
Abstract
Background:
Bone substitutes should ideally promote rapid vascularization, which could be accelerated if these substitutes were vitalized by autologous cells. Although adequate engraftment of porous poly(L-lactide-co-glycolide) (PLGA) scaffolds has been demonstrated in the past, it has not yet been investigated how vascularization is influenced by vitalization or, more precisely, by seeding PLGA scaffolds with osteoblast-like cells (OLCs). For this reason, we conducted an in vivo study to assess host angiogenic and inflammatory responses after the implantation of PLGA scaffolds vitalized with isogeneic OLCs.
Materials and Methods:
OLCs were seeded on collagen-coated PLGA scaffolds that were implanted into dorsal skinfold chambers in BALB/c mice (n = 8). Two further groups of animals received either collagen-coated (n = 8) or uncoated PLGA scaffolds (n = 8). Animals that received chambers without implants served as controls (n = 8). Angiogenesis, neovascularization, and leukocyte–endothelial cell interaction were analyzed for 14 days using intravital fluorescence microscopy.
Results:
PLGA scaffolds with and without OLCs showed a temporary increase in leukocyte recruitment. At day 3 after implantation, a marked angiogenic host tissue response was observed in close vicinity of all scaffolds studied. At days 6 and 10, the angiogenic response was significantly higher (p < 0.05) in PLGA scaffolds vitalized with OLCs than in uncoated or collagen-coated PLGA scaffolds. The majority of OLCs, however, died within 14 days after implantation.
Conclusion:
Our study demonstrates that PLGA scaffold vitalization with OLCs accelerates the angiogenic response in the surrounding host tissue. Bone substitutes created by tissue engineering may thus be superior to nonvitalized substitutes although the seeded cells do not survive for long periods.
Introduction
Although previous studies demonstrated an adequate engraftment of poly(L-lactide-co-glycolide) (PLGA) scaffolds,11,12 the influence of vitalization with osteoblast-like cells (OLCs) on vascularization is still poorly understood. In particular, the fate of OLCs seeded on PLGA scaffolds is unknown. The aim of this study was therefore to assess angiogenic and inflammatory responses to the seeding of OLCs on 3D PLGA scaffolds that are manufactured using a rapid prototyping 3D Bioplotting technique. 13
Materials and Methods
Animals
All animal procedures were approved by the responsible animal care committee and were conducted in accordance with the German Protection of Animals Act and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication #85-23, revised 1985). The study included 12- to 16-week-old female BALB/c mice weighing 22–25 g. The animals were housed singly in cages at a room temperature of 22°C–24°C and a relative humidity of 60%–65% with a 12-h day/night cycle. They received tap water and a commercial pellet diet (Altromin, Lage/Westphalia, Germany) ad libitum.
Anesthesia
Both surgery and repeated intravital fluorescence microscopy were performed under anesthesia by intraperitoneal injection of saline (0.1 mL/10 g body weight) containing ketamine hydrochloride (90 mg/kg body weight, Ketavet®; Parke Davis, Freiburg, Germany) and dihydroxylidinothiazine hydrochloride (25 mg/kg body weight, Rompun®; Bayer, Leverkusen, Germany).
Dorsal skinfold chambers
A dorsal skinfold chamber contains a single layer of striated muscle, subcutaneous tissue, and skin and allows microcirculation to be studied in the anesthetized animal over an extended period of time using intravital microscopy. The chamber and its implantation have been described in detail previously. 14 Two symmetrical titanium frames were implanted on the extended dorsal skinfold of the mice in such a way that the double layer of skin was sandwiched between the frames. A circular area with a diameter of 15 mm that consisted of one layer of skin, subcutaneous tissue, and striated skin muscle tissue was then removed. The remaining layers containing striated skin muscle tissue, subcutaneous tissue, and skin were covered with a removable coverslip incorporated into one of the titanium frames. After this procedure, the animals were allowed to recover from anesthesia and surgery for at least 48 h before scaffold implantation. Both the procedure and the chamber were well tolerated by the animals, which displayed normal feeding and sleeping behavior.
Scaffolds
The scaffolds were produced in the Freiburg Institute for Material Research and Macromolecular Chemistry using a 3D Bioplotter™ and PRIMCAM software (PRIMUS DATA, Einsiedeln, Switzerland) as described in detail previously. 13
Isolation and cultivation of murine OLCs
Primary cultures of OLCs were isolated from male BALB/c mice aged 6–8 weeks as described before. 15
Detection of alkaline phosphatase
Cells were washed thrice with phosphate-buffered saline (PBS; PBS Biochrom, Berlin, Germany) and incubated for 15 min in 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (Roche, Mannheim, Germany). They were then examined with light microscopy (DM4000B Leica Mikrosysteme, Wetzlar, Germany) without counterstaining. 16
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde (Sigma Aldrich, Taufkirchen, Germany) for 10 min and then blocked in PBS solution (PBS Biochrom) containing 5% fetal calf serum and 0.2% Triton X-100 (Sigma Aldrich) for 20 min. They were incubated with primary antibodies overnight at 4°C and subsequently with secondary antibodies for 1 h. After each step, cells were washed thrice with PBS for 5 min. Rabbit anti-mouse collagen I (Biotrend, Cologne, Germany), rabbit anti-mouse osteocalcin (Antibodies Online, Aachen, Germany), and rat anti-mouse SPARC (osteonectin; R&D Systems, Wiesbaden, Germany) were used as primary antibodies. Cy-2-conjugated goat anti-rabbit antibody and Cy-3-conjugated goat anti-rat antibody (both from Dianova, Hamburg, Germany) were used as secondary antibodies. The cells were examined by fluorescence microscopy (DM4000B Leica Mikrosysteme). 17
Measurement of vascular endothelial growth factor release under normoxic and hypoxic conditions
OLCs were exposed to hypoxic culture conditions to analyze vascular endothelial growth factor (VEGF) protein expression. About 2 × 105 OLCs were plated in each well of a six-well plate (Greiner, Frickenhausen, Germany) in the culture medium (Dulbecco's modified Eagle medium [DMEM]; 10% fetal calf serum, 20 mM HEPES, 1000 IU/mL penicillin, and 0.1 mg/mL streptomycin, PAA [PAA Laboratories GmbH, Cölbe, Germany]) and incubated at 37°C in a humidified atmosphere containing 91.5% air and 8.5% CO2 for 3 days. The culture medium was removed, cells were washed with Hank's balanced salt solution (PAA), equal amounts of the serum-free culture medium (1 mL) were added to each well, and plates were placed in normoxic (21% O2) or hypoxic conditions. Hypoxic culture conditions were created using an anaerobic jar with in-put and out-put Schrader valves (Oxoid, Cambridge, United Kingdom). The anaerobic jar was de-oxygenated by infusion of standardized gas mixtures of known oxygen content. We used mixtures with oxygen contents of 1% O2 and 5% O2. Additional components were 8.5% CO2 and N2 up to 100% (Linde Gas, Pullach, Germany). Hypoxic cultures and normoxic controls were incubated at 37°C in a humidified atmosphere containing 91.5% air and 8.5% CO2. At the indicated time points, the conditioned medium was collected. VEGF levels in the conditioned medium were determined with an ELISA kit (mouse VEGF DuoSet development system; R&D Systems) according to the manufacturer's protocol. Values were corrected for cell number by crystal violet staining of plated cells by an adopted method from Kueng et al. 18 Briefly, cells were washed thrice with PBS, fixed with 4% paraformaldehyde for 10 min, permeabilized with 20% methanol for 20 min, and stained with 0.5% crystal violet (both from Sigma Aldrich) in 20% methanol for 30 min. Destaining was performed with three gentle washes with deionized water, and residual crystal violet was eluted with 10% acetic acid (Sigma Aldrich) for 30 min. Optical density was measured spectrophotometrically at 650 nm (Tecan Group Ltd., Maennedorf, Switzerland).
