
Abstract
Tendons transmit tensile loads from muscle to bone. They consist primarily of parallel collagen fibers between longitudinally oriented rows of tendon fibroblasts. In this study, we describe a novel scaffoldless dialysis–roller culture system that allows tendon cells to form large, organized, tendon-like structures. We compare cell and collagen orientation and synthesis in these cultures with that of monolayer and high-density pellet cultures. Monolayers are unable to deposit a substantial matrix, losing most of their secreted collagen to the medium. High-density pellet cultures deposit more matrix, lose less to the medium, and become organized at their periphery but show signs of nutritional compromise in the center core. In the novel system, cells formed highly organized structures resembling embryonic tendons, synthesized much more collagen, and incorporated around 70% of the secreted collagen into the tendon-like extracellular matrix. The three-dimensional cultures appear to allow substantial cell–cell interactions and may mimic important aspects of the early development of tendons, including the formation of membrane-bound extracellular spaces to contain and organize the secreted collagen.
Introduction
In adult tendons, cells have intimate associations with one another and with the collagen fiber bundles.4–6 A given tendon cell has associations around its longitudinal axis with several fiber bundles, partially enclosing them in channels delineated by membranous processes. The channels are aligned along the cell row; hence, the fiber bundles are passed from one cell to the next along the cell row.4,7 These channels relate to those described in early collagen deposition by Birk and Trelstad 8 and more recently by Canty et al. 9 Tendon cells maintain their position within rows using actin stress fibers linked from cell to cell by adherens junctions, the abundance of these components being mechanically regulated. 5
Monolayer culture procedures are routinely used to grow many different cell types. They are an effective way of increasing cell numbers, but are not generally associated with maintenance of differentiated function of connective tissue cells. This is well known in cartilage cell biology10–12 but is less obvious with fibroblasts, as there are no convenient markers of differentiation and dedifferentiation. Tendon fibroblasts do seem to dedifferentiate in monolayer cultures. They produce tissue-appropriate extracellular matrix components, typically type I and type III collagens, but most of these are released to the culture medium rather than being deposited as matrix in the cell layer. 13 Also, there are changes in the amount and type of matrix they synthesize—in vitro, less collagen is produced relative to other cell and matrix proteins compared to the in vivo situation14–16 and the proportion of type III collagen increases.17,18 The type I collagen produced also shows abnormalities with changes in the ratios of α1(I) and α2(I) chain synthesis. 15
More complex three-dimensional culture systems may be able to maintain a more tissue-like phenotype, although details of the parameters outlined above for monolayers have not been investigated in such systems. Schulze-Tanzil et al. 19 describe a system in which tendon fibroblasts suspended at high cell density are placed on a membrane filter, on top of a stainless steel grid at the culture medium–air interface and maintained for 14 days. Under these circumstances cells retained the ability to produce normal tendon matrix and expressed scleraxis, which can be a marker of some embryonic tendons in development, 20 although matrix orientation was not addressed. Fibroblasts grown in collagen gels will form oriented cell arrays appropriate to their tissue of origin; in the case of tendon fibroblasts, these are parallel arrays of cells; the cell arrays deposit a certain amount of oriented matrix.21,22 Thus, there is clearly some intrinsic ability of fibroblasts to form organized structures in vitro under appropriate conditions. Variants of cell-seeded collagen gel cultures have been developed in a tissue engineering/mechanical testing context to form oriented structures, along with culture systems using synthetic scaffolds seeded with tendon cells to produce tendon-cell-rich constructs, which can form tendon-like tissue.23–28
The purpose of the work described here is to determine the extent to which tendon cells can intrinsically form organized structures, without the involvement of an exogenous scaffold—connective tissue cells are, after all, capable of making their own scaffolds in vivo. Certainly, mesenchymal stem cells can differentiate toward a ligament fibroblast phenotype and self-assemble appropriate cell and matrix structures under appropriate culture conditions. 29 In the current paper, we compare tendon cell behavior in three distinct cell culture systems: monolayers, as a baseline for comparison; high-density pellet cultures, to allow cells to interact in three dimensions; and a novel three-dimensional dialysis-tube-based roller culture system to allow large numbers of cells to interact over an extended culture period without disturbance from medium changes. We show that monolayers deposit a little matrix, pellet cultures an extensive extracellular matrix, and roller cultures form complex, well-ordered structures with tendon-like arrangements of tendon cells and collagen fibers. The products of the roller cultures have potential to be developed into useable structures for tissue repair, without the use of exogenous scaffolding materials.
