
Abstract
This study addresses synergistic effects of bone morphogenetic protein-2 (BMP-2) and transforming growth factor-β3 (TGF-β3) in the induction of chondrocytic differentiation of bone marrow multipotent mesenchymal stromal cells (BM MSCs) in vitro for potential use in intervertebral disc (IVD) repair. Human BM MSCs encapsulated in alginate beads were induced to differentiate in serum-free medium containing BMP-2 and TGF-β3. The expression of chondrocytic genes and proteins was analyzed by real-time PCR, western blot, histological, and immunohistochemical assays. This differentiation system showed a potent induction of chondrocytic phenotypes. The expression of chondrocytic markers, such as aggrecan (ACAN) and type II collagen (COL2A1), was upregulated at higher levels than using TGF-β3 alone. Blocking BMP-2 by noggin completely suppressed BMP-2–enhanced gene and protein expression, confirming a crucial input of BMP-2 signaling in this differentiation process. Inhibition of extracellular signal-regulated kinases 1 and 2 signaling resulted in an increase in ACAN and COL2A1 gene expression, suggesting a negative regulatory role of this pathway. In conclusion, BMP-2 enhances TGF-β3–mediated chondrogenesis of MSCs. The combination of BMP-2 and TGF-β3 in alginate culture is superior to the standard differentiation method using TGF-β alone. This potent induction system may provide an alternative cell source for IVD and cartilage regeneration in clinical practice.
Introduction
Bone marrow–multipotent mesenchymal stromal cells (BM MSCs) are capable of differentiating into various cell lineages, including osteoblasts, chondrocytes, and adipocytes. BM MSC transplantation showed effective in decelerating disc degeneration in experimental models, providing us a new hope to utilize BM MSCs as an alternate cell source for NP regeneration.6,7 Directed differentiation of MSCs into chondrocytes in vitro has been studied by means of exposure to exogenous growth factors, coculture with cartilage or NP cells, 8 and overexpression of specific genes to promote chondrocytic gene expression. 9 Transforming growth factor-β (TGF-β)–based techniques have been extensively used by supplementation of TGF-β isoforms in serum-free induction medium applied to high-density MSCs in three-dimensional (3D) cultures, such as cell pellets, agarose gels, alginate beads, synthetic polymers, and other biomaterials.10–12 Since NP cells are chondrocyte-like cells, it is rational to apply similar principles of chondrogenic differentiation of MSCs to generating NP-like cells for the potential use in IVD repair. In the limited studies aiming to generate NP-like cells, differentiation of MSCs was induced in vitro using TGF-βs as the chondrogenic morphogens.13,14 In recent years, bone morphogenetic proteins (BMPs) have emerged as key regulators of stem cell commitment. 15 BMP-2 plays an essential role in osteoblast and chondrocyte differentiation, inducing the formation of bone and cartilage. BMP-2 has also been shown to enhance the expression of chondrogenic matrix components, such as type II collagen and aggrecan, and maintain chondrocytic phenotype in vitro in human articular chondrocytes16,17 and IVD cells. 18 It has been reported that BMP-2 can augment chondrogenic differentiation of human embryonic stem cells in 3D culture. 19
In the present study, we hypothesized that a combination of BMP-2 with TGF-β3 in alginate bead 3D culture is more effective than TGF-β3 to induce chondrocytic differentiation of BM MSCs. The chondrocytic marker gene expression of differentiated BM MSCs was compared with that of human NP tissue and cultured NP cells. The possible molecular control and signaling pathways involved in the differentiation process were also studied. Our results demonstrated that BMP-2 could synergistically enhance TGF-β3–mediated chondrogenic differentiation of BM MSCs with the phenotypes similar to NP cells, which provides the cells with a potential to be used for IVD repair.
Materials and Methods
Tissue samples
All tissue samples were collected following written informed consent under ethical approval from Human Research Ethics Committees of St. Vincent's Hospital and St. George Hospital Sydney, Australia. Human BM was collected from iliac crest of six hematologically normal donors (age range 27–64 years). Human IVD tissue was collected from eight patients (age range 18–66 years) with degenerated IVD and undergoing lumbar disc replacement. The NP tissue was immediately separated from AF after surgery. Half of NP tissue sample was used for RNA extraction, and the other half was for NP cell isolation.
Cell isolation and cultivation
BM MSCs were isolated by immunodepletion, Ficoll-Paque density gradient centrifugation, and plastic adhesion. 20 Briefly, fresh BM specimens were incubated for 20 min with the antibody cocktail (StemCell Technologies, Vancouver, Canada) to remove mature lineage-committed cells. Ficoll-Paque (GE Healthcare, Uppsala, Sweden) density gradient centrifugation was then performed to separate mononuclear cells from antibody cross-linked cells, and enriched cells from the interface were seeded in plastic cultureware. The cells were cultured in growth medium consisting of 51% Dulbecco's modified Eagle's medium–low glucose (DMEM-LG), 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA), 34% MCDB-201 medium, 1% insulin transferrin selenium (ITS), 1% linoleic acid/bovine serum albumin (BSA), 1 nM dexamethasone, and 32 μg/mL ascorbic acid 2-phosphate (Sigma-Aldrich, St. Louis, MO) and incubated at 37°C with 5% CO2. After 3 days, nonadherent cells were discarded, and adherent BM MSCs were cultured to 80% confluence with medium changed twice weekly. The NP cells were isolated by overnight digestion with 0.025% collagenase solution and collected by centrifugation. NP cells were cultured in DMEM-LG containing 32 μg/mL ascorbic acid 2-phosphate and 10% FBS. Passage 0 cells were used as positive control.
Flow cytometry analysis
MSCs were trypsinized and washed with PBS containing 10% FBS and incubated with human AB plasma at 4°C for 30 min. After washing with FACS buffer (PBS containing 13.6 mM Tri-sodium citrate and 1% BSA), MSCs (1 × 105 per tube) were resuspended in 50 μL FACS buffer and labeled with 5 μL of fluorescein isothiocyanate, phycoerythrin, or peridinin chlorophyll protein conjugated monoclonal antibodies in dark at 4°C for 30 min. Antibodies used included CD29, CD73, CD45, CD14, CD34, CD166, HLA Class I, HLA Class II (BD Biosciences Pharmingen, San Jose, CA), CD44, and CD105 (Chemicon, Temecula, CA). The cells were analyzed on an FACSCalibur flow cytometer (BD Biosciences).