Cell labeling
Murine OLCs were labeled using the PKH26 Red Fluorescent Cell Linker Kit (Sigma, Deisenhofen, Germany) according to the manufacturer's instructions. This kit allows the labeled cells to be tracked in subsequent experiments. OLC viability was determined by observing cell morphology. Viable cells display an annular pattern of fluorescence.
Collagen-coated PLGA scaffolds
The four-layer PLGA scaffolds were cut aseptically into pieces of approximately 3 × 3 × 1 mm, strung on a thread, placed into rat collagen type I (0.2%; PAN, Aidenbach, Germany) for 1 min, and air-dried for 1 h. This procedure was repeated a second time. Collagen-coated scaffolds were washed with PBS for 1 h and air-dried. They were stored at 4°C for no longer than 1 week before use.
Seeding of murine OLCs on PLGA scaffolds
Murine OLCs were seeded onto collagen type I–coated PLGA scaffolds for in vivo experiments as described before. 15
Detection of viable cells
We prepared four additional chambers to prove the suitability of PKH26 for detecting viable OLCs. For the detection of living cells on implanted scaffolds, the mice were anesthetized by intraperitoneal injection of saline (0.1 mL/10 g body weight) containing ketamine hydrochloride (90 mg/kg body weight, Ketavet; Parke Davis) and dihydroxylidinothiazine hydrochloride (25 mg/kg body weight, Rompun; Bayer). Subsequently, the coverslip was removed and the open chamber was flushed thrice with PBS. Living cells on the scaffold were stained with calcein acetoxymethyl (calcein AM) diluted in PBS (8 μg/mL; Invitrogen, Karlsruhe, Germany) for 30 min. After incubation, the chamber was flushed thrice with PBS and sealed with a new coverslip. Viable cells were identified by the capacity of intracellular esterases to convert nonfluorescent calcein AM into green-fluorescent calcein.18,19 Images were acquired with a confocal laser scanning microscope (TCS SP2 AOBS; Leica Mikrosysteme) at the four corners and at the center of the scaffolds. Living green-fluorescent cells were counted at 10× magnification and at 1024 × 1024 pixel resolution on days 0, 3, 6, 10, and 14.
Intravital fluorescence microscopy
For in vivo microscopy, the mice were again anesthetized as described above and immobilized in a custom-made stereotactic frame. After intravenous retrobulbar injection of 0.1 mL 5% fluorescein-isothiocyanate-labeled dextran (150,000) for contrast enhancement of blood plasma and 0.1 mL 0.1% rhodamine 6G (Sigma) for direct observation of leukocytes, intravital fluorescence microscopy was performed using a modified Zeiss Axiotech microscope equipped with a 100-W mercury lamp and an illuminator with blue, green, and ultraviolet filter blocks (Zeiss, Jena, Germany) for epi-illumination. The microscopic images were recorded by a charge-coupled device video camera (FK-6990, COHU; Prospective Measurements, San Diego, CA) and were transferred to a DVD recorder (LQ-MS 800; Panasonic, Hamburg, Germany) for off-line evaluation. Long-distance lenses (10× and 20× magnifications) were used for displaying the images on a 14-inch video screen (Panasonic, DVD video recorder LQ-MD 800E; Matsushita Electronic Industrial Co. Ltd., Osaka, Japan).
Image analysis
Quantitative off-line analysis of the recorded data was performed by means of a computer-assisted image analysis system (CapImage; Zeintl, Heidelberg, Germany). Leukocyte–endothelial cell interaction, microhemodynamics, and macromolecular leakage were assessed at a magnification of 20× in four different microvascular regions of interest (ROIs) at the periphery of the scaffolds. In the control animals, four comparable ROIs were analyzed in the dorsal skinfold chambers, which were unaffected by material implantation. In each ROI, one to three venules (internal diameter 20–40 μm) were chosen for measurement. Depending on their interaction with vascular endothelium, leukocytes were classified as adherent, rolling, or free-flowing cells as described previously. 11 Adherent leukocytes were defined in each vessel segment as cells that did not move or detach from the endothelial lining within a specified observation period of 20 s. They are expressed as the number of cells per square millimeter of endothelial surface, calculated from the diameter and length of the vessel segment. Cylindrical vessel geometry was assumed. Rolling leukocytes were defined as cells that moved with a velocity less than two fifths of centerline velocity. They are expressed as the number of cells per minute that passed a defined reference point in a microvessel. Diameters, centerline velocity, volumetric blood flow, and wall shear rate were determined in those venules in which leukocyte–endothelial cell interaction was analyzed. Diameters (d) were measured in micrometers perpendicularly to the vessel path. Centerline red blood cell velocity (v) was analyzed using a computer-assisted image analysis system and the line shift method. Volumetric blood flow was calculated using the following formula: Q = π ×(d/2) 2 × v/1.6 picoliter/second (pl/s), where 1.6 represents the Baker–Wayland factor to correct for the parabolic velocity profile in microvessels with a diameter >20 μm. Wall shear rate (y) was calculated based on the Newtonian definition: y = 8 × v/d. Macromolecular leakage served as an indicator of microvascular permeability. After intravenous injection of the macromolecular fluorescent dye fluorescein-isothiocyanate-labeled dextran (150,000), it was assessed by the densitometric measurement of gray levels in the tissue directly adjacent to the venular vessel wall (E1) and in the marginal cell-free plasma layer within the vessel (E2). Extravasation (E) was then calculated using the following formula: E = E1/E2. Angiogenesis was analyzed using a 10× long-distance lens in four different microvascular ROIs at the periphery of the scaffolds as described above.
The fluorescent-labeled OLCs that were seeded on PLGA scaffolds were counted immediately as well as on days 3, 6, 10, and 14 after implantation in two different ROIs (0.1 mm2).