Materials and Methods
Cell isolation and expansion
Flexor digitorum longus tendons were aseptically dissected from the feet of 50–55-day-old broiler chickens, obtained within 1 h of slaughter; 6–10 tendons used at each harvest. Tendon fibroblasts were isolated by collagenase digestion 30 ; plated on 100 mm culture dishes in high-glucose DMEM supplemented with 5% FBS, L-glutamine, and antibiotic–antimycotic (all tissue culture reagents from Invitrogen–Gibco, Paisley, United Kingdom); and incubated at 37°C, 100% humidity, 5% CO2 in air. Cells were grown to confluence, passaged with trypsin–EDTA, and used for experiments at passage 3. Some of these were maintained as monolayers in the above culture medium containing 1 mg/mL ascorbic acid for 7 days as a baseline for analysis of results from the culture systems described below. Spent media from these and the culture systems outlined below were retained for analysis of collagen content.
High-density pellet culture
Cell suspensions were adjusted to 1 × 106 cell/mL in DMEM/5% FBS, and 1 mL aliquots transferred to sterile 1.5 mL Eppendorf tubes. The tubes were centrifuged in a microcentrifuge (2000 rpm, 2 min) to form cell pellets. The supernatant was gently replaced with 1 mL DMEM–5% FBS as above, with the addition of 12.5 mM HEPES buffer and 1 mg/mL ascorbate. Pellets were incubated for up to 14 days at 37°C, and the medium was changed at 3-day intervals. Pellet cultures were grown on many different occasions from different cell isolates and showed similar behavior in terms of DNA content and matrix synthesis and deposition; in the experiments described here, there were three replicates for immunohistochemical studies, three for ultrastructural studies, and seven for biochemical studies.
Electron microscopy
Some pellet cultures were prepared for transmission electron microscopy (TEM) and scanning electron microscopy (SEM). TEM samples were fixed in Karnovsky's fixative 31 and osmium tetroxide, dehydrated, and embedded in Spurr Resin. Silver–gold sections were collected on Formvar coated grids, stained with uranyl acetate and lead citrate, and examined using a Phillips EM208 transmission electron microscope. SEM samples were fixed in glutaraldehyde in 0.1 M phosphate buffer, dehydrated in ethanol, critical-point dried, sputter coated with gold, and examined on a Philips XL20 scanning electron microscope.
Large-scale dialysis tubing–roller cultures
The culture apparatus is shown in Figure 1. About 1 mL of aliquots of cell suspension containing 25 × 106 cells was pipetted into sterile Spectra/Por® CE DispoDialyzer tubes (Medicell Int., London, United Kingdom). These are 5 cm lengths of 5-mm-diameter cellulose ester dialysis tubing sealed at either end with plastic caps, one of which is fixed and the other a screw cap. Before the addition of the cell suspensions, 200 μL of 2% agarose (Sigma-Aldrich, Dorset, United Kingdom) was placed into the closed end of the tube forming a flat nonadherent surface to ensure all cells remain in suspension—preliminary studies showed accumulation of cells in the plastic cap at this end of the tube in the absence of this agarose surface. Initial experiments used tubes with 300 kDa molecular weight cut-off (MWCO) membranes; however, changes in the product range offered after the experimental series had started resulted in replacement with 100 kDa MWCO and 50 kDa MWCO membranes. After filling with cell suspension, the dialysis tube screw cap was closed and the now sealed dialysis tube placed in a standard 50 mL sterile centrifuge tube (Corning, Schipol-Rijk, The Netherlands), which were then filled with DMEM supplemented with 5% FBS and 1% antibiotic–antimitotic, 12.5 mM HEPES buffer (Invitrogen–Gibco), and ascorbate (1 mg/mL). The cultures were incubated at 37°C with the large tubes running horizontally on rollers rotating at 35 rpm. The HEPES buffer controls pH in the absence of continual exchange of CO2 with the incubator atmosphere as the centrifuge tube has to be fully sealed to run on the roller without leakage. Medium in the large tube was changed every 3 days, leaving the medium and cells in the dialysis tube undisturbed. Cultures were maintained for up to 14 days. In addition, in a preliminary study some cultures were grown in DMEM containing 1% serum and 1% serum supplemented with TGF-β (10 ng/mL) or IGF-1 (50 ng/mL; both from Sigma-Aldrich). The large-scale cultures were grown on six different occasions from different cell isolation runs and produced substantial cell–matrix aggregates, although with some variation in the precise morphology of culture products, as described in detail in the Results section; in the experiments described here, there were three replicates for immunohistochemical studies and three for biochemical studies.