Chondrogenic differentiation in alginate bead 3D culture
MSCs at passages 3–6 were trypsinized and suspended in a solution of 1.2% (w/v) low-viscosity sodium alginate (Sigma-Aldrich) in 150 mM NaCl, pH 7.4, at the density of 5 × 106/mL for differentiation and 1 × 106/mL for undifferentiated control. Alginate beads were made by gently pressing the cell suspension dropwise into 102 mM CaCl2 solution through a syringe with a 19 G needle. The hydrogel beads formed instantly and were placed in 12-well plates after washing three times with 150 mM NaCl solution.
Chondrogenic differentiation was induced by adding serum-free media containing DMEM-high glucose supplemented with 100 nM dexamethasone, 50 μg/mL ascorbate 2-phosphate, 40 μg/mL L-proline, 1.25 mg/mL BSA, 5.35 μg/mL linoleic acid, 1% ITS solution, and 10 ng/mL of recombinant human (rh) TGF-β3 alone or in combination with 100 ng/mL of rhBMP-2 (TGF-β3&BMP-2; R&D Systems, Minneapolis, MN). In some parallel experiments, MSCs were treated with 200 ng/mL recombinant mouse noggin (R&D Systems) to block BMP-2 signaling or with 20 μM U0126 (Sigma-Aldrich) to block extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling. Undifferentiated MSCs were cultured in parallel in growth medium. Cells were kept at 37°C and 5% CO2 for up to 21 days. The media were changed twice weekly. For cell recovery, the cell beads were washed twice in PBS and incubated in 55 mM of Na-citrate solution, pH 7.4, at 37°C for 10 min. The solubilized alginate was removed by centrifugation, and the cell pellet was washed with PBS.
RNA extraction, cDNA synthesis, and real-time PCR
Total RNA was isolated from MSCs, NP tissue, and cultured NP cells using TRIzol reagent (Invitrogen) and RNeasy kit (Qiagen, Düsseldorf, Germany) following manufacturer's instructions. cDNA was prepared using SuperScript III first-strand synthesis system (Invitrogen). Total RNA (1 μg) was reverse transcribed in a final volume of 20 μL using M-MLV reverse transcriptase (200 units) and a mixture of random hexamers (50 ng) and Oligo(dT)20 (50 pmol) as primers. Samples were incubated at 25°C for 10 min, 50°C for 50 min, and then heated to 85°C for 5 min. The 1:40 diluted cDNA was used in 20 μL reactions for real-time PCR analysis in a Rotor-Gene RG3000 system (Corbett Life Science, Sydney, Australia). All primers were designed using published mRNA sequences. To exclude possible genomic DNA contamination, the RNA was treated with DNase and primers were designed to be intron spanning. The thermal profile for all reactions was as follows: 5 min at 95°C, followed by 40 amplification cycles of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C. The GenBank access numbers, real-time PCR primers, and product sizes are listed in Table 1. Relative expression levels were calculated as a ratio to the average value of housekeeping genes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and hypoxanthine phosphoribosyltransferase 1 (HPRT1).
Histology and immunohistochemistry
Alginate beads encapsulated with MSCs were fixed in 10% neutral buffered formalin for 1 h and embedded in paraffin. Sections of 4 μm thickness were cut and mounted on Super Plus slides (Lomb Scientific, Sydney, Australia). Sections were dewaxed in xylene and hydrated with graded ethanol before staining. Hematoxylin-eosin staining was carried out for general histological examinations. For Alcian blue staining, slides were stained in 1% Alcian blue solution for 15 min, and nuclei were counterstained with 0.1% nuclear fast-red solution. For immunohistochemical staining, slides were equilibrated in Tris-HCl (pH 7.6) buffer. Endogenous peroxidases were scavenged with 3% (v/v) H2O2, and unspecific binding was blocked by incubation in 10% skim milk in Tris-HCl buffer. The sections were incubated with primary goat anti-human type II collagen (1:100 dilution) and goat anti-human SOX9 (1:200 dilution) polyclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature. Slides were treated with Multilink solution (DAKO, Sydney, Australia) followed by streptavidin-conjugated peroxidase incubation. The sections were visualized with 3,3′-diaminobenzidine hydrochloride solution and counterstained with hematoxylin. The primary antibody was omitted for the negative controls.
Western blot analysis
Cells were rinsed with cold PBS and lysed in CelLytic-M solution containing protease inhibitors (Sigma-Aldrich). Equal amount of proteins (20 μg) were electrophoresed on 8–12% gradient SDS-polyacrylamide gels (Invitrogen). Proteins were transferred by electroblotting to polyvinylidene fluoride (PVDF) membranes, which were then blocked with 5% skim milk in Tris-HCl buffered saline (TBS; 20 mM Tris [pH 7.6], 0.15 M NaCl) overnight at 4°C. Membranes were incubated with goat anti-human type II collagen and rabbit anti-human SOX9 polyclonal antibodies (Santa Cruz Biotechnology) in TBS buffer containing 0.1% Tween-20 (TTBS) for 2 h at room temperature. Alpha-tubulin was detected as reference protein. After washing and incubation with fluorescent dye-conjugated secondary antibodies, immunolabeling was detected using the Odyssey infrared imaging system (LI COR Biosciences, Lincoln, Nebraska).
35S-sulfate incorporation
The cell function of differentiated MSCs was investigated in vitro by detecting the biosynthesis of proteoglycans using 35S-sulfate incorporation assay. 21 In brief, the alginate beads containing differentiated MSCs were incubated with 20 μCi/well of 35S-sulfate (GE Healthcare) at 37°C for 24 h. Following release from alginate beads, the cells were harvested and resuspended in papain digestion buffer containing 2 μL of papain suspension per 1 mL of PBS (pH 6.2), 5 mM L-cysteine, and 10 mM EDTA at 60°C for 3 h to release glycosaminoglycans. An aliquot was separated for DNA determination. Newly synthesized 35sulfated glycosaminoglycans were separated from free 35SO4 by a precipitation procedure. Samples were then counted in an automated Scintillation Analyser and normalized by DNA concentration. The fold change of relative counts represents the change in proteoglycan synthesis.
Statistics
Data were presented as the mean ± SEM. Statistical analysis was performed using Student's t test for comparison of the TGF-β3&BMP-2–treated group with the TGF-β3 alone–treated group in gene expression analysis. Differences between TGF-β3&BMP-2 and TGF-β3 were considered statistically significant at *p < 0.05 and **p < 0.01.