Immunohistochemical detection of VEGF, osteocalcin, CD31, and smooth muscle actin
For immunohistochemical detection of VEGF, osteocalcin (as evidence of the presence of OLCs), CD31 (as evidence of the presence of endothelial cells), and smooth muscle actin (as evidence of the presence of smooth muscle cells) at various time points in the scaffolds, formalin-fixed specimens from the dorsal skinfold chamber from four additional groups at days 3, 6, 10, and 14 were embedded in paraffin and cut into 5-μm-thick sections. A chamber containing PLGA scaffolds without OLCs was added as a control. Consecutive sections were incubated with a rabbit anti-human VEGF antibody (Acris Antibodies GmbH, Hiddenhausen, Germany), a rabbit anti-mouse osteocalcin antibody (Antibodies Online), a rabbit anti-mouse CD31 antibody (Acris Antibodies GmbH), or a rabbit anti-human smooth muscle actin antibody (Dianova), respectively, as primary antibodies. A biotin-conjugated goat anti-rabbit antibody (Dianova) was used as a secondary antibody. Incubation with streptavidin–horseradish peroxidase (Dianova) was followed by color development with aminoethylcarbazole substrate (Axxora Deutschland GmbH, Loerrach, Germany) at room temperature. Color development was stopped under microscopic control by washing with water. The sections were counterstained with hemalaun and examined by light microscopy (DM4000B Leica Mikrosysteme). For negative controls, the primary antibody was omitted. All control stainings were negative.
Histology
At the end of the in vivo experiments, histological examinations were performed. For light microscopy, formalin-fixed specimens from the dorsal skinfold chamber were embedded in paraffin. Four-micrometer-thick sections were cut and stained with hematoxylin and eosin according to standard procedures.
Experimental protocol
For intravital fluorescence microscopy, dorsal skinfold chambers were implanted into a total of 40 BALB/c mice. Uncoated PLGA scaffolds (n = 8), collagen-coated PLGA scaffolds (n = 8), or OLC-seeded, collagen-coated PLGA (n = 8) were inserted. Special care was taken to avoid contamination, mechanical irritation, or damage to the chamber. In eight additional control animals, the glass coverslip of the dorsal skinfold chamber was temporarily removed without scaffold implantation (sham implantation). The macroscopic appearance of the skinfold chambers and the implanted scaffolds was documented daily. Intravital fluorescence microscopic analysis of leukocyte–endothelial cell interaction, microhemodynamics, macromolecular leakage, and angiogenesis was performed immediately and on days 3, 6, 10, and 14 after scaffold or sham implantation. An additional group of animals (n = 8) received scaffolds with fluorescent-labeled (PKH26) OLCs for the detection of viable cells during the observation period. In addition, four chambers were prepared for intravital staining with calcein AM to prove the suitability of PKH26 for detecting OLCs.
Statistical analysis
Results are expressed as means ± standard error of the mean. Differences between groups were assessed by one-way analysis of variance; differences within each group were analyzed by one-way repeated measures analysis of variance. Student–Newman–Keuls or Dunn's post hoc tests were used to isolate specific differences. Results with p < 0.05 were considered significant.
Results
In vitro characterization of OLCs
For the characterization of cultivated OLCs, alkaline phosphatase was detected with a colorimetric assay (Fig. 1A). Additional immunocytochemical detection of collagen I (Fig. 1B), osteocalcin (Fig. 1C), and osteonectin (Fig. 1D) using fluorescence microscopy confirmed that the cells were bone cells and of high purity.
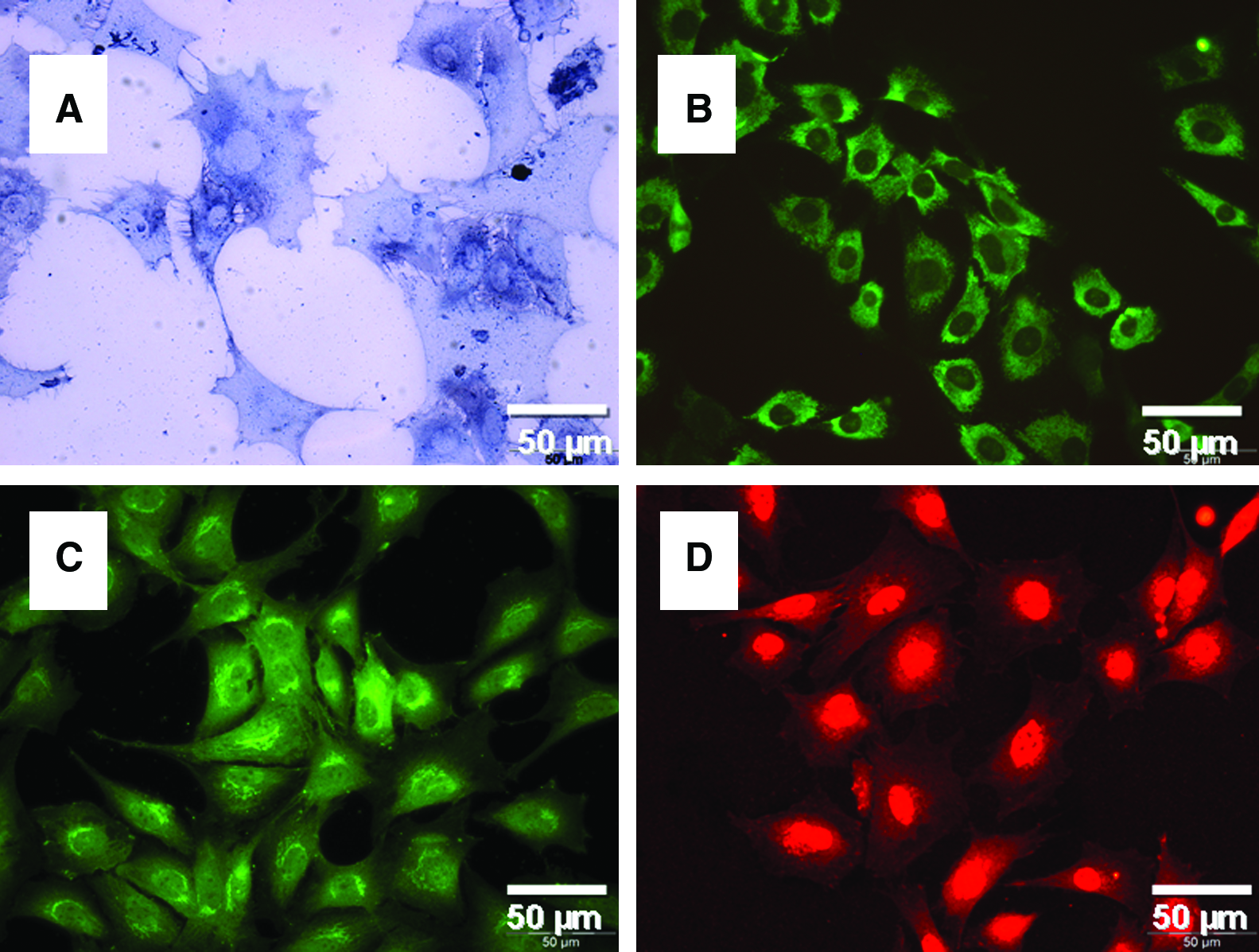
Characterization of cultivated OLCs: detection of alkaline phosphatase by light microscopy (
Measurement of VEGF release under normoxic and hypoxic conditions
Exposure of OLCs to hypoxic culture conditions revealed significantly enhanced VEGF protein expression. At 1% oxygen content, a threefold increase of VEGF could be measured compared to the control after 24 and 48 h. Using 5% oxygen content, the difference between both groups was less marked but still significant (Fig. 2)

VEGF released by OLCs exposed to hypoxic conditions. Hypoxic (1% (
Microhemodynamics
In all experimental groups, analysis of microhemodynamics revealed constant venular diameters ranging between 20 and 40 μm (data not shown). Moreover, volumetric blood flow and wall shear rates of venules in the immediate vicinity of the scaffolds were not significantly different from those obtained for the control group throughout the entire 14-day observation period.