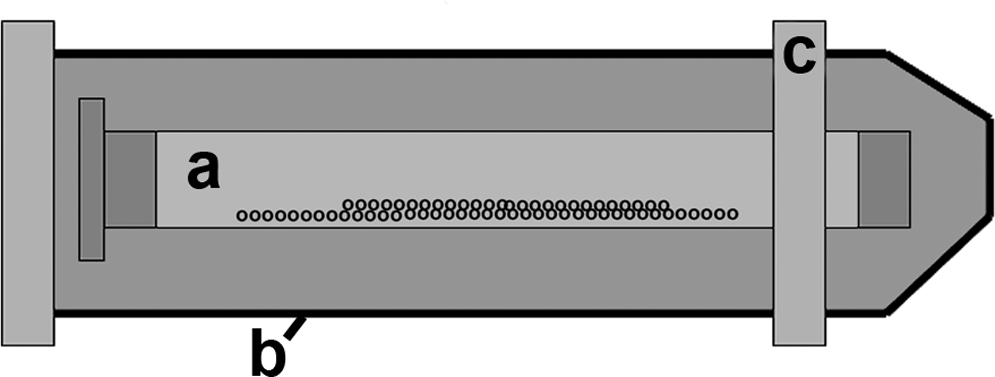
Diagram illustrating the large-scale dialysis tube–roller culture system. A 1 mL Spectra/Por DispoDialyser tube containing around 25 million cells in 1 mL of culture medium (
Immunohistochemical labeling
Cell cultures to be used for immunolabeling procedures were fixed in 95% alcohol at 4°C for differing time intervals. For comparative purposes, digital flexor tendons were dissected from the feet of 17-day chicken embryos and prepared as for large cultures below. Fibroblast monolayers were fixed for 2 min; pellets and suspension roller cultures were fixed for 5–15 min dependant on size. Monolayers were labeled directly; the other cultures were frozen onto cryostat chucks in OCT compound (Tissue Tek; Sakura Finetek, Thatcham, United Kingdom) using dry ice and cryosectioned at 15 μm. Specimens were labeled using standard procedures for indirect immunofluorescence. Briefly, they were blocked with 5% goat serum, incubated for 1 h with primary antibodies to type I collagen (rabbit anti-chicken type I collagen, 1:200 dilution; Millipore, Livingston, United Kingdom), or type III collagen (monoclonal 3B2, 32 10 μg/mL; obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, IA), 32 and then incubated with goat anti-mouse or rabbit antibody–AlexaFluor488 conjugates (Invitrogen Molecular Probes, Paisley, United Kingdom) as appropriate. Controls had primary antibodies omitted or replaced with 10 μg/mL nonimmune mouse or rabbit immunoglobulins. Labeled specimens were mounted in Vectashield with or without propidium iodide as nuclear counterstain (Vector Laboratories, Peterborough, United Kingdom).
Collagen production
Collagen content of cultures and culture medium was determined using a hydroxyproline microassay assay 33 (all reagents from Sigma-Aldrich), using pellet and roller cultures as we have already published results from monolayers. 13 We chose to compare cultures at 7 days, because at 14 days there was evidence of nutritional compromise in the pellet cultures (see Results, pellets and monolayers). Cell pellets or aggregates were digested in 125 μg/mL papain at 60°C and hydrolyzed with 500 μL of 6 N hydrochloric acid. Proteins were precipitated with ammonium sulfate in the presence of protease inhibitors phenylmethanesulphonylfluoride, ethylenediamine tetraacetic acid, and N-ethylmaleimide. Samples were centrifuged, and the residue was dissolved in 1 mL PBS and hydrolyzed again with hydrochloric acid. Acid was removed by vacuum drying, and samples were reconstituted in water and centrifuged again to remove insoluble material. Triplicate 30 μL aliquots of sample or standard were pipetted into a 96-well plate, followed by assay buffer, chloramine-T, and 4-Dimethylaminobenzaldhyde, and plates were incubated at 70°C for 10–20 min. Samples were read immediately on a plate reader at an absorbance wavelength of 540 nm. To allow comparison of hydroxyproline data between different culture procedures, results were normalized to DNA content. 34 Triplicate 50 μL papain-digested sample or plasmid DNA standard were added to 1 mL of 100 μg/mL Hoechst 33258 dye solution (Sigma-Aldrich). Results were measured fluorimetrically at A350 nm excitation and A450 nm emission.
Statistical analysis
Total collagen, cell pellet and dialysis–roller culture aggregate, and culture medium collagen content, relative to DNA content, were compared using a one-way ANOVA followed by the Tukey multiple comparison pair-wise test, with a 95% confidence limit.
