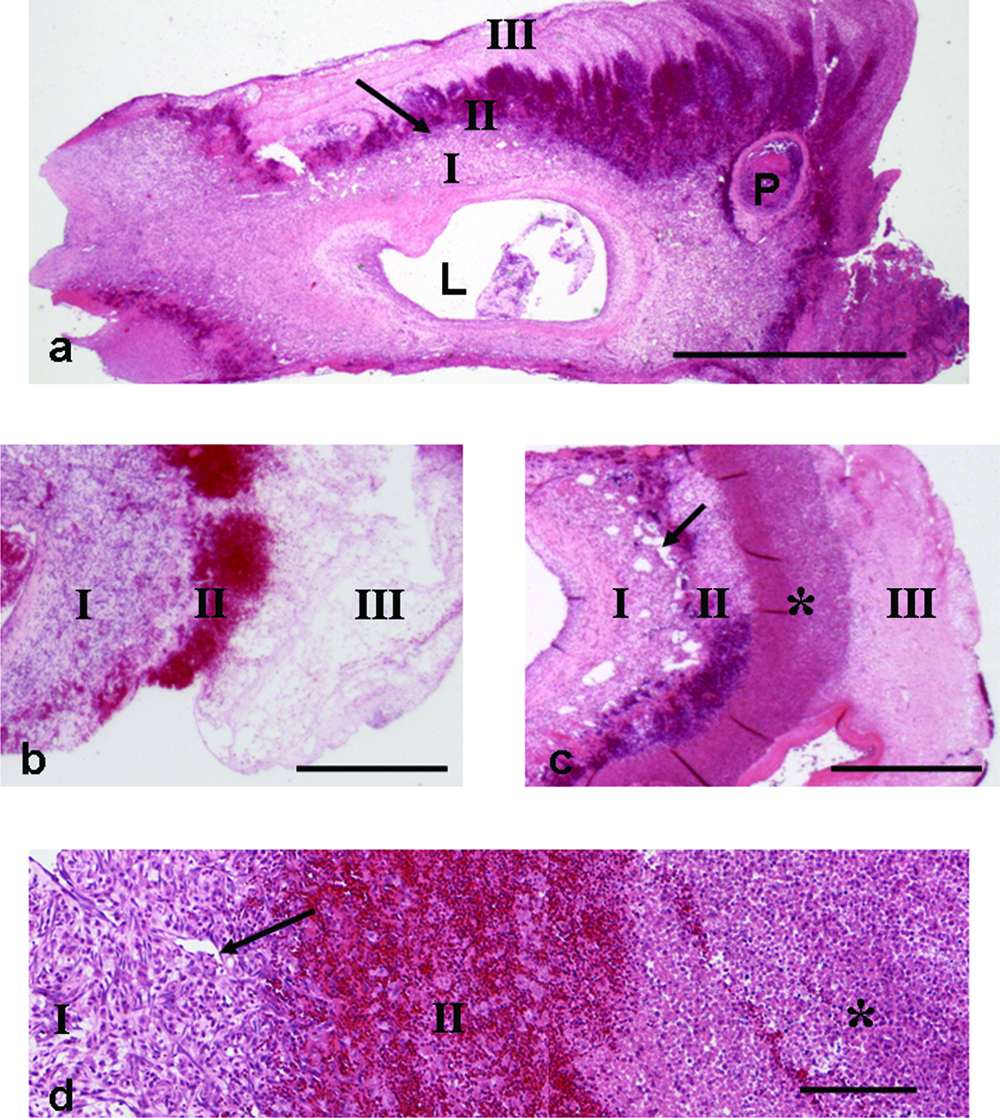
Representative figures of coronal histological sections through the AVL construct. (
Morphometric determination of the volume proportion of the original AVL was similar across all groups (mean ± SEM; AVL proportion was between 0.058 ± 0.016 and 0.020 ± 0.004; Fig. 3a), and no significant difference was observed between groups. At 13 days, more fibrovascular tissue had developed than at 8 days regardless of treatment (Fig. 3a). The mean new tissue proportion at 13 days was approximately 0.6 of the whole construct, but was only 0.35–0.40 at 8 days. More new tissue had formed in the groups that had been treated with CXCL12 infusion; however, this difference was not significant (Fig. 3a). In each case the new tissue formed at the expense of the fibrin component—the proportion of which declined significantly between 8 and 13 days, regardless of treatment (p = 0.002). The mean fibrin component at 8 days was approximately 0.55–0.60 of the whole construct, but at 13 days the fibrin component was approximately 0.25–0.30 of the whole construct (Fig. 3a).