Inflammatory response
A comparison with control animals showed that the implantation of uncoated PLGA scaffolds as well as collagen-coated and OLC-seeded, collagen-coated PLGA scaffolds resulted in a transient activation of leukocytes as demonstrated by an increased number of rolling leukocytes in postcapillary venules at the periphery of the scaffolds (Fig. 3A). After 10 days, however, the number of rolling leukocytes was comparable to control values (Fig. 3A). In addition, the number of leukocytes adherent to the endothelium of postcapillary venules was found to be slightly increased in the immediate vicinity of the scaffolds (Figs. 3B and 4). This finding was associated with a moderate increase in microvascular permeability, which, however, did not reach the starting value over the entire 14-day observation period (Fig. 3C).

Numbers of rolling leukocytes are expressed as cells/min (

Intravital fluorescence microscopy of postcapillary venules at the periphery of uncoated (
Angiogenesis and neovascularization
In the control group, no angiogenesis was observed in response to the implantation of a dorsal skinfold chamber at any time point, neither in the center of the observation window nor in the immediate vicinity of the titanium frame (Fig. 5G, H). In all scaffolds types, first signs of angiogenesis were detected at the periphery of the scaffolds at day 3 after implantation and consisted of capillary dilation and the formation of capillary buds and sprouts primarily originating from venular segments of striated muscle capillaries and postcapillary venules of the host tissue. These sprouts interconnected to each other and formed new red blood cell–perfused microvascular