Results
Morphology and characterization of BM MSCs
BM MSCs presented a typical fibroblast-like morphology in monolayer expansion culture (Fig. 1A) and a rounded morphology when encapsulated in alginate beads (Fig. 1B). The MSC phenotype characterized by flow cytometric analysis showed a standard pattern of surface markers for human MSCs, namely positive for CD29 (beta 1 integrin), CD44 (hyaluronan receptor), CD105 (endoglin), CD166 (ALCAM), and human leukocyte antigen class I (HLA-I), and negative for hematopoietic antigens, such as CD14 (LPS receptor), CD34 (hematopoietic stem cell marker), CD45 (leukocyte common antigen), and human leukocyte antigen class II (HLA-II) (Fig. 1C).
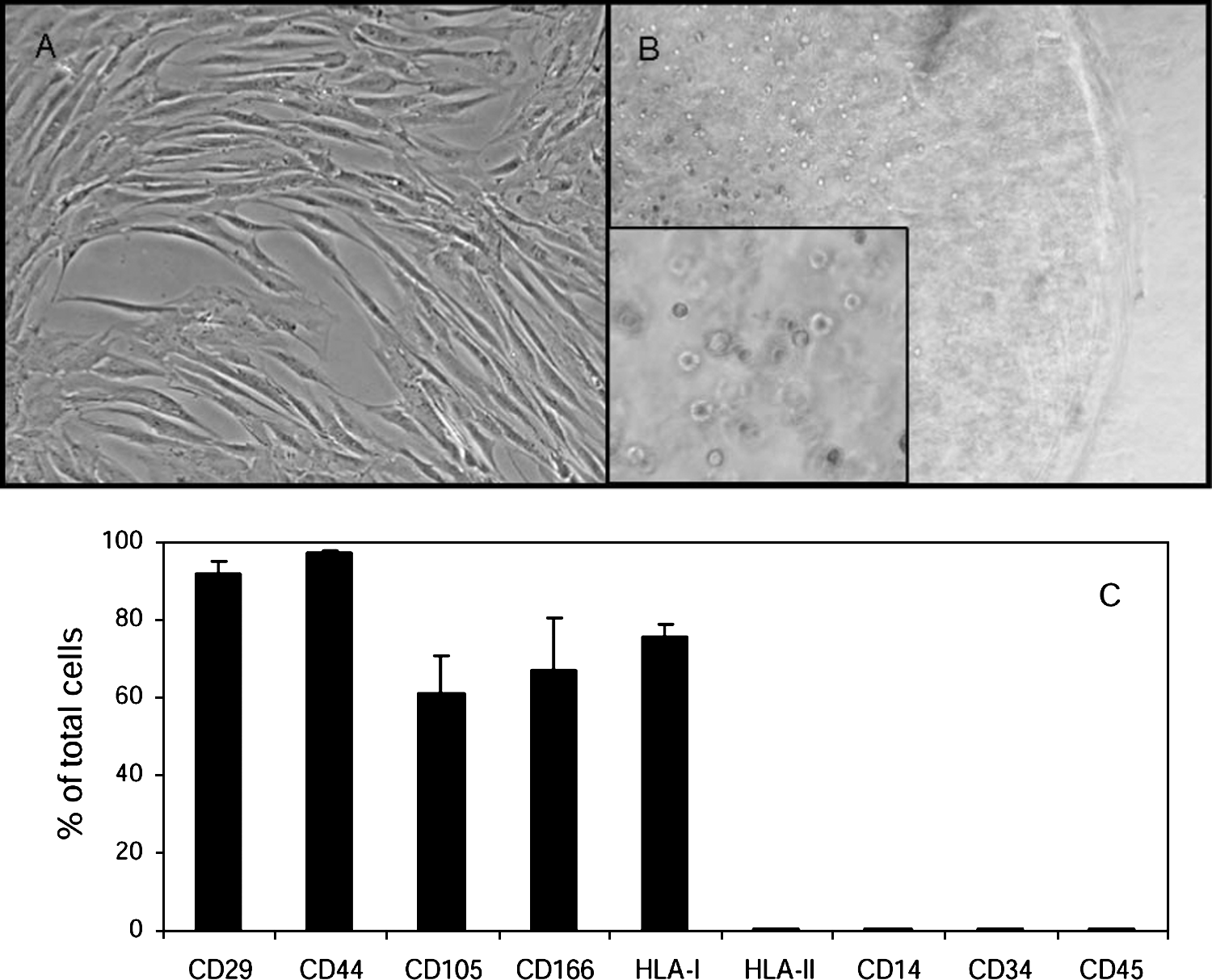
Morphology and characterization of BM MSCs. MSCs adopt a fibroblast-like morphology in monolayer culture (
Specific gene expression in chondrogenic differentiation
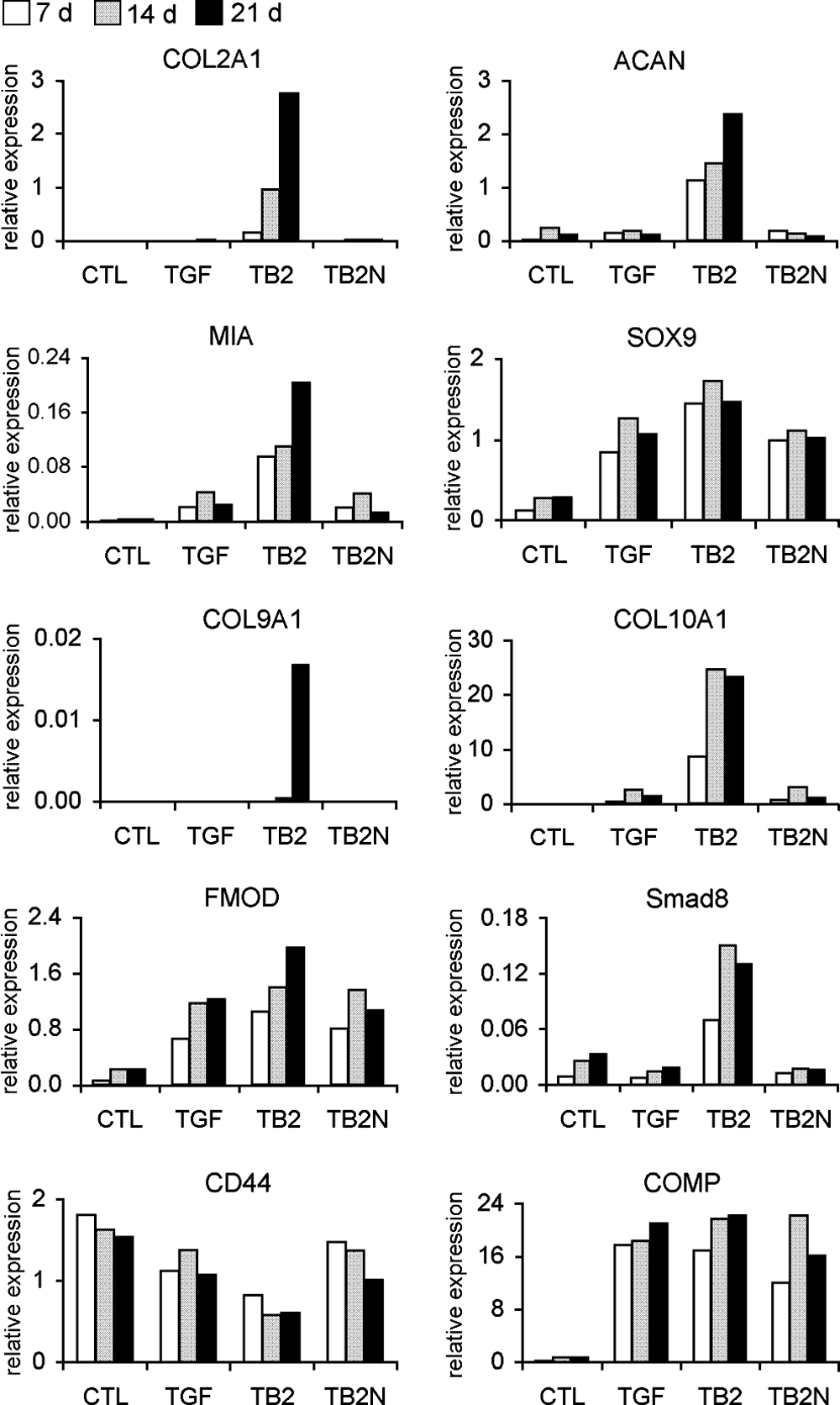
At the initial stage of this study, the differentiation of MSCs was induced with TGF-β3, BMP-2, or TGF-β3&BMP-2, respectively, in alginate beads for 21 days. TGF-β3&BMP-2 showed a much more potent induction of chondrocytic gene expression than TGF-β3 or BMP-2 alone (Fig. 2). Subsequent experiments were carried