(
WBC infiltrate
The most obvious difference in the CXCL12-treated tissue compared to untreated was the large number of WBCs, including neutrophils and macrophages. This was particularly obvious in the peripheral fibrin clot surrounding the new tissue where an obvious band was apparent (Fig. 2c, d). In the proliferative zone, increased numbers of leucocytes were also observed. The leucocytes were predominantly neutrophils. They included immature neutrophils in the proliferative zone and mature neutrophils in the adjacent fibrin, many of which demonstrated evidence of necrosis with fragmented nuclei (Fig. 4a). There were large numbers of ED1-positive macrophages within the immature fibrovascular tissue in CXCL12-treated AVL tissue (Fig. 4a, b). These were more numerous in CXCL12-treated tissue than in nontreated tissue.

Representative figures of ED1 immunohistochemical labeling of an 8-day AVL construct receiving a CXCL12 infusion. (
Using the fibrin clot as the reference compartment, the proportion of fibrin volume that was infiltrated by leucocytes was quantified by point counting. Statistical analysis by two-way ANOVA (time and treatment) showed no significant difference between the two time points (p = 0.9) but significant differences between treatment groups (p < 0.0005), the addition of CXCL12 increasing the infiltrate significantly from the BSA group (p = 0.004) and the addition of EPCs increasing the proportion again (p = 0.021) (Tukey's post hoc) (Fig. 3b).
Vascularity