Results
Pellets and monolayers: Morphology and collagen deposition
Cells in monolayer adopted the elongated and flattened morphology typical of fibroblasts, and rapidly formed confluent monolayers (not shown; see immunohistochemical labelling, below). Cells in pellet culture formed dense, rounded aggregates with a layer of flattened cells at the periphery (Fig. 2a–d). At day 1 (Fig. 2b) peripheral cells showed no particular organization, although at day 7 and beyond (Fig. 2c) they were oriented circumferentially. Within the pellets, TEM showed that there was no obvious fibrillar banded collagen between cells at 1 day (Fig. 2e, f), although there was grayish fibrillar material between cells that had extensive cell processes, some with membrane-bound cavities within them. Obvious fibrils were present between cells at 2 days (Fig. 2g) and more were present at 7 days (Fig. 2h), although the fibers were widely spaced at a fairly low density. Fibers tended to be in small, oriented groups, although there was no overall direction of orientation, with the fiber bundles being interwoven.
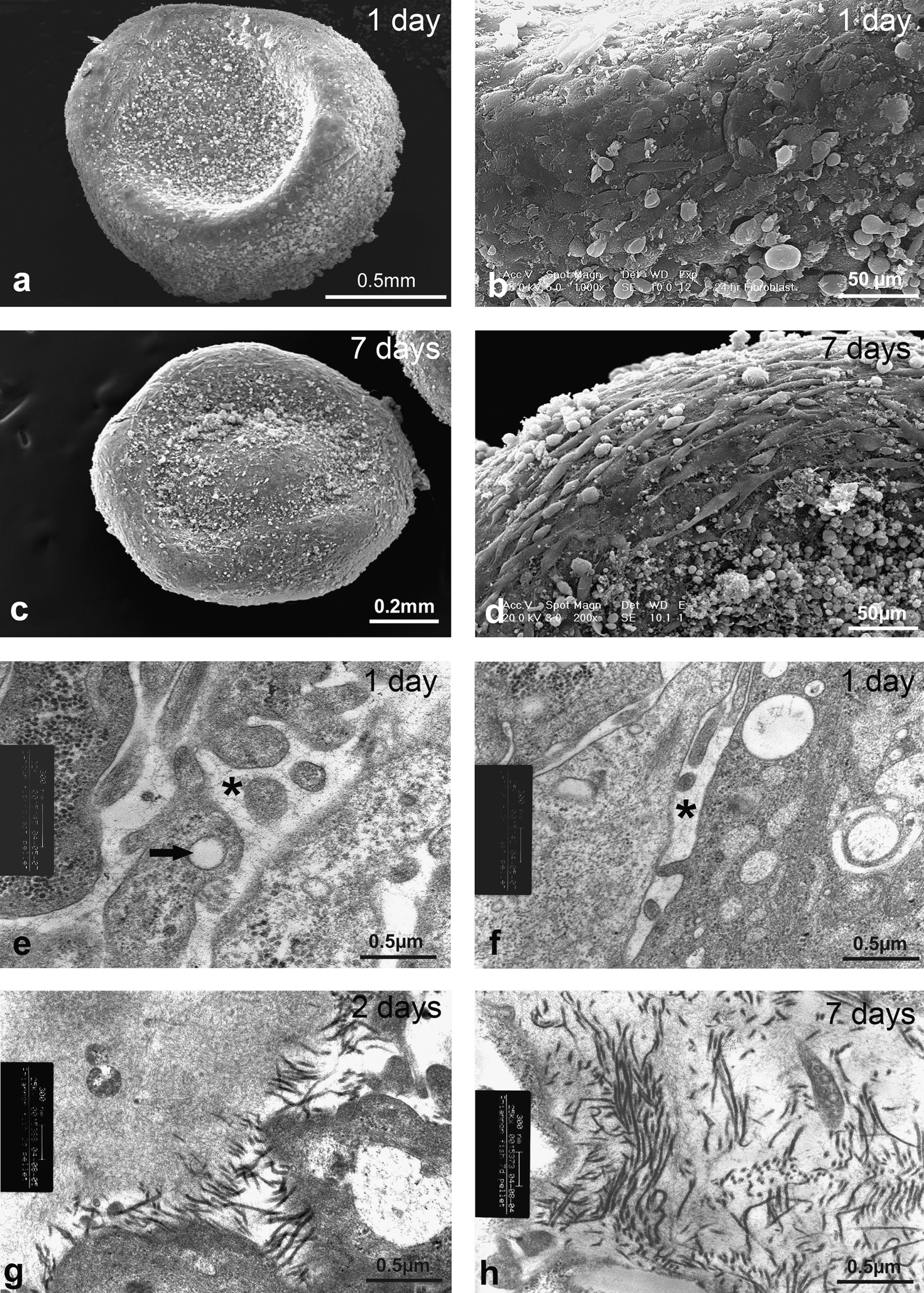
Electron microscopy studies of pellet cultures. (
Immunohistochemical labeling of monolayers showed the presence of sparse label for type I and type III collagens in monolayers—type I label with a streaky appearance that may be intercellular (Fig. 3a), whereas type III collagen was intracellular (Fig. 3b). In pellet cultures type I and type III collagens were present from 1 day of culture onward, with type III label being most prominent in the early stages. Initially, label appeared as small extracellular foci and became progressively more extensive and fibrous in appearance over time (Fig. 3c–f). However, by 14 days of culture, label was generally diminished in the central part of the pellet, and this region became fragile, tending to disintegrate during cryosectioning. One striking feature of the pellets was at the periphery, where the cells that became oriented circumferentially as shown by SEM studies above, appeared to lay down a collagenous matrix with discernable circumferential orientation (Fig. 3g, h).