out comparing TGF-β3&BMP-2 with standard TGF-β3 method. To follow MSC differentiation, the gene expression of markers for chondrocytic cells, MSCs, and osteoblasts was analyzed on days 7, 14, and 21 by means of real-time PCR using undifferentiated MSCs as negative control (Fig. 3).
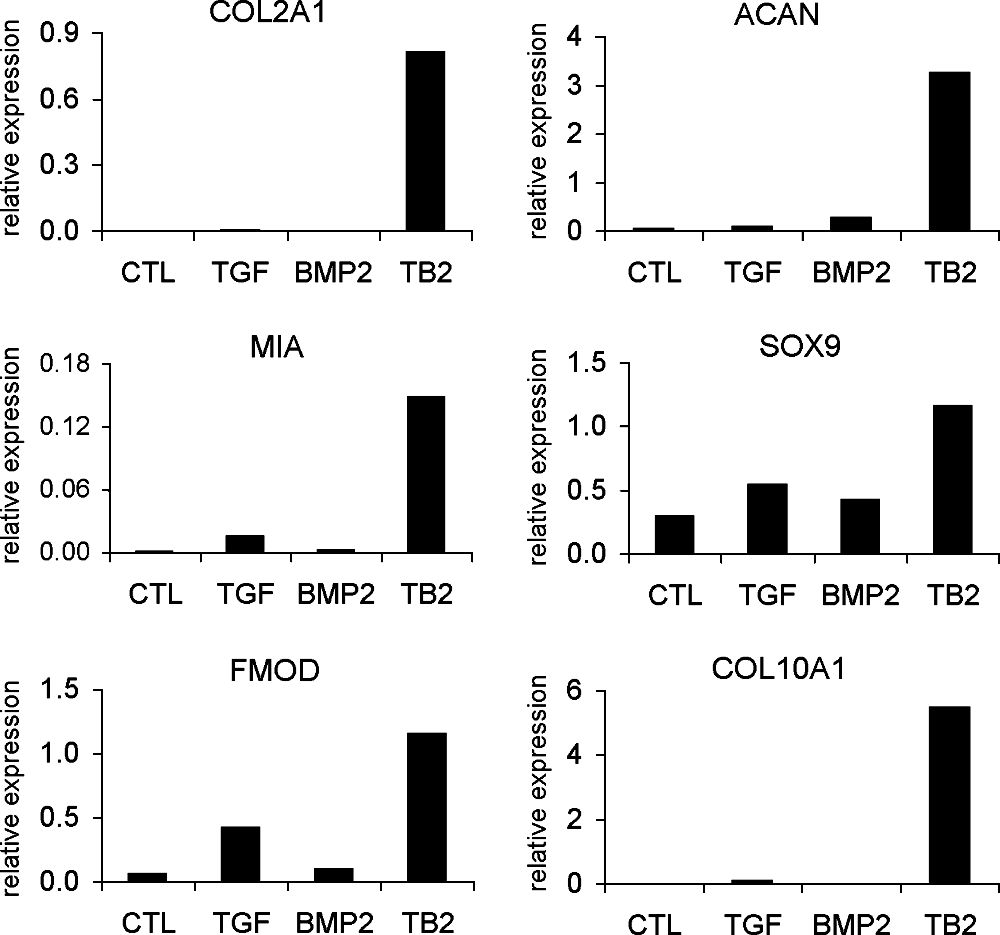
Gene expression analysis of chondrocytic markers in differentiated BM MSCs induced by TGF-β3 alone, BMP-2 alone, and their combination for 21 days by real-time PCR. Relative expression (in all relevant figures) was calculated as a ratio to the average value of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and hypoxanthine phosphoribosyltransferase 1 (HPRT1). Undifferentiated MSCs were used as negative control. Representative of two independent experiments. CTL = undifferentiated control; TGF =TGF-β3; TB2 = TGF-β3&BMP-2, for all figures.

Gene expression analysis by real-time PCR. The gene expression of the specific markers for chondrocytic cells, COL2A1, ACAN, MIA, SOX9, FMOD, COMP, COL9A1, COL10A1; for osteoblasts, BGLAP; and for MSCs, CD44, was detected in undifferentiated and chondrogenic differentiated BM MSCs induced by TGF-β3 alone or TGF-β3&BMP-2 for 7, 14, and 21 days (n = 6). *p < 0.05; **p < 0.01.