Lectin immunohistochemistry clearly demonstrated large numbers of new endothelial-lined blood vessels in all groups. Vascular maturity was also similar, with all groups demonstrating arteriole development at 8 days and beyond. Both control and CXCL12-treated tissue were highly vascular, and no difference in overall vascularity was apparent between groups (Fig. 5b, c).
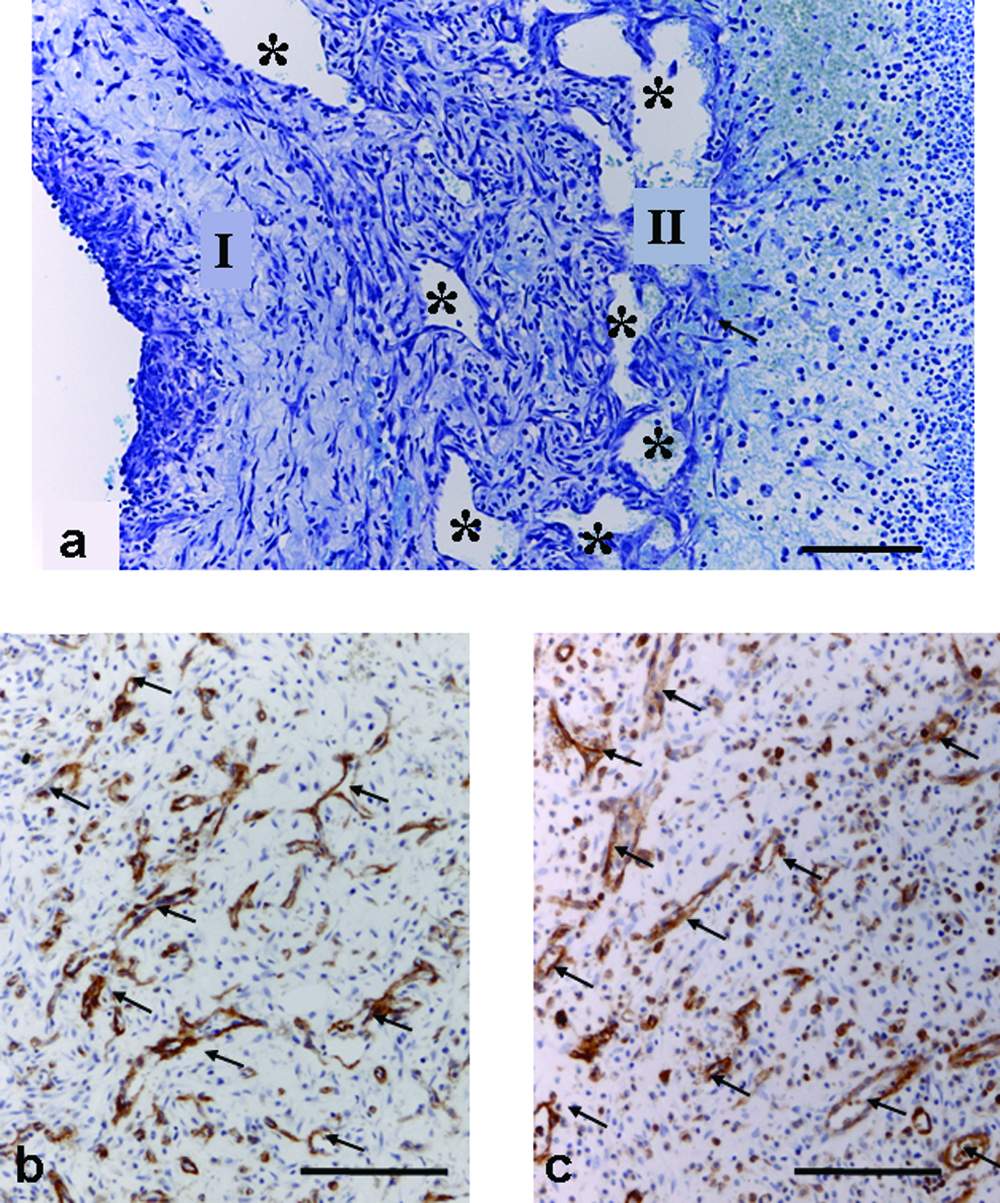
Vascularity of constructs. (
Large endothelium-lined vascular channels were evident in CXCL12-treated tissue at 8 days (Fig. 5a). These channels were smaller and less numerous at 13 days. Vascular channels were not apparent in untreated tissue (compare Fig. 2b and c). Lectin labeling indicated that they were mainly endothelial lined; however, some channels did not appear to be lined with endothelium, particularly when located near the proliferative zone.
RNA analysis
RT-PCR results were normalized in each sample against a housekeeping gene (ribosomal L32). Greater RNA levels are expressed numerically as a smaller negative number (dCt) and graphically as a shorter bar. RNA expression of the CXCL12 gene in AVL tissue from all groups at both 8 days and 13 days was high compared to other gene products that have been measured in our chamber (e.g., transforming growth factor β and VEGF; Z. Lokmic, personal communication). VEGF and CXCL1 RNA levels appeared higher in the CXCL12-infused groups than in the noninfused groups at both time points measured (Fig. 6); however, statistical analysis by one-way ANOVA showed no significant differences in the levels of CXCL1, CXCL12, and VEGF, between the CXCL12 infused and non-CXCL12-infused tissue.
