
Abstract
In the present study, we investigated the ex vivo expansion of human adipose tissue–derived mesenchymal stromal cells (ATSCs) to identify factors that promoted efficient expansion while preserving stem cell potential. We examined several growth factors and steroids, and found that the combination of a low concentration of fibroblast growth factor-2 (FGF-2) (1 ng/mL) and dexamethasone (DEX) or betamethasone (BET) enhanced the proliferation of ATSCs by approximately 30–60% as compared to control. Enhanced proliferation under these conditions was confirmed using ATSCs isolated from three independent donors. ATSCs that were expanded in the presence of FGF-2 and DEX for 5 days were capable of differentiating into either osteoblastic or adipogenic cells, and the cells were positive for the mesenchymal stem cell markers such as CD29, CD44, CD90, CD105, and CD146, suggesting that the stem cell potential of the ATSCs was preserved. Analysis of signaling pathway revealed that tyrosine phosphorylation of Src kinase was dramatically increased in response to FGF-2 and DEX, suggesting the involvement of Src-dependent pathways in the stimulatory mechanism of proliferation of ATSCs by FGF-2 and DEX. Moreover, Src family kinase inhibitors (SU6656 and Src kinase inhibitor I) substantially reduced the FGF-2 and DEX–induced proliferation of ATSCs. SU6656 also inhibited the osteogenic and adipogenic differentiation of ATSCs. The results of the current study demonstrate that FGF-2 in combination with DEX stimulates the proliferation and osteoblastic and adipogenic differentiation of ATSCs through a Src-dependent mechanism, and that FGF-2 and DEX promote the efficient ex vivo expansion of ATSCs.
Introduction
Several growth factors have been implicated in the regulation of MSC proliferation. In particular, fibroblast growth factors (FGFs) regulate multiple biological processes, including bone and cartilage formation. 8 The effect of FGF-2 on MSC proliferation varies, and depends largely on the stage of differentiation of the MSCs.9–11 Similarly, variable effects of FGF-2 on the osteoblastic differentiation of BMSCs have also been reported. FGF-2 significantly increases the proliferation of ATSCs and enhances chondrogenesis in a three-dimensional micromass culture. 12 However, FGF-2 also reportedly inhibits osteogenesis in mouse ATSCs and sustains their proliferative and osteogenic potential. 13 Recently, FGF-2 has been shown to maintain the self-renewing state human ATSCs. 14
Steroids can regulate multiple biological processes, including bone formation. Dexamethasone (DEX), a synthetic corticosteroid, is a potent modulator of the osteogenic differentiation of MSCs. DEX supports osteogenic lineage differentiation15–17 by binding to specific regulatory proteins and activating the transcription of osteoblast-specific genes. DEX increases alkaline phosphatase (ALP) activity, which is required for matrix mineralization. 16 The combination of FGF and DEX induces more profound effects on the osteoblastic differentiation of MSCs derived from bone marrow. For example, FGF-2 in combination with DEX increases the colony size and DNA content of human BMSCs as compared to FGF-2 alone. However, the underlying molecular mechanism of action of the two factors is unknown.18–20 BMSCs that are expanded in the presence of FGF-2 and DEX stimulate bone formation in nude mice. 20 In some cases, such as with rat marrow stromal cells, treatment with FGF-2 and DEX decreases DNA content as compared to FGF-2 alone. 21
In the present study, we developed an efficient method for the ex vivo expansion of ATSCs using FGF-2 in combination with steroids, and showed that ATSCs that are expanded in the presence of FGF-2 and DEX maintain their multi-lineage potential. We also investigated the stimulatory mechanism of proliferation of ATSCs by FGF-2 and DEX.
Materials and Methods
Isolation and culture of ATSCs
Fat tissue was obtained from patients undergoing total hip replacement surgery. Informed consent was obtained from the patients before surgery. Fat tissue was minced with a scalpel and washed with phosphate buffered saline (pH 7.4) to remove contaminating blood, and then digested with collagenase type I 2 mg/mL (Warthington Biochemical, Lakewood, NJ) for 45 min at 37°C with constant agitation. Enzyme activity was neutralized by the addition of α-modified Eagle's medium (α-MEM) (Gibco BRL, Gaithersburg, MD) containing 10% fetal bovine serum (FBS) (Biowhittaker, Walkersville, MD). Cells were obtained by centrifugation at 1200 g for 10 min. Cell pellets were resuspended in α-MEM and filtered through a 250-μm nylon mesh to remove debris. The filtered cell suspension was incubated with α-MEM supplemented with 10% FBS for 24 h. After incubation, nonadherent cells were removed, and adherent cells, comprised of ATSCs, were cultured and expanded for further analysis. ATSCs that were passaged less than three were used for all experiments. The culture medium was changed every other day.
Analysis of ATSC proliferation
ATSCs were seeded into 96-well plates at a density of 700cells/well. Cells were treated with the indicated concentrations of FGF-2 (R&D Systems, Minneapolis, MN), betamethasone (BET), DEX, hydrocortisone (HYD), prednisolone (PRE) (Sigma, St. Louis, MO), or a combination of FGF-2 and steroid for 3, 5, or 7 days. Proliferation was measured using a proliferation assay kit (Promega, Madison, WI), according to manufacturer's instructions. Optical density at 490 nm was measured with an ELISA plate reader (BioRad, Hercules, CA). Cell number was obtained using a hemocytometer.