The results showed that undifferentiated BM MSCs expressed low or undetectable levels of chondrocytic genes. Upon differentiation for 7–21 days, the chondrocytic gene expression was upregulated, and TGF-β3&BMP-2 showed more potent induction than TGF-β3 alone. When compared to undifferentiated MSCs, for instance, type II collagen (COL2A1) expression was increased a maximal 22,000-fold by TGF-β3&BMP-2 treatment, while TGF-β3 alone only induced a maximal 1200-fold increase. ACAN expression was increased by 25-fold with TGF-β3&BMP-2, and 6-fold with TGF-β3 alone. Only the expression of cartilage oligomeric matrix protein (COMP) was highly increased with no difference between TGF-β3 and TGF-β3&BMP-2 stimulation. The expression level of type X collagen (COL10A1) was also upregulated by a maximal 21,000-fold by TGF-β3&BMP-2 treatment and a maximal 6000-fold by TGF-β3 alone. The MSC marker gene CD44 was downregulated up to day 21. TGF-β3&BMP-2 did not significantly upregulate osteogenic marker gene bone gamma-carboxyglutamate protein (BGLAP, osteocalcin) compared to either TGF-β3 alone or undifferentiated MSCs (Fig. 3).
Chondrocytic gene expression of MSCs differentiated by TGF-β3&BMP-2 for 21 days was compared with that of cultured NP cells and fresh NP tissue (Table 2). Dramatically decreased gene expression was observed in cultured NP cells compared to NP tissue. The TGF-β3&BMP-2–treated MSCs showed much higher expression level than cultured NP cells, but lower than that found in NP tissue, except for COL9A1 and COL10A1 expression.
Proteoglycan deposition and protein expression in chondrogenic differentiation
As assessed by histological/immunohistochemical analysis (Fig. 4A) and western blot (Fig. 4B), differentiated MSCs produced specific proteins and proteoglycans of chondrocytic cells. BMP-2 significantly enhanced TGF-β3–induced protein expression at day 21 of differentiation. Alcian blue staining revealed a strong blue coloration, indicating proteoglycan deposition in the extracellular matrix of TGF-β3&BMP-2–treated MSCs, similar in appearance to human NP tissue (Fig. 4A). The protein expression of type II collagen and SOX9 was also higher in TGF-β3&BMP-2–treated MSCs than TGF-β3 alone (Fig. 4A, B).
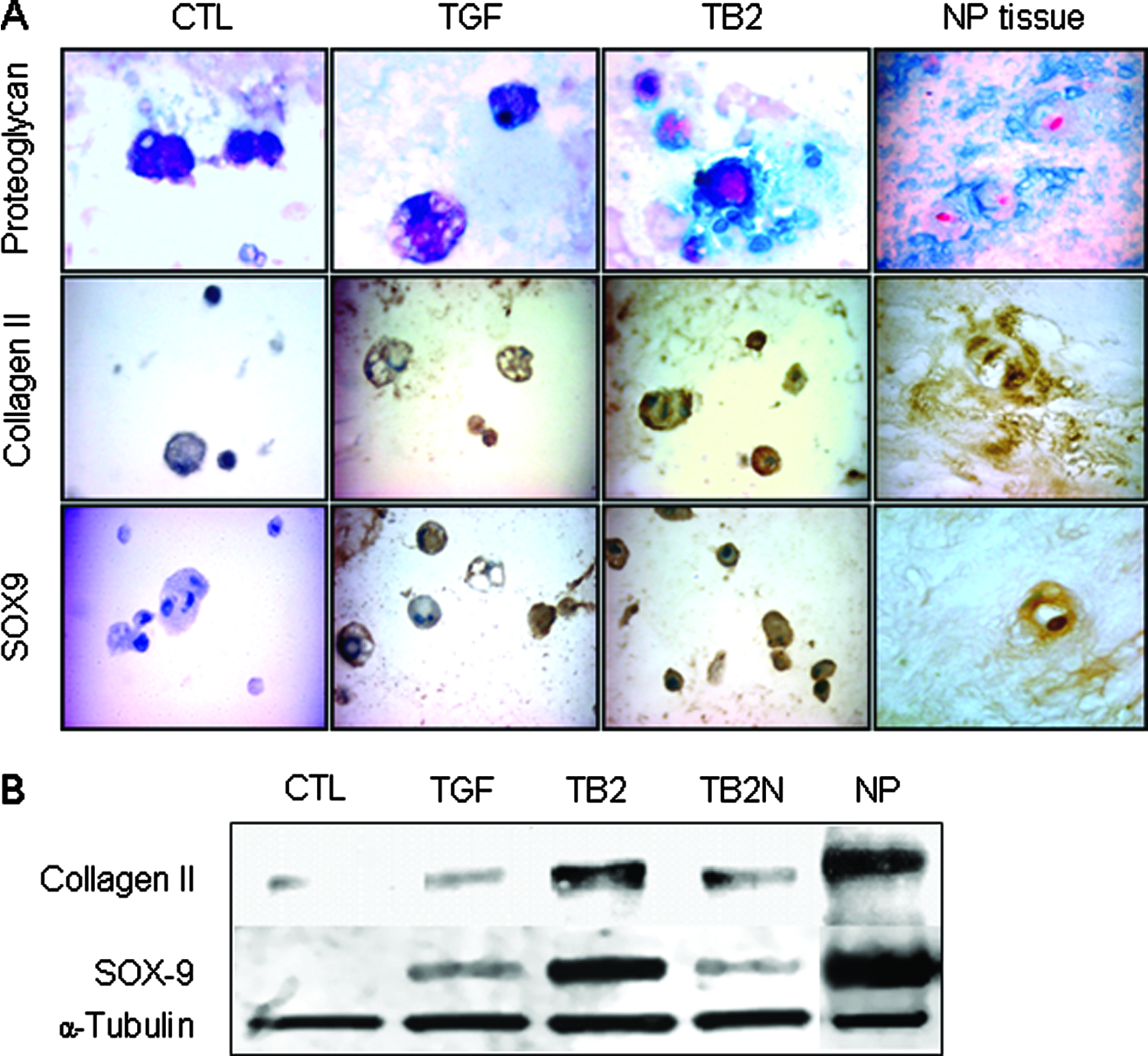
Protein expression and proteoglycan deposition of differentiated BM MSCs. After differentiation for 21 days, the MSCs were fixed for histological and immunohistochemical assays (
Differentiated MSCs are capable of synthesis of proteoglycans
In addition to detecting proteoglycan accumulation in the extracellular matrix by Alcian blue staining, the rate of synthesis of the glycosaminoglycan chain in proteoglycans was estimated by 35S-sulfate incorporation assay. Significantly elevated activity of cells for proteoglycan synthesis was observed, with increased 35S-sulfate incorporation in 24 h prior to the termination of chondrogenic differentiation. Slightly higher levels of proteoglycan synthesis were detected in cells treated by TGF-β3&BMP-2 than by TGF-β3 alone (Fig. 5).