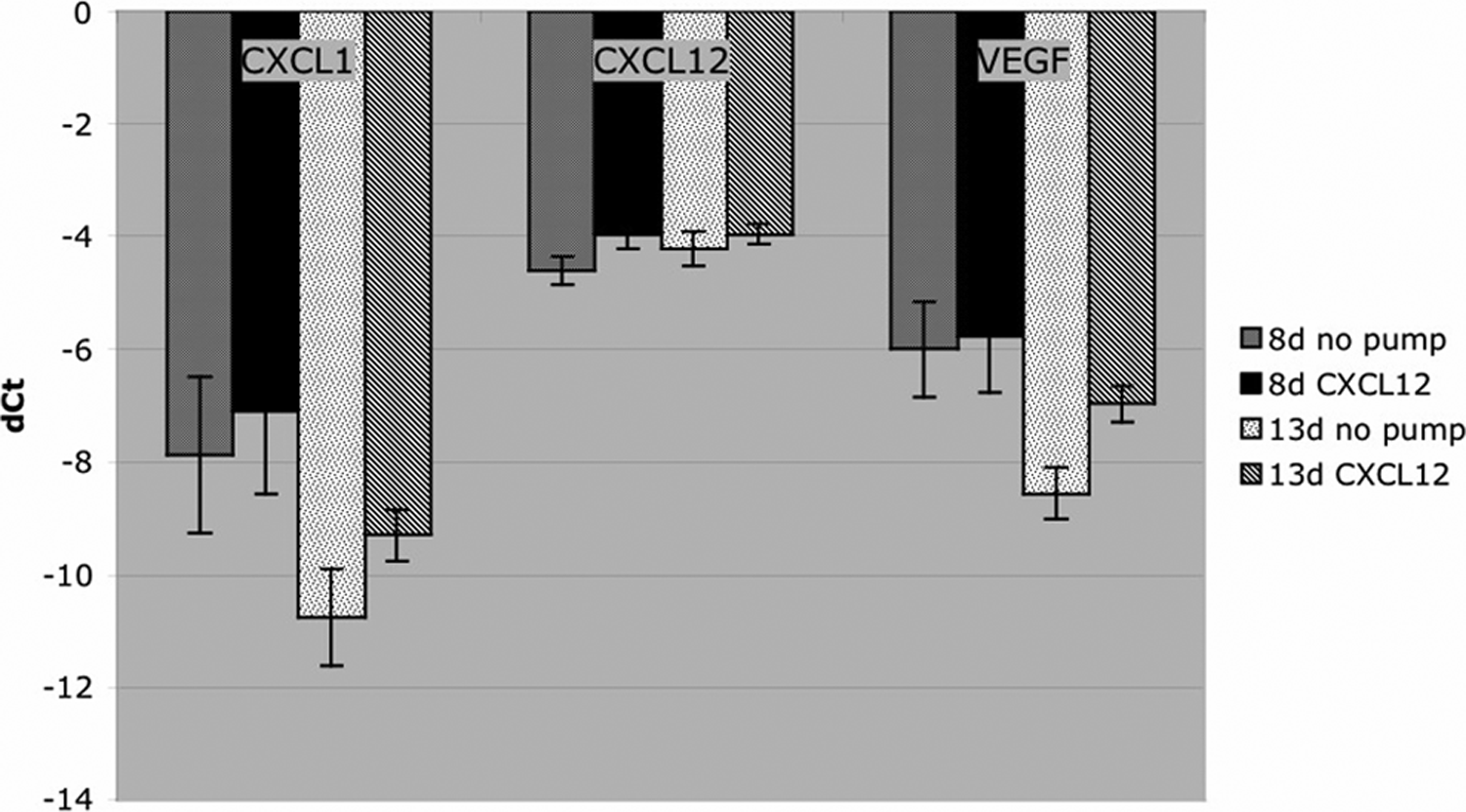
RT-PCR for RNA expression of CXCL1, CXCL12, and VEGF in AVL tissue. There was no significant change in steady-state mRNA levels (mean ± SEM) for all three genes in the CXCL12-infused tissue although a trend to higher levels is indicated.
EPC counts
CM-DiI–labeled EPCs were readily visualized within the AVL tissue by fluorescence microscopy, and every animal that was infused with labeled EPCs demonstrated EPCs within the construct, although the numbers varied according to whether or not the chamber was infused with CXCL12. Labeled EPCs were seen both within the leucocyte infiltrate of the fibrin and within the newly developed fibrovascular tissue (Fig. 7a, b) in all tissue sections of constructs that received CXCL12, although they were not observed as part of the endothelial lining of blood vessels, other than in the femoral vein wall in one specimen. Occasional cells were seen in all control chambers (no pump and BSA + EP), but their numbers were significantly less than in chambers receiving CXCL12. Fewer labeled cells were seen within the tissue that received the carrier protein (0.1% BSA) only (Fig. 7c). Differences in cell numbers between groups were significant (p < 0.0005; exact Kruskal–Wallis). The number of DiI-labeled EPCs in CXCL12-infused chambers at 8 days (p = 0.03) and 13 days (p = 0.008) was significantly higher than controls. Fewer EPCs were seen in the CXCL12 group when harvested at 7 days after EPC injection (total 13 days) as opposed to 2 days after EPC injection (total 8 days) (p = 0.03).