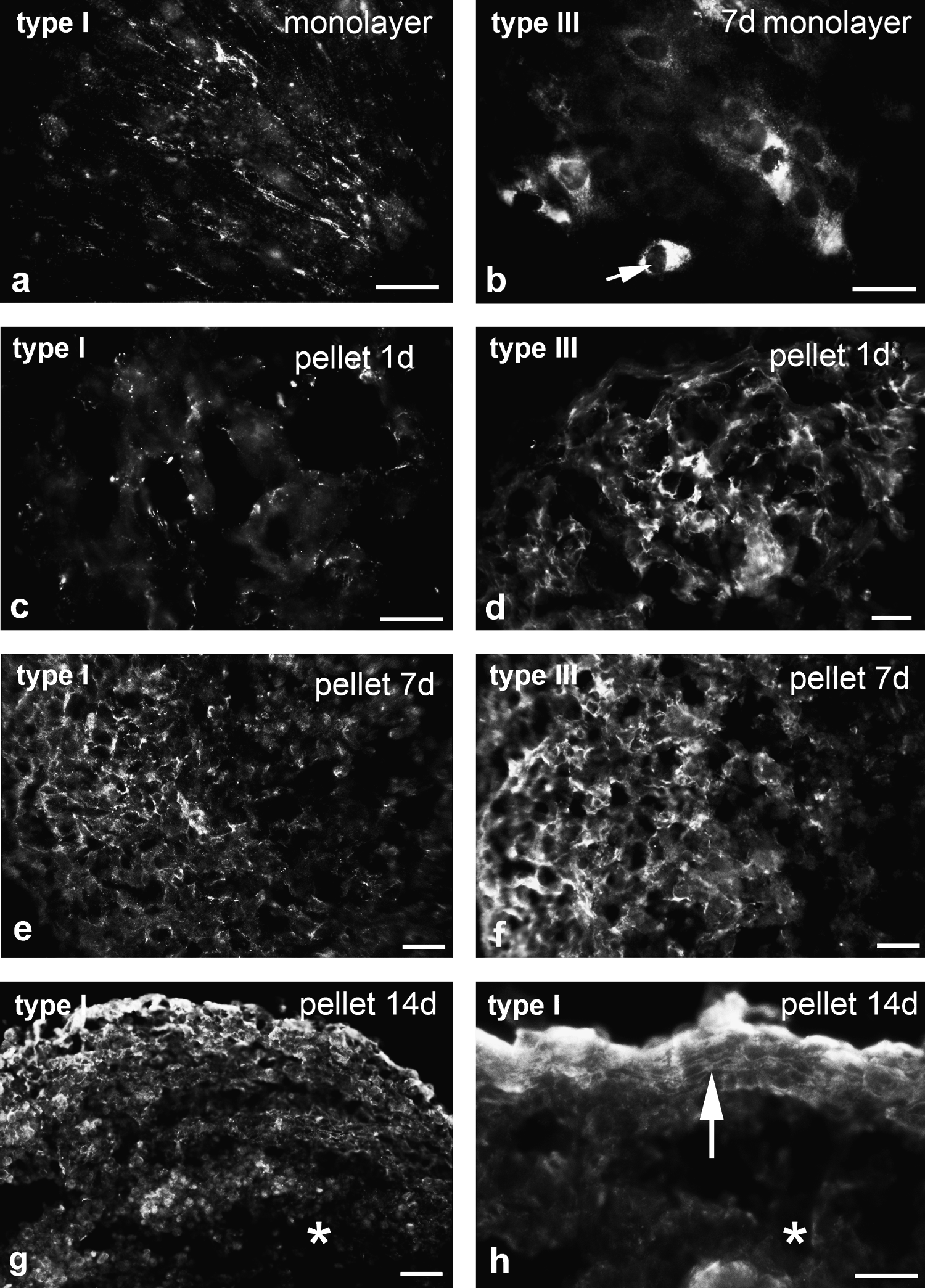
Immunofluorescence labeling of monolayer (
Dialysis roller culture system: morphology and collagen deposition
Cells cultured in DMEM–5% FBS within the large-scale system formed visible elongated aggregates inside the dialysis tube within 24 h. Initially, these were so fragile that they could not be removed from the tubes and handled with forceps without disintegrating, but over the following 7–14 days the aggregates became larger and could be handled easily while remaining intact. These older structures took a variety of forms. In most cultures there were elongated rod-shaped aggregates (Fig. 4a, b), although funnel-shaped sheets or a solid sphere of cells were sometimes formed; one or all could occur within any given culture. The elongated structures could reach relatively large sizes, on occasions being over 3 cm in length and around 0.5 mm in diameter (Fig. 4a, b). These structures formed in all the dialysis tubes used regardless of the different MWCO membranes. Cultures grown in DMEM–1% FBS formed no aggregates—loose cellular material was visible in the tube, but it did not aggregate.