networks within 14 days (Fig. 5A–F). The quantitative analysis of functional microvascular density demonstrated a significantly increased density of the microvascular network that grew into OLC-seeded scaffolds compared to uncoated or collagen-coated PLGA scaffolds at days 6 and 10 after implantation (Fig. 6).
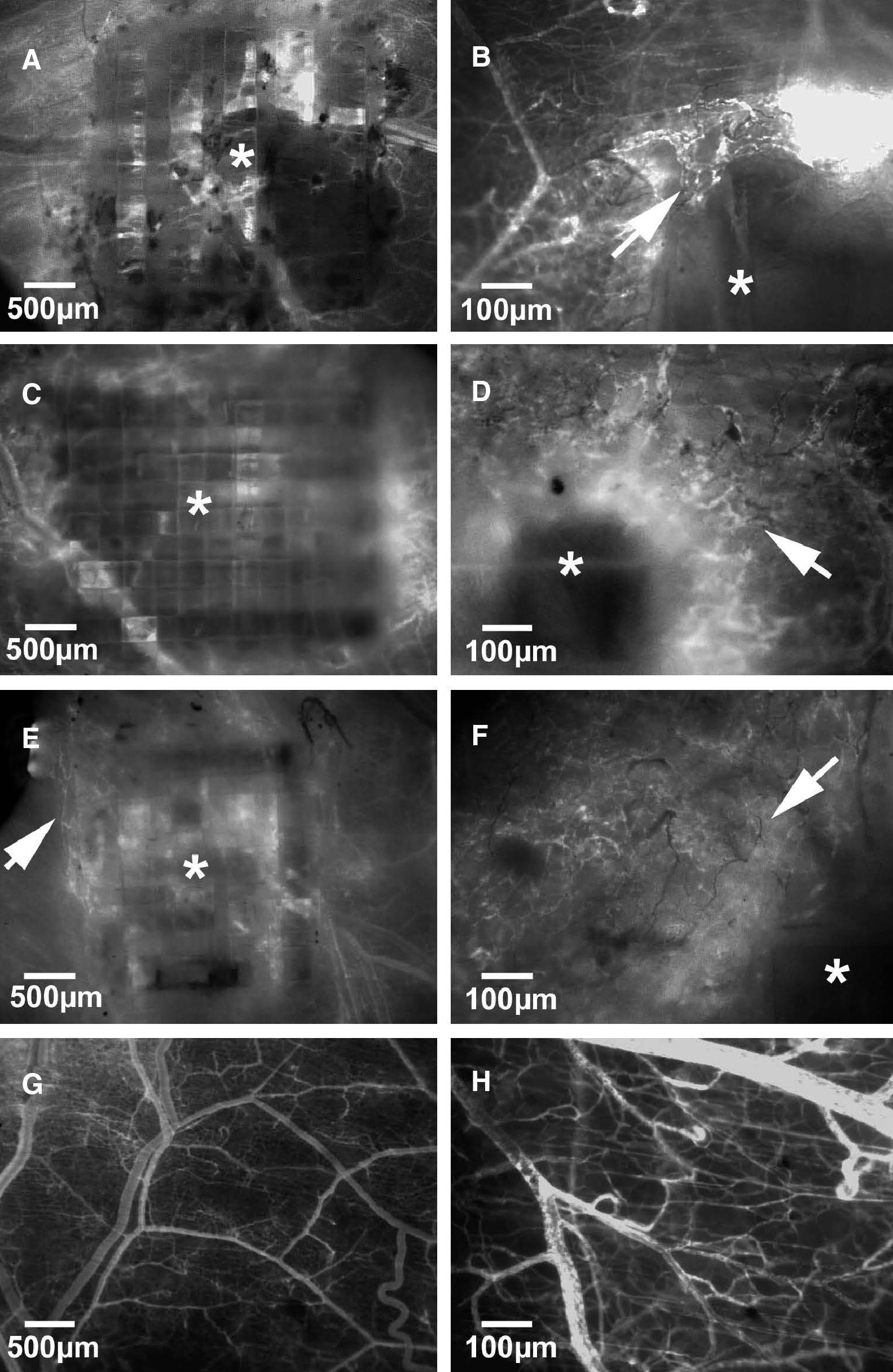
Intravital fluorescence microscopy of uncoated (
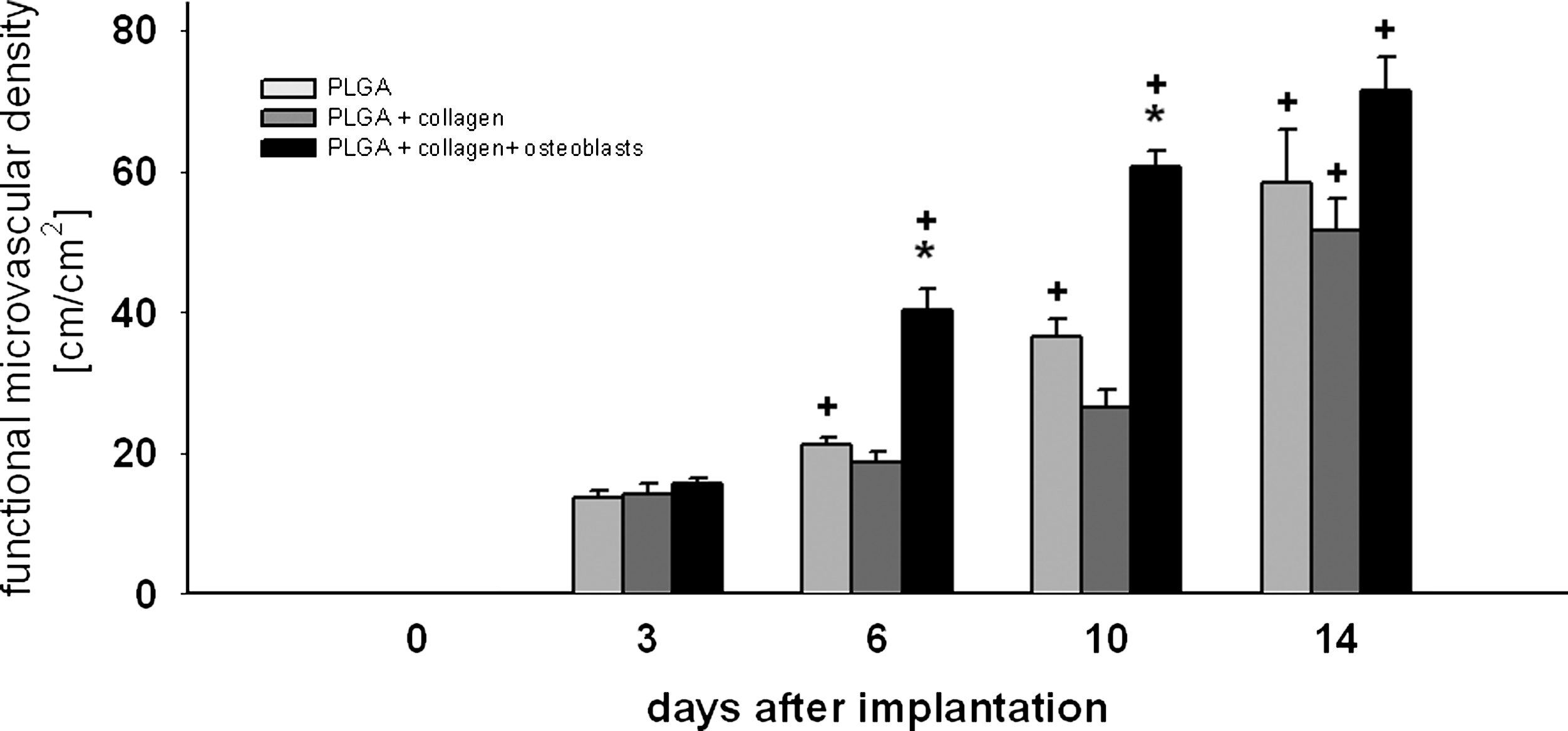
Functional microvascular density of newly formed microvessels expressed in cm/cm2 in regions of interest of uncoated (light gray bars), collagen-coated (dark gray bars), and OLC-seeded, collagen-coated (black bars) PLGA scaffolds immediately (day 0) and on days 3, 6, 10, and 14 after implantation into dorsal skinfold chambers in BALB/c mice. There was no angiogenesis observed in sham controls. Accordingly, this group is not included in the present figure. Means ± SEM. *p < 0.05 versus uncoated and collagen-coated PLGA scaffolds; +p < 0.05 versus day 0 and day 3.
OLC survival
An examination of PKH26 fluorescent-labeled OLCs showed a decrease in surviving OLCs as early as 6 days after implantation. Only a very small number of OLCs survived until day 14. At the end of the experiment, major parts of the scaffolds were covered with cell detritus (Fig. 7). For validation of these results, intravital staining with calcein AM was performed. Using a confocal laser scanning microscope, the decrease in seeded OLCs over time was confirmed (Fig. 8).
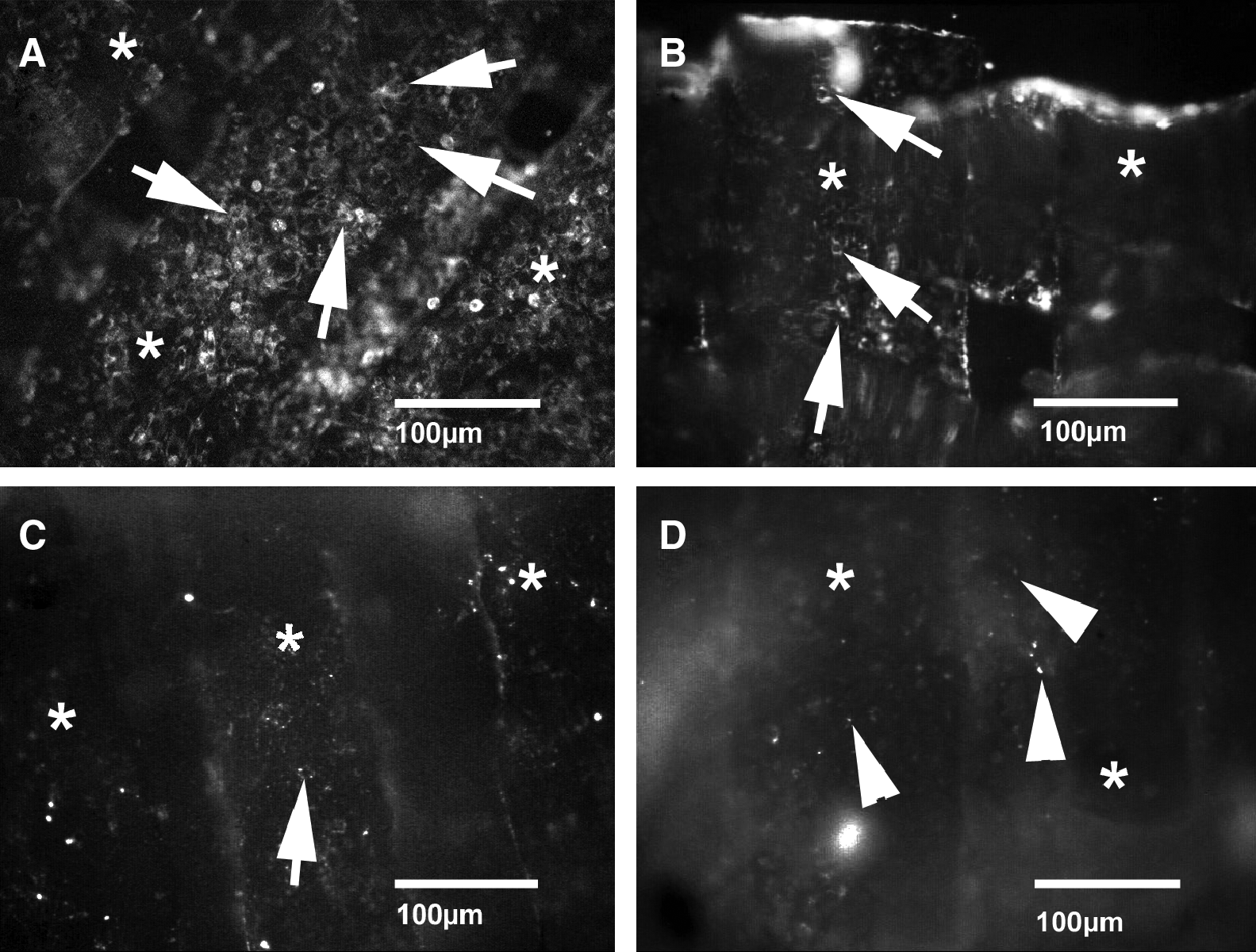
Intravital fluorescence microscopy of labeled OLCs attached to a PLGA scaffold immediately (

Intravital confocal laser scanning microscopy of calcein AM-labeled OLCs attached to a PLGA scaffold immediately (