Proteoglycan biosynthesis of differentiated BM MSCs analyzed using 35sulfate incorporation assay. MSCs were induced in differentiation medium containing TGF-β3 alone or TGF-β3&BMP-2 for 21 days and then incorporated with 35sulfate for 24 h. Relative radioactive counting represents the rate change of proteoglycan biosynthesis (n = 4). Undifferentiated BM MSCs were used as negative control, and defined as 1.
BMP-2 signaling is required to promote chondrogenic differentiation
Smad pathway was examined as it plays a central role in TGF-β and BMP signal transduction. The results showed that gene expression of the receptor-regulated Smad 8 was gradually upregulated from day 7 to 21 as responsiveness to TGF-β3&BMP-2 stimulation. The inhibitory Smads, Smads 6 and 7, were upregulated in TGF-β3&BMP-2–treated MSCs, largely due to negative feedback regulation of BMP-2 signaling. There was no significant change in the gene expressions of Smads 1, 2, 4, and 5, while Smad 3 was downregulated in differentiated MSCs (Fig. 6).
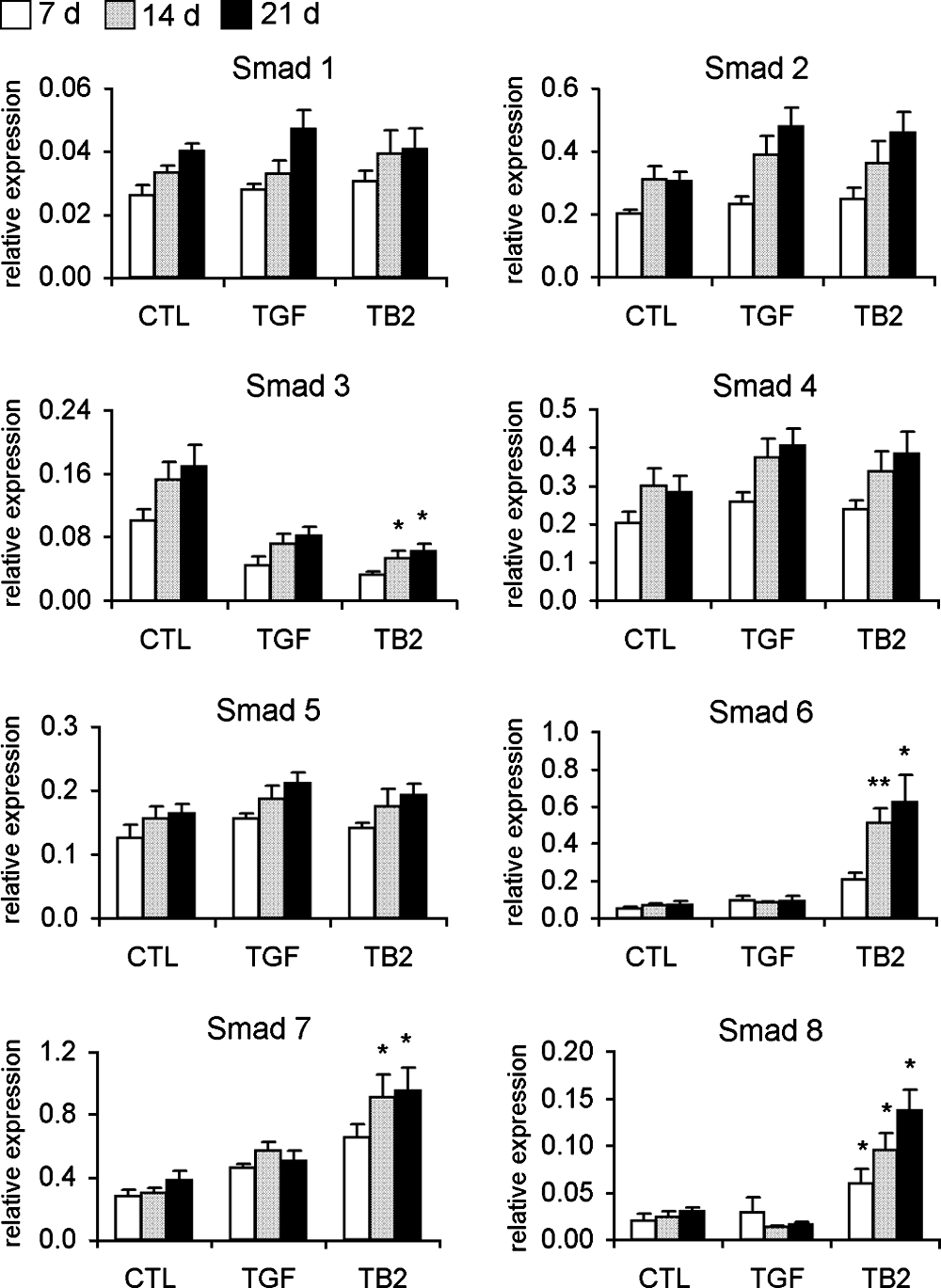
Regulation of Smad gene expression during chondrogenic differentiation of BM MSCs by real-time PCR analysis. Gene expression of Smad signaling molecules was analyzed at days 7, 14, and 21 of MSC differentiation induced by TGF-β3 alone or TGF-β3&BMP-2. Undifferentiated MSCs were used as negative control (n = 6). *p < 0.05; **p < 0.01.
The contribution of BMP-2 in TGF-β3&BMP-2–induced differentiation was evaluated by inhibiting BMP-2 signaling using the BMP antagonist, noggin. Noggin inhibition completely suppressed BMP-2–enhanced gene expression, while it reversed CD44 gene expression to the levels obtained with TGF-β3 alone and did not affect COMP gene expression in TGF-β3&BMP-2–treated MSCs (Fig. 7), confirming that COMP expression was not effected by BMP-2 (Fig. 3). Western blot analysis also showed that BMP-2–enhanced protein expression of type II collagen and SOX9 was suppressed by noggin (Fig. 4B).
Effects of BMP-Smad signaling on chondrocytic gene expression by real-time PCR analysis at days 7, 14, and 21 of MSC differentiation. BMP-2–enhanced gene expression was completely suppressed by addition of noggin. Representative of two independent experiments.