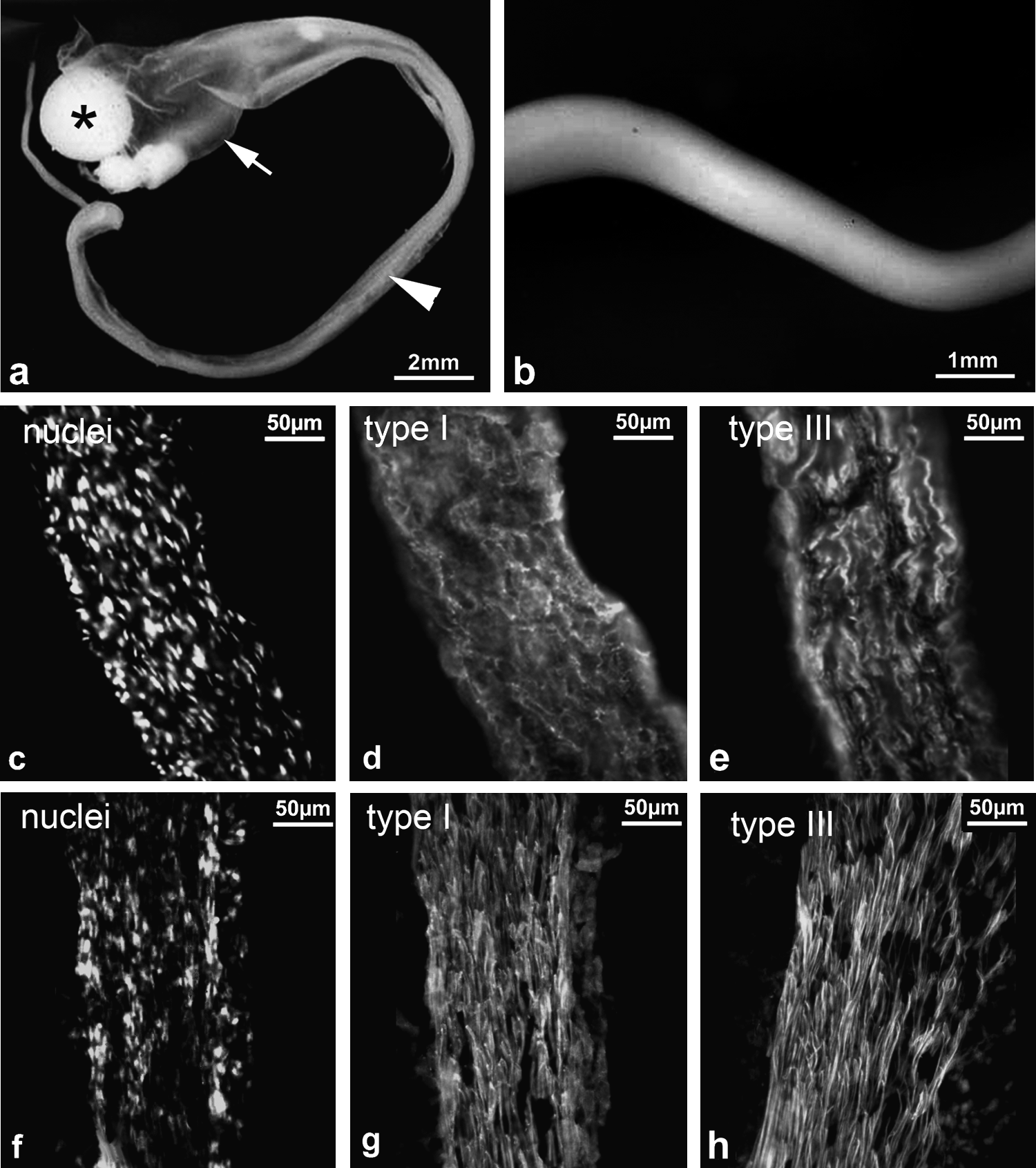
Appearance and collagen immunohistochemistry of dialysis–roller cultures; comparison with embryonic tendons. Macroscopic views of 14-day cultures show the formation of large structures. (
Cell distribution within aggregates could be observed in all of the immunolabeled material because of the nuclear counterstain. This showed a degree of orientation in the thinner rod-shaped and sheet-like aggregates. Cells were arranged in parallel rows separated by ECM, reminiscent of the in vivo organization of tendon cells (Fig. 4c). Type I and type III collagens were detected in the aggregates at all stages. Type I collagen label was positioned between orientated rows of tendon cells in the rod-like and thin sheet cell aggregates, with some indication of a predominant longitudinal orientation (Fig. 4d). Type III collagen labeled very brightly and formed long, relatively widely spaced fibers after 24 h, which persisted through the culture period. In elongated aggregates it was highly organized with bright longitudinal fibers positioned between cell rows (Fig. 4e). There were no effects of different MWCO pore sizes of dialysis tube membranes on cell aggregate structure and immunolabel characteristics. The cultures showed striking similarities in cell and matrix orientation between these cultures and the digital tendons of 17 day chicken embryos, with the latter showing a higher degree of matrix organization, particularly for type I collagen (Fig. 4f–h).
DNA content and hydroxyproline analysis: comparison of pellet and 100 kDa MWCO dialysis–roller cultures after 14 days
Comparison of total collagen produced by the cultures (cell aggregate + medium content) shows that normalized to DNA content, the roller cultures made about sevenfold more collagen than the pellet cultures (Fig. 5). In pellet cultures, 62% of collagen was released to the culture medium, leaving 38% in the cell aggregate. By comparison, the roller cultures retained 70% in the cell aggregate and released 30% to the culture medium. Of the latter, 7% was in the dialysis tube and 23% had passed through it into the surrounding medium. Mean ± SD of DNA content of the cultures was 3.1 ± 0.31 μg/pellet and 41.8 ± 6.0 μg/roller culture.