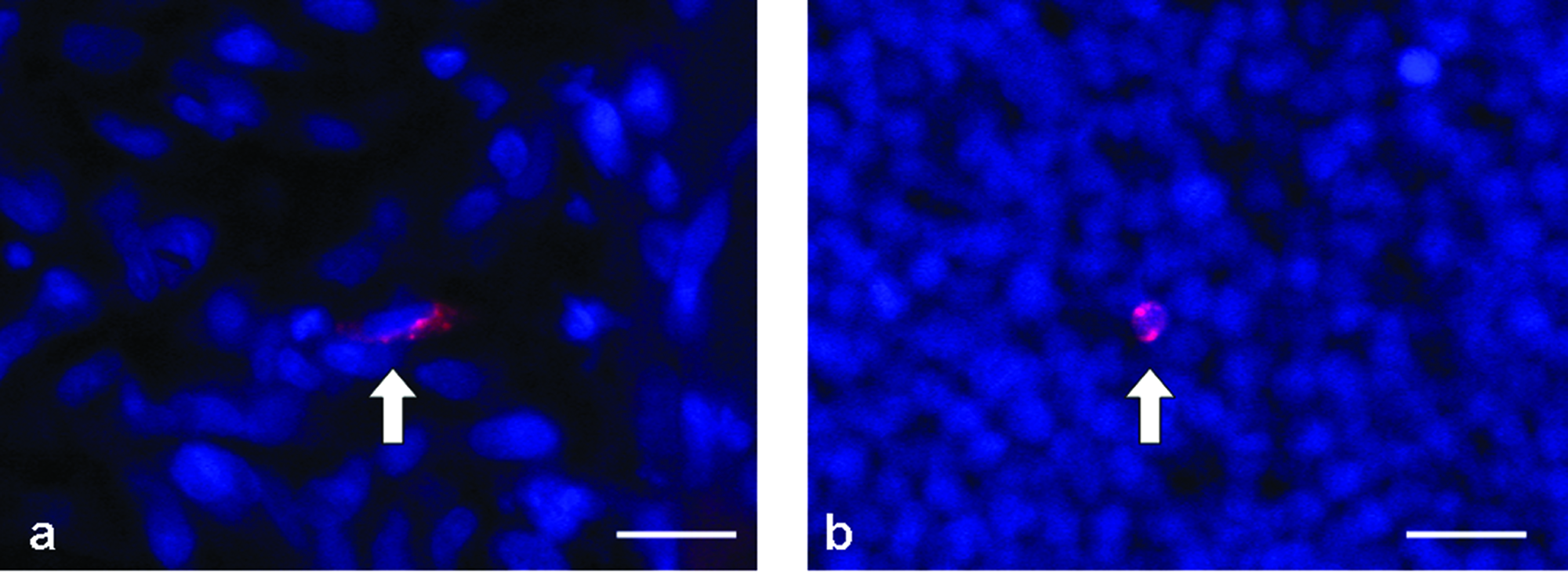
Representative figures of CM-DiI–labeled CD34+ cells 2 days following injection. CM-DiI–labeled cells (arrows) were seen within new fibrovascular tissue (
Discussion
Tissue engineering implies the creation of whole tissue consisting of cells, matrix, and a blood supply. Bringing the three components together in the same place at the same time is one of the central challenges of tissue engineering. Either cells are combined with matrix in a bioreactor and the blood supply must grow into the construct once it is placed in the body with an inevitable period of hypoxia, or a matrix is vascularized in the body and cells must be added to an already fixed structure. One novel way to deal with this conundrum is to build a vascularized structure, and deliver new pluripotent or specialized cells to it via the growing microcirculation. Such a strategy will require a method of attracting specific cell types into the growing structure.