ERK1/2 MAPK signaling is involved in chondrogenic differentiation
To examine the involvement of the ERK1/2 pathway in the chondrogenic differentiation of BM MSCs, the MEK1/2 inhibitor U0126 was used to block downstream ERK1/2 signaling. ERK1/2 inhibition further increased BMP-2–enhanced chondrocytic gene expression of COL2A1, ACAN, FMOD, and COL9A1 in TGF-β3&BMP-2–treated MSCs. However, there was weak increase in COL10A1 and decrease in SOX9 expression (Fig. 8). ERK1/2 inhibition in MSCs treated with TGF-β3 alone showed either no change or decreased expression of all genes examined (Fig. 8).
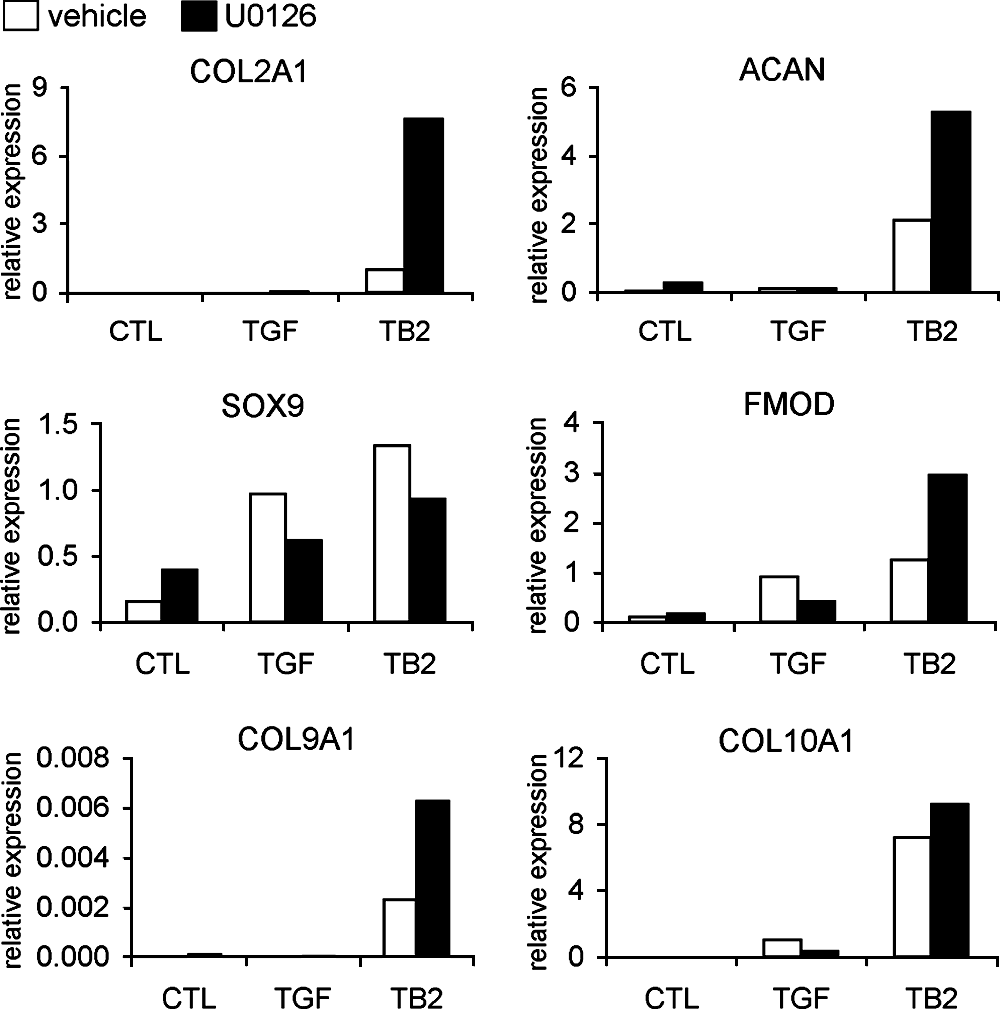
Effects of ERK1/2 MAPK signaling on chondrocytic gene expression by real-time PCR analysis at day 21 of MSC differentiation. Involvement of the ERK1/2 signaling was observed by upregulation of BMP-2–enhanced chondrocytic gene expression when upstream MEK1/2 activity was blocked by U0126. Representative of two independent experiments.
Discussion
In this study, we used the combination of TGF-β3 and BMP-2 under defined serum-free conditions in alginate bead culture to generate chondrocyte-like cells from a relatively homogeneous population of BM MSCs. The results demonstrated that TGF-β3&BMP-2 in our induction system provides the cells with better chondrocytic phenotype than the standard TGF-β–based method. BMP-2 acts synergistically with TGF-β3 as an enhancer rather than an initiator of chondrocytic lineage development. In addition, our results confirmed that predifferentiated BM MSCs have advantages over cultured NP cells in both quantity and quality for potential use in IVD regeneration.
We considered the efficient chondrogenic induction of MSCs as a net outcome of optimal exposure to defined growth factors in an appropriate 3D culture. Most of the published reports utilize pellet culture systems for chondrogenic differentiation of MSCs. The differentiated cells could not be retrieved from the pellet culture, limiting subsequent usage. Alginate beads, initially used for culturing chondrocytes to maintain the phenotype, have been recently used for chondrogenic differentiation of BM MSCs.12,22,23 In our study, alginate bead culture was chosen, because cells in nonadhesive alginate hydrogel remain in a natural round morphology and the 3D culture system resembles the physiological microenvironment of IVD tissue. Moreover, the differentiated cells can be readily liberated from the beads for further studies or cell-based transplantation therapies.