Collagen produced by pellet and dialysis–roller as measured by hydroxyproline analysis of day 7 cultures. Values were normalized to DNA content; error bars show standard deviations. Large-scale cultures produce more collagen overall, and retain much more of it in the cell aggregate than do pellet cultures (n = 3 large-scale cultures, 7 for pellets; p < 0.01% all comparisons aggregate vs. pellet cultures).
Discussion
This study shows that three-dimensional culture of tendon cells substantially enhances their ability to deposit and organize an extracellular matrix compared to monolayer culture. Indeed, the large-scale dialysis–roller cultures develop a matrix that resembles embryonic tendon in both cell and matrix composition and organization.
Monolayer cultures contained detectable collagen in the cell layer, but extracellular material was very sparse and type III collagen intracellular. It has been shown that only around 15% of total collagen is retained in the cell layer, the remainder being released to the medium. 13 Our results suggest that even measured amounts of collagen contained in the cell layer are likely to be an overestimate of the collagen actually deposited as a extracellular matrix—much remains intracellular. The lack of matrix deposition may result in qualitative and quantitative matrix synthetic changes consistent with dedifferentiation in monolayer culture,14–18 allied with the absence of substantial three-dimensional cell–cell interactions, discussed further below.
Collagen production and deposition is considerably enhanced in pellet cultures, with collagen deposited extracellularly in measurably and observably greater quantities than in monolayer—some 38% of the total collagen produced is retained in the cell pellet, and ultrastructural and immunohistochemical studies show considerable deposition of extracellular collagen fibrils and the presence of type I and type III collagens in the matrix. Peripherally, in the cultures, cells become oriented and deposit an oriented collagenous matrix, showing a link between cell orientation and matrix orientation as demonstrated in vivo 35 and fibroblasts grown in three-dimensional collagen gels.21,22 Also, the projection of cell processes, some with hollow cores, by fibroblasts into their surroundings just before the appearance of collagen fibrils is suggestive of the formation of fibropositors,7,9 structures associated with early oriented fibril deposition. In longer-term pellet cultures, the amount of matrix did not appear to increase, and this is taken, along with evidence of degeneration in central regions of pellets, as evidence of nutritional compromise.