Immunohistochemical detection of VEGF, osteocalcin, CD31, and smooth muscle actin
Pronounced expression of VEGF was immunohistochemically detected on day 3 in scaffolds seeded with OLCs (Fig. 9F). In scaffolds without cells we could detect only a minor VEGF expression (Fig. 9E). Positive immunohistochemical staining for osteocalcin confirmed the presence of OLCs on day 3 in OLC-seeded scaffolds (Fig. 9H), in contrast to collagen-coated scaffolds, where no osteocalcin expression was detected (Fig. 9G). At day 14, CD31 (Fig. 9A, B) and smooth muscle actin–positive microvessels could be detected in OLC-seeded scaffolds and, to a lesser extent, in control scaffolds (Fig. 9C, D). Additional immunohistochemical staining for CD31 in OLC-seeded scaffolds at days 3, 6, 10, and 14 after scaffold implantation allowed us to follow the time course of microvessel growth (Fig. 10). The increase in stained microvessels was comparable to the increase in functional microvascular density observed by intravital fluorescence microscopy.

Immunohistochemical demonstration of CD31 (
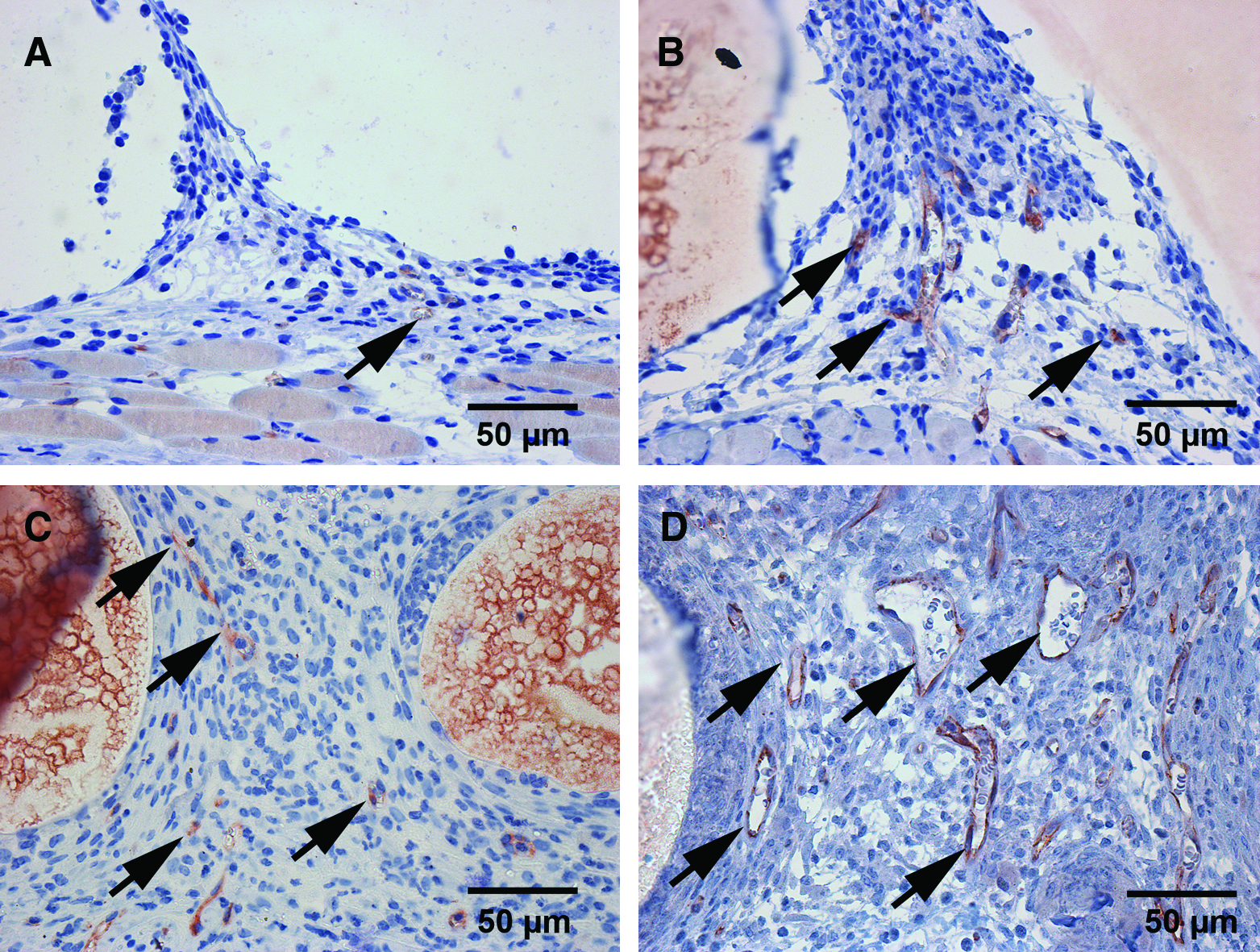
Immunohistochemical demonstration of CD31 in OLC-seeded, collagen-coated PLGA scaffolds on day 3 (
Histological examination
A histological examination on day 14 after scaffold implantation confirmed our intravital microscopic findings. All scaffolds exhibited a vascular host tissue response with formation of densely vascularized granulation tissue despite a distinct infiltration of inflammatory cells and also some giant cells. This host tissue response is evidence of adequate engraftment of the scaffolds in host tissue (Fig. 11).