The TGF-β–based differentiation method is well established and has been extensively utilized for in vitro chondrogenic induction of MSCs.13,14 We found that BMP-2 was not a lineage determinant with limited effect on chondrogenic gene expression, but it could synergistically enhance TGF-β3–mediated chondrogenesis of MSCs. The mechanism of synergism has not been well elucidated. This might be partially attributed to the potential involvement of BMP receptor activation by TGF-β3. 24 Schmitt et al. reported that BMP-2 alone could induce chondrogenic lineage development of BM MSCs in pellet culture, and showed week effects in combination with TGF-β3. 25 The differences between our studies might be related to different 3D cultures. Our finding that a combination of growth factors improves chondrogenic differentiation was also supported by previous reports that used combinations of BMP-6, growth differentiation factor-5 (GDF-5), or insulin-like growth factor-1 (IGF-1) with either TGF-β1 or TGF-β3 in pellet cultures.24,26 During the preparation of this manuscript, Mehlhorn et al. published their findings that simultaneous stimulation of adipose-derived MSCs with TGF-β1 and BMP-2 achieved a synergistic effect on chondrocytic gene expression in alginate bead culture. 27
In this study, the co-upregulation of COL2A1 and COL10A1 gene expression was observed even at early stages of TGF-β3– or TGF-β3&BMP-2–induced differentiation of MSCs. The concept that COL10A1 is expressed by chondrocytes when they become hypertrophic has been challenged recently by contradictory findings. Our observations were consistent with previous studies that gene expression of type X collagen was initiated and upregulated at the same time point or even before type II collagen during chondrogenic differentiation of BM MSCs. Mwale et al. suggested that caution must be exercised when using type X collagen as a marker for chondrogenesis and chondrocyte hypertrophy in association with adult BM MSC differentiation.12,13,28–30 Taken together with recent observations from other groups, our results indicated that COL10A1 gene expression might be regulated by different mechanisms as in chondrocyte hypertrophy. We also observed that the expression of MIA, a chondrocytic marker gene, was enhanced by the addition of BMP-2. MIA favors chondrogenesis of MSCs by supporting a chondrogenic phenotype, while inhibiting osteogenic differentiation, and has been postulated to be an important regulator during chondrogenic differentiation of MSCs. 31 Under our chondrogenic conditions, osteoinductive activity of BMP-2 was restricted, with little change in gene expression levels of osteoblastic marker BGLAP, when compared to TGF-β3–treated or undifferentiated MSCs. This restriction, as a result of TGF-β3 inhibition, also possibly related to upregulation of MIA expression.
Each individual member in TGF-β superfamily exhibits specific as well as overlapping activities by signaling through their receptors that directly regulate the intracellular Smad pathways. Smads 1, 5, and 8 comprise the BMP-responsive group of Smads, while Smads 2 and 3 constitute the TGF-β-activin–responsive group. 32 Our results showed an increase in Smad 8 gene expression along with chondrocytic genes. However, it is not clear whether these two events are correlated. The downregulation of Smad 3 gene expression during the differentiation process has also been previously reported by Sekiya et al. 28 It has been reported that overexpression of Smad 3 gene could enhance the osteoblastic differentiation of rat BM MSCs. 33 In this study, the downregulation of Smad 3 expression might imply a lineage choice for chondrogenesis. Apart from gene expression, increased activation of BMP-2–responsive Smads might account for the enhanced gene and protein expression of extracellular matrix components, which was not addressed in this study. The blocking of BMP-2 signaling by BMP antagonist, noggin, resulted in complete suppression of BMP-2–enhanced gene expression and a recovery of BMP-2–inhibited gene expression, confirming a critical role of BMP-2–activated Smad signaling pathway in the differentiation.
We further examined the involvement of the ERK1/2 signaling pathway by pharmacological inhibition of MEK1/2 during the differentiation process. Blocking ERK1/2 signaling further elevated TGF-β3&BMP-2–enhanced chondrogenic gene expression. Our results are consistent with previous reports of enhanced chondrogenic differentiation of goat MSCs 34 and restored chondrocyte phenotype due to ERK1/2 inhibition. A recent report that growth arrest-specific 6 (Gas6) negatively regulates BMP-2–mediated chondrogenic differentiation of murine mesenchymal cells through activation of ERK1/2 pathway 35 also supports our findings that ERK1/2 signaling negatively modulated TGF-β3&BMP-2–induced chondrogenic differentiation and the chondrocyte-specific gene expression was regulated by a cross talk between Smad and ERK signaling pathways. However, Risbud et al. inhibited MAPK signaling by pretreatment of BM MSCs with ERK and p38 MAPK inhibitors, respectively, before TGF-β1 stimulation, leading to reduced type II collagen, aggrecan, and SOX9 gene expression. Lee et al. have reported that MEK inhibition resulted in complete downregulation of type II collagen, while there was no effect on aggrecan gene expression during TGF-β3–induced chondrogenic differentiation of BM MSCs.14,36 The inconsistency of marker gene regulation under different circumstances reveals the complexity of the cellular response to concerted multiple signaling pathways.
In this study, to characterize whether differentiated MSCs possess NP-like phenotype, human NP tissue and cultured NP cells were examined for gene and protein expression. A significant difference in gene expression levels was observed between native NP cells in tissue and cultured NP cells. Reduced gene expression of cultured NP cells was a consequence of removal of the cells from their matrix environment, altering the range of signals the cells received. The rationale to compare the differentiated MSCs with cultured NP cells is that both cells are in a nonmatrix environment, while with fresh NP tissues, their gene expression is close to in vivo levels. Our study showed that expression of all NP matrix genes in differentiated MSCs was higher than that of cultured NP cells and lower than NP tissue with similar expression pattern of NP tissue. However, there were sample limitations in this study. The NP tissues and NP cells were obtained from surgically discarded tissues with various grades of degeneration. The gene expression information from these samples might not be the same as that from normal NP. In addition, it was difficult to separate NP accurately from inner AF due to fibrosis of NP. Currently, there is a lack of unique marker characteristic to distinguish NP and cartilage-derived chondrocytic cells. Fujita et al. reported that CD24 was expressed specifically in NP cells and concluded that CD24 could be utilized as a molecular marker of NP. 37 However, we were not able to detect either CD24 gene expression by RT-PCR or CD24 protein expression by flow cytometry in human NP cells. We noted that their positive findings were obtained from NP of 8-week-old rats likely to contain notochordal cells, and human chordoma derived from notochordal cells. It thus appears that high CD24 expression is associated with notochordal cells, which disappear in human NP after the first decade of life. Since NP chondrocytic cells and hyaline cartilage chondrocytes share many characteristics with similar extracellular matrix components, our view is that the BM MSC–derived chondrocytic cells generated by our induction system also provide therapeutic potential for cartilage repair.
We conclude that the combination of TGF-β3 and BMP-2 in serum-free medium and alginate bead 3D culture is a potent chondrogenic induction system for human BM MSCs. BMP-2 enhances TGF-β3–mediated chondrogenic differentiation of MSCs with both Smad and MAPK pathways involved in this process. This induction system provides a potential means to supply BM MSC–derived chondrocyte-like cells for IVD and cartilage regeneration, and also can be used as an in vitro model for investigating the molecular mechanisms involved in chondrogenic differentiation of BM MSCs.
Footnotes
Acknowledgments
We thank Shane Whittaker for technical assistance and David Connor for advice on flow cytometric analysis. This study was supported by grants from The Arrow Bone Marrow Transplant Foundation and St. Vincent's Clinic Foundation.
Disclosure Statement
No competing financial interests exist.