Large-scale roller–dialysis cultures produced large, often highly organized cell–matrix constructs, with extensive oriented matrix deposition—around 70% of the collagen produced being incorporated into the cell aggregate. This high efficiency in matrix deposition is accompanied by a high degree of tissue organization, whereas the pellets were mostly nonorganized aggregates of cells and matrix, the roller–dialysis cultures produced structures with a high degree of cell and matrix organization, similar to fetal tendons. At early stages of culture (24 h) the aggregation of cells into cellular rods may be perhaps analogous to embryonic cell condensation, which precedes tendon development in vivo. 2 It is interesting that tendon cells were unable to aggregate in DMEM–1% FBS. This shows that some specific components of serum are required for cell aggregation, and preliminary experiments have shown that supplementation of DMEM–1% FCS with TGF-β or IGF-1 allows cells to form the aggregates.
Why should the three culture systems offer such major differences in collagen deposition into an extracellular matrix? In developing fibrous connective tissues, collagen is secreted into a series of membrane-enclosed compartments.8,9 It may be that in monolayers the opportunity for forming these is limited, with the two-dimensional array of cells resulting in little possibility of forming compartments—only a small area of cell–cell membrane contact is possible around the periphery of the flattened cells. Three-dimensional cultures allow cell–cell contacts all around cells and thus allow the formation of more cell-enclosed space for matrix deposition, as our ultrastructural studies suggest. Longitudinal alignment of tendon cells and the collagen fibers they produce is a requirement for oriented tendon matrix development. 7 The roller–dialysis system clearly allows the self-assembly of substantial organized structures, and therefore simulates the in vivo development of tendons—there are striking similarities in cell and matrix organization in the aggregates and in early developing tendons. The shape of the initial cell aggregates as elongated rods is probably due to the movements of the roller culture tube. Cell aggregates will be exposed to certain mechanical and hydrodynamic loads—they will impact and be rolled by the rolling dialysis membrane, and the rotary motion will also cause a flow of medium around the cell–matrix aggregates. These stimuli may have been influential in producing the aggregate shapes and could promote matrix synthesis and assembly, given the importance of mechanical stimulation to production of matrix components by tendon cells.13,36–38
Three-dimensional, high-density culture systems are important for other connective tissue types, being used most commonly with cartilage, where pellet and other culture techniques allow chondrogenic differentiation and secretion of cartilage-like matrix by chondrocytes or mesenchymal stem cells.39–41 As with tendon development in vivo, chondrogenesis requires a three-dimensional cell aggregation phase that is followed by overt differentiation and matrix synthesis.39,42 However, there are fundamental differences in differentiation and subsequent morphogenesis: chondrocytes become isolated from one another by the matrix they secrete and essentially live as individuals, whereas tendon cells retain extensive cell–cell contacts,4,13 which are clearly important to matrix organization and subsequent maintenance.7,13 Notably, when we attempted to grow chondrocytes within the roller–dialysis system, they were unable to form aggregates, suggesting that they are unable to form sufficiently strong cell–cell attachments to maintain contact with one another and thus are fundamentally different from tendon cells even when isolated from their matrix.
The culture system presents opportunities for development of a scaffoldless tissue engineering culture system for fibrous tissues. Although we have not measured mechanical parameters of the culture aggregates, they can be handled carefully without damage, and it would be possible therefore to take aggregates from the system and mechanically or biochemically condition them using cyclic tensile loading and/or growth factor treatments to enhance its development and strength. One might expect this to be successful based on existing understanding of mechanical loading and fibrous connective tissues13,36–38 and also from further comparison with embryology, which shows that tendons initially develop but only progress to fully formed structures if the associated muscle cells migrate and start to mechanically stimulate them.2,43 The roller system also needs to be developed further to concentrate all of the cells into the rod-shaped aggregates rather than allowing formation of other structures, generally spherical ones, as well. This is likely to be a function of the culture environment within the rotating tube, relating to roller speed and medium agitation and should be improvable with further development work. The system lends itself to the use of different cell types, such as various fibroblasts, including corneal and dermal, or mesenchymal stem cells,44–47 and we will explore how it could be modified and applied to other tissue organizations. Finally, the system has a significant advantage in being based on cheap, sterile, disposable, and readily available consumables.
Footnotes
Acknowledgments
This work was supported by an EPSRC quota studentship. We gratefully acknowledge Dr. Ilyas Khan and Dr. Rebecca Williams for advice and assistance with the biochemical assays.
Disclosure Statement
No competing financial interests exist.