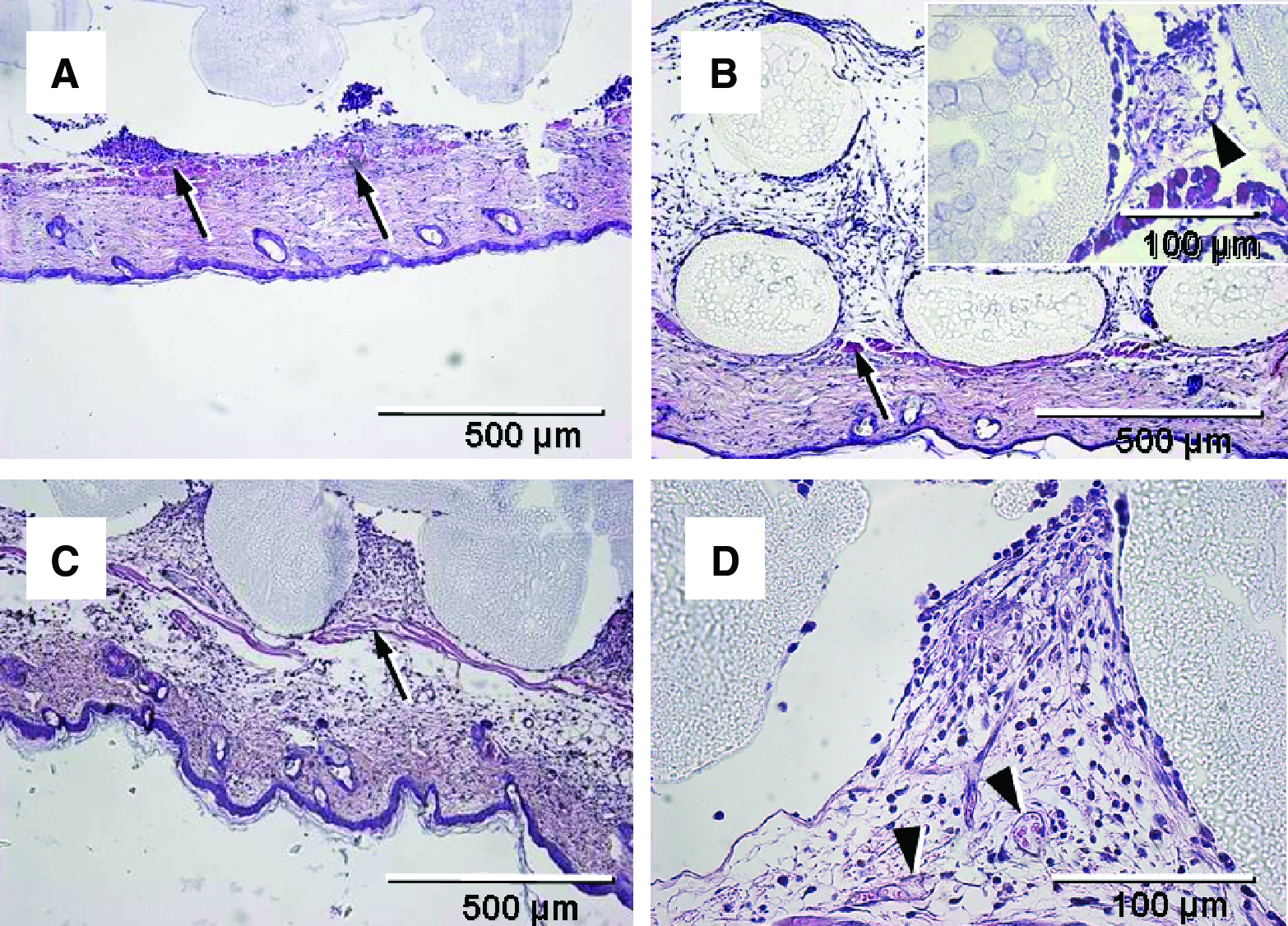
Hematoxylin and eosin–stained cross sections of uncoated (
Discussion
A bone transplant is seldom completely embedded in bone tissue. Transplants used for bone reconstruction also come into contact with surrounding soft tissue. For this reason, the clinical success of bone transplantation depends on safe and effective soft tissue coverage. In this study, we demonstrated a significant enhancement of PLGA scaffold neovascularization in vivo after seeding with OLCs for vitalization. For our in vivo microscopic studies of the host tissue response following scaffold implantation, we used a dorsal skinfold chamber, which offers several distinct advantages over other models. The dorsal skinfold chamber is an established method to analyze the proliferation of vessels for 3 weeks and has recently been adopted for the analysis of microcirculatory parameters in tissue engineering constructs.12,14,15,19,20
Angiogenesis and neovascularization of different vascular graft materials was demonstrated in vivo showing that a network of microvessels developed that originated from the capillaries of the perigraft tissue. Our results underline the feasibility of the chosen approach, as we could verify the ingrowth of capillaries by detection of CD31-positive cells and by further recruitment of smooth muscle actin–positive mural cells, pointing to the development of a functional and well-perfused microvascular network. The chamber allows us to compare our results with the findings of previous studies since this model was used in the past to investigate the inflammatory and angiogenic host tissue response to different biomaterials.11,12,21 Using this model, future studies can thus be conducted in an attempt to improve the biocompatibility and successful engraftment of materials by modifying scaffolds with different factors that regulate angiogenesis and inflammation. The suitability of PKH26 for detecting seeded OLCs was assessed by intravital staining with calcein and the subsequent search for OLCs using a confocal laser scanning microscope.22,23 Immunohistochemistry staining of the OLC marker osteocalcin was performed at day 3. The results proved that PKH26 staining is a reliable method of detecting OLCs using intravital microscopy.
The question arises as to what extent the chamber-frame material induces inflammation or angiogenesis. In the sham-operated controls, we found no signs of angiogenesis or inflammation. The skinfold chamber allows the observation of the striated muscle microcirculation. During the preparation of the chamber a portion of skin and muscle tissue is removed to open the view to the observation area. The preparation method guaranties that the observation area is not influenced, leaving the microcirculation untouched. Because of this lack of stimulus, no angiogenesis is observed in the sham-operated animals. 24 Previous studies10,12 reported further that titanium frames do not induce any inflammatory or even angiogenic responses that might interfere with an assessment of neovascularization of the implanted scaffolds. In particular, it was demonstrated that titanium implants inserted in striated muscle tissue caused only a transient increase in leukocyte–endothelial cell interaction within the first 120 min, which was not associated with a significant change in macromolecular leakage, leukocyte extravasation, and venular diameter. 25 To prevent interactions of these temporary inflammatory responses, we allowed the animals to recover from chamber implantation and inserted the scaffolds not earlier than 2 days after the procedure.
A 3D scaffold as a guiding structure for new bone formation is the basis of all bone tissue engineering constructs. In this study we analyzed scaffolds that were fabricated from resorbable PLGA, because this material is well characterized and used for many tissue engineering applications. In case of bone tissue engineering, PLGA is often combined with hydroxyapatite or other ceramics/glasses to provide mechanical properties similar to bone. However, the mechanical properties of tissue engineering constructs are of minor significance since in clinical practice bone defects are usually stabilized by titanium plates. In fact, these plates take the mechanical load from the tissue construct and guarantee an adequate stabilization of the bone defect until the tissue construct is fully incorporated into the surrounding tissue. Recently, we could demonstrate that PLGA scaffolds induce an angiogenic host tissue response, which is comparable to implanted hydroxyapatite and isogeneic bone.10–12 This is an important finding because angiogenesis and neovascularization play an important role in the process of bone healing. A major prerequisite for the ingrowth of new blood vessels into implanted PLGA scaffolds is an adequate porosity. Different methods of producing porous scaffolds are described in the literature.13,26–28 The PLGA scaffolds of this study could be fabricated by rapid prototyping with a standardized pore size of 250 μm, which has previously been shown to support a rapid microvascular ingrowth. 29 A essential element of cell survival is a sufficient delivery of nutrients usually guaranteed by an adequate vascular system. In this way, the preexisting studies suggested the suitability of the used scaffolds. As the results presented here and the results of Rücker et al. showed, PLGA scaffolds were able to promote angiogenesis. In addition, the PLGA scaffolds were characterized by a good in vitro and in vivo biocompatibility. 12 For these reasons, these PLGA scaffolds represent a promising scaffold type for tissue engineering of bone.
Shortly after the implantation of PLGA scaffolds into dorsal skinfold chambers, inflammatory parameters increased in the postcapillary and collecting venules at the periphery of the scaffolds. This result was in line with other studies reporting an acute short-term inflammatory tissue response after implantation of foreign materials into the chamber.25,30,31 Our study further demonstrates that there is no difference between vitalized and nonvitalized scaffolds in terms of the inflammatory response initiated.
Apart from an inflammatory tissue response, leukocyte activation can also induce angiogenesis by producing a number of angiogenic factors.32,33 In particular, VEGF is believed to be of crucial importance to the formation of granulation tissue and the successful engraftment of implanted scaffolds into host tissue. 34 The impact of VEGF signaling and the regulation of subsequent factors on angiogenesis is well understood and described in detail.35–37
In our study, macromolecular leakage increased moderately after implantation of PLGA scaffolds, collagen-coated PLGA scaffolds, and OLC-seeded, collagen-coated PLGA scaffolds. This may be interpreted as a result of both inflammation and VEGF-driven neovascularization. To investigate this hypothesis, we performed immunohistochemical staining for VEGF at day 3. Compared to PLGA scaffolds without cells, OLC-seeded scaffolds showed enhanced expression of VEGF. This result supports the hypothesis that neovascularization is driven by VEGF. It would therefore be interesting to evaluate systemic levels of angiogenic factors like VEGF. The region where VEGF is expressed is, however, characterized by the presence of newly formed vessels. It appears unlikely that VEGF is transported through these new vessels, and if this was the case, the quantity of systemic VEGF levels would be too low to be detected systemically.
It was interesting to note that OLC-seeded scaffolds showed a significantly increased angiogenic response although the number of vital OLCs declined after the implantation of the scaffolds into dorsal skinfold chambers. This indicates that the process of angiogenesis may also be driven by the decrease in OLCs. Due to hypoxia, however, even vital OLCs increase transcription factors such as hypoxia-inducible factors, which can induce angiogenic growth factors.38–41 Disintegration of OLCs may have increased VEGF release. 42 VEGF is a heparin-binding growth factor specific for vascular endothelial cells and is able to induce angiogenesis in vivo.43,44 Therefore, we performed additional in vitro experiments with OLCs under hypoxic conditions. Our results demonstrated that these cells increased expression of VEGF significantly under hypoxic conditions compared to normoxic controls. These findings are in line with the results of Steinbrech et al., who showed that MC3T3-E1 OLCs increased VEGF production as a response to hypoxia in an extent comparable to the VEGF values we could detect. 45 Although our in vitro experiments were performed with cells in monolayer, the results can be transferred to the in vivo situation where cells are in a 3D topology on the scaffold. Jarrahy et al. demonstrated that MC3T3-E1 OLCs exhibit enhanced gene expression and protein secretion of VEGF when cultured in a 3D environment compared to 2D conditions. 46 Osteogenic-differentiated stem cells derived from processed lipoaspirate were also influenced by the culture topology. Cells cultured on 3D PLGA scaffolds showed higher VEGF expression. 47 Further, MC3T3-E1 OLCs upregulated VEGF as an answer to cyclic strain and fluid shear stress.48,49 Altogether, the information available supported our conclusions, as they prove that the switch from 2D to 3D culture conditions enhances VEGF expression instead of reducing it. These endogenous properties of OLCs appear to be the reason why vitalized scaffolds showed a higher density of newly formed microvascular networks than nonseeded PLGA scaffolds. Obviously, the acceleration of vascularization at days 6 and 10 was not rapid enough to ensure the survival of seeded OLCs. Endothelial cell migration and physiological growth of new blood vessels has been demonstrated not to be faster than ∼5 μm/h.50,51 Because the amount of oxygen, which is required for cell survival, is limited to a diffusion distance of only ∼150–200 μm from a supplying blood vessel.52,53 Cells lying beyond this physiological border suffer from hypoxia, resulting in VEGF release and therefore the onset of angiogenesis from the scaffold surrounding tissue. It is thus not surprising that most of the OLCs seeded on the scaffolds died within the observation period of 14 days, because ingrowing blood vessels could not reach the center of the scaffolds within this short time solely via the process of angiogenesis. Therefore, it is of major interest for the field of tissue engineering to establish novel strategies that guarantee the survival of seeded cells within tissue constructs in the initial phase after implantation. These strategies may include preconditioning methods to increase the ischemic tolerance of cells inside a tissue construct, 54 inosculation of preformed microvascular networks, 55 or the use of arteriovenous loops for promoting prevascularization and overcoming hypoxia.56,57 Coculture strategies employing OLCs and differentiated or progenitor cells could also be successful in the development of an appropriate tissue engineering construct. Mesenchymal stem cells from different species express higher levels of osteogenic markers when cultured together with OLCs.58,59 Further endothelial cells from different sources migrate and lead to tubular network structures when cocultured with osteogenic cells. 60
The initial period of scaffold implantation is decisive for success, and the main point in the further development will be the acceleration of angiogenesis and the enhancement of cell survival. Mammoto et al. reported recently about a possible control of angiogenesis by the surrounding tissue microenvironment. 61 This signaling pathway controls VEGFR2 promoter activity and expression and is sensitive to extracellular matrix elasticity as well as soluble VEGF. These results open the opportunity of surface modifications of the PLGA scaffolds, which ideally should enhance angiogenesis and provide the fundament for a longer cell survival.
In this study, we have demonstrated that neovascularization of OLC-seeded scaffolds is significantly accelerated compared to nonvitalized scaffolds although the number of vital OLCs declined after implantation. Future studies should investigate options to improve the survival of seeded OLCs and further accelerate the vascularization of implanted tissue substitutes. These studies could address prevascularization with arteriovenous loops and the addition of angiogenic cytokines.
Footnotes
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance provided by Marie Luise Jenzer and Stefanie Rausch. This research was conducted with financial support from the Deutsche Forschungsgemeinschaft (German Research Foundation) (RU 1224/1-1, GE 820/6-1).
Disclosure Statement
No competing financial interests exist.