Flow cytometric analysis
ATSCs were incubated with FGF-2 and DEX in α-MEM supplemented with 10% FBS for 5 days. Cells were harvested with 0.25% trypsin/EDTA and incubated in 1% bovine serum albumin (BSA) containing 100 μL of a solution of monoclonal antibody directed against CD29, CD44, CD90, CD105, CD146, or CD34 (BD Pharmingen, Palo Alto, CA) for 30 min at 4°C. The cells were centrifuged at 1200 g for 5 min and then washed with 1% BSA. The resuspended cells were fixed for 30 min in ice-cold 4% formaldehyde. After fixation, the cells were washed with 1% BSA, and then analyzed using a FACSAria Sorting Flow Cytometer (Beckton Dickinson, Franklin Lakes, NJ).
Multilineage differentiation of ATSCs
ATSCs that were expanded in the presence of FGF-2 and DEX for 5 days were replated into 48-well plates at a density of 1 × 104 cells/well. On the following day, osteoblastic differentiation was induced by treatment with osteogenic medium (α-MEM supplemented with 10% FBS, 50 μg/mL α-ascorbic acid, 10 mM β-glycerophosphate, and 10 nM DEX) for 14 days. Osteoblastic differentiation was assessed using alizarin red S (AR-S) staining. AR-S deposits were recovered by incubating the cells in a solution of 10% cetylpyridinium chloride (Sigma) in 10 mM sodium phosphate (pH 7.0) for 15 min at room temperature. The amount of AR-S was determined by measuring absorbance at 570 nm. Adipogenic differentiation was induced by treatment with adipogenic medium (α-MEM supplemented with 10% FBS, 1 μM DEX, 111 μg/mL isobutylmethylxanthine, 0.2 mM indomethacin, and 10 μg/mL insulin) for 14 days. Adipogenesis was assessed using Oil red O staining. To obtain quantitative data, 300 μL of isopropyl alcohol was added to the stained cells, and the amount of extracted dye was determined by absorbance at 540 nm using an ELISA plate reader.
Western blot analysis
Total cell lysate was prepared in RIPA buffer (50 mM Tris, pH 7.4, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, pepstatin 1 g/mL, and aprotinin 1 g/mL), and equal amounts of protein (20 μg) were separated by 8% SDS-PAGE. Proteins were transferred to a nitrocellulose membrane. The membrane was incubated in a solution of 5% skim milk in Tris buffered saline containing Tween-20 (20 mM Tris, pH 7.6, 137 mM NaCl, and 0.1% Tween-20) and then incubated with primary antibodies directed against phosphorylated (phospho)-p38 (BD Pharmingen), phospho-Src, Src (Cell signaling Technology, Danvers, MA), extracellular signal–regulated kinase (ERK), c-Jun N terminal kinase (JNK), or p38 (Santa Cruz, Santa Cruz, CA), as indicated. The membranes were incubated with peroxidase-conjugated secondary anti-mouse or anti-rabbit immunoglobulin (IgG) (Zymed, South San Francisco, CA), and immunoreactive proteins were detected using chemiluminescence and X-ray film.
Statistical analysis
Statistical analysis was performed using SPSS 11.0. Data were analyzed using one-way ANOVA, and a Duncan's Multiple Range Test was also adapted as a post hoc test. A p-value of less than 0.05 was considered statistically significant.
Results
Effect of steroids and FGF-2 on the proliferation of ATSCs
To investigate the effect of steroids alone or in combination with a low concentration of FGF-2 (1 ng/mL) on the proliferation of ATSCs, cells were treated with varying amounts of steroid, with or without FGF-2, for 7 days. Treatment of ATSCs with 1 or 10 nM BET, DEX, HYD, or PRE had no obvious stimulatory effect on proliferation. However, when combined with FGF-2, 1 nM BET or DEX enhanced optical density (OD) by 29.3% and 30.3%, respectively (p < 0.05), as compared to control (Fig. 1), indicating stimulation of the proliferation of ATSCs by these combinations. The stimulatory effect of steroids plus FGF-2 was greater in the presence of 10 nM BET and DEX (increases of OD by 62.9% and 61.7%, respectively, p < 0.05) (Fig. 1). The effect of FGF-2 plus other steroids (HYD and PRE) on the proliferation of ATSCs was not prominent. These results suggested that FGF-2 and DEX or BET act in a cooperative manner to stimulate the proliferation of ATSCs.

Induction of proliferation of adipose tissue-derived mesenchymal stromal cells (ATSCs) by fibroblast growth factor-2 (FGF-2) and steroids. ATSCs were cultured in the presence of FGF-2 (1 ng/mL) and the indicated concentrations of steroid (BET, DEX, HYD, and PRE) for 7 days. Proliferation assays were performed as described in Materials and Methods. *p < 0.05 as compared to untreated control cells. BET, betamethasone; DEX, dexamethasone; HYD, hydrocortisone; PRE, prednisolone; OD, optical density.
Effect of FGF-2 and DEX on the proliferation of donor-derived ATSCs
To confirm the stimulatory effect of FGF-2 and steroids on the proliferation of ATSCs, ATSCs were isolated from three independent donors and treated with FGF-2 (1 ng/mL), DEX (10 nM), or FGF-2 plus DEX for 3, 5, or 7 days. Treatment with FGF-2 alone increased the proliferation of ATSCs by an average of 22% and 20% at days