This study demonstrates that an angiogenic chemokine (CXCL12) can be successfully infused into a growing tissue-engineered construct to exert bioactive effects, including significantly increased EPC homing, a concomitant influx of leukocytes predominantly neutrophils into the fibrin clot, and of macrophages into the growing tissue. None of these increased cell infiltrations had any effect on normal angiogenesis and connective tissue growth and maturation that occurs in the AVL construct up to 13 days. CXCL12 has previously been associated with increased numbers of lymphocytes 23 ; however, this was not seen in our tissue, presumably because nude rats are T-lymphocyte deficient.
Postnatally, the main function of CXCL12 is leucocyte homeostasis. Both mature and progenitor hematopoietic cells are controlled by the CXCL12/CXCR4 axis. As well as playing a central role in the interactions of hematopoietic stem cells, lymphocytes, and developing neutrophils in the bone marrow, the CXCL12/CXCR4 axis has recently been shown to be critical in circulating neutrophil homeostasis. In particular, CXCR2-binding inflammatory chemokines release neutrophils from the bone marrow by reducing CXCR4 bone marrow expression. 24
The CXCL12/CXCR4 axis has effects both on endogenous angiogenesis (either directly or via certain secondary cytokines, e.g., VEGF-A) and on vasculogenesis (via EPC mobilization and homing). The mechanism by which circulating EPCs home to areas of neovascularization such as that occurring in the AVL construct is a CXCL12/CXCR4-dependant process requiring both CXCR4 surface receptor on the EPCs and CXCL12 on the luminal surface of the endothelium.11,15 CXCL12 induces a rapid but transient activation of integrins on the circulating EPCs, thus facilitating the binding of the cells to endothelium and their egress from the circulation. 25
There are many reported secondary effects of CXCL12 as an angiogenic chemokine, including increasing expression of angiogenic cytokines. CXCL12 is proinflammatory and proangiogenic. The coordination of angiogenesis and inflammation is a result of the ability shared by endothelial cells and leucocytes to respond to chemokines via their specific receptors.26,27 This study demonstrates the combined effects of CXCL12 on the recruitment of EPCs and macrophages and neutrophils to the chamber. These cells home down CXCL12 gradients via the CXCR4 receptor, and the addition of exogenous CXCL12 into the chamber “bioreactor” increases that gradient toward the chamber and therefore promoted the recruitment of all three cell types. Certainly, macrophages and neutrophils produce more angiogenic factors that may have influenced the angiogenesis in the chamber although no increase in angiogenesis was observed in CXCL12-treated chambers. It should also be acknowledged that the normal “untreated” chamber growth is highly angiogenic and always includes many macrophages. There was no obvious tissue death in the new tissue within the chamber; despite the increase in macrophages. However, there was significant necrotic death in neutrophil infiltrates within the fibrin. Thus most of the invading neutrophils die soon after reaching the chamber and may have very little influence on other cell movements.
VEGF can be produced by many of the cells involved in the AVL construct's angiogenic system, including endothelial cells, macrophages, and neutrophils.28,29 The role of CXCL12 is supported by a regulatory loop between VEGF-A and the CXCL12/CXCR4 axis. CXCL12 upregulates VEGF-A production, and VEGF-A upregulates CXCR4 expression, thus generating an amplification circuit crucially influenced by hypoxia. 7 We have previously demonstrated that the AVL chamber is hypoxic between days 3 and 14.5,30 This level of hypoxia was not effective in promoting EPC recruitment, whilst a specific infusion of CXCL12 as demonstrated in this study provided a localized target to circulating CXCR4+ cells.
CXCL12-infused tissue demonstrated large, irregular vascular channels not seen in untreated chambers 5 previously. These channels may either have been generated due to the chemokine itself or, more likely, by the significant numbers of additional leukocytes attracted to the chamber circulation, which then underwent diapedesis into the new tissue and surrounding fibrin clot. These spaces did not persist following the end of the infusion and did not appear to exert any adverse effects on the tissue. We did not see irregular large vascular channels or increased numbers of leucocytes in tissue where only the carrier protein solution was infused, attributing these effects specifically to CXCL12 rather than a nonspecific inflammatory response to the carrier protein, catheter, or pump.
Infusion of CXCL12, an angiogenic chemokine, may be expected to increase vascularity in the growing tissue. Unlike the studies of subcutaneous CXCL12 injection that resulted in increased vascularity, 7 we were unable to demonstrate any marked increase in vascularity in CXCL12-treated constructs. This may be due to the fact that, unlike mature subcutaneous tissue, the tissue growing from the AVL was already highly vascular, 5 considerably more so than normal skin or healing skin wounds5,21 and that there was therefore limited capacity for it to increase. The fact that the AVL construct tissue was highly angiogenic could also explain why the CXCL12 infusion did not result in a significant increase in VEGF RNA expression in the tissue. It is possible that since the level of VEGF expression was already high in the AVL tissue, there may have been limited capacity for it to increase. The lack of statistical significance in the trend toward increased VEGF RNA expression could also be due to variations in measurement due to tissue heterogeneity, as reflected in the relatively large error margins.
In addition to the leucocyte infiltrate described above, EPCs were observed to spontaneously home to the untreated chamber tissue in very small numbers; however, human EPCs homed in significantly increased numbers to the growing construct tissue in response to the CXCL12 infusion. This finding is consistent with the successful use of CXCL12 to augment EPC homing in ischemic models. 31 There are a number of differences between the two models, including that the ischemic limb skeletal muscle has lower vascularity than the AVL construct and so interventions in the ischemic limb model aim to enhance neovascularization. It also has an existing extensive capillary network through which EPCs may home to the tissue immediately following onset of ischemia. In contrast, our model of VTE does not produce an abundant capillary network until 5–14 days. 5 Therefore, the AVL model required a CXCL12 infusion over time rather than a single injection. Preliminary studies showed that no EPCs homed to the AVL until the capillary network had developed (data not shown).
Enhancing the vascularity of the AVL engineered tissue was not a major objective of this study, rather enhancing EPC recruitment to the AVL tissue was undertaken with the aim that, in future experiments, a specific differentiation signal could be introduced into the early chamber tissue. This study demonstrates that CXCL12 may be utilized to enhance EPC homing to rapidly growing tissue. CXCL12 has specific transient histomorphologic effects on the tissue, but has no detrimental effects on either quantity or quality of the tissue grown.
The use of the osmotic pump with the AVL construct is a major advancement. This is the first time the AVL construct has been manipulated whilst in vivo to modify the growth of the construct. At the moment, CXCL12 is required to be infused for a number of days to produce a response in terms of increased EPC homing. Although desirable, the technology to incorporate CXCL12 into a controlled drug release system within the construct is currently not available, but one can envisage advancements in minipumps and/or drug delivery systems and/or matrices that would allow this in the future, not just for the administration of CXCL12, but for any number of chemicoattractant, proliferative, or differentiation factors, depending on the tissue being engineered.
In summary, in VTE, exogenous CXCL12 significantly enhanced homing of EPCs to an angiogenic site developing around an AVL inserted in a tissue engineering chamber, thus enabling this population of precursor cells to be used in future experiments to carry a differentiation message specifically to the engineered construct.
Footnotes
Acknowledgments
The authors wish to acknowledge EMSU staff, St. Vincent's Hospital—Sue Mc Kay, Liliana Pepe, Anna Deftereos, and Amanda Rixon—for assistance in animal surgery, Jason Palmer for assistance in histology, Effie Keramidaris for assistance in EPC preparation, and Tony Blicks for assistance with RT-PCR. The EPCs were kindly donated by the laboratory of Prof. Silviu Itescu, University of Melbourne, Department of Medicine, St. Vincent's Hospital; J. Simcock was the recipient of a University of Melbourne Scholarship and a Royal Australasian College of Surgeons Scholarship. The project was also supported by a St. Vincent's Hospital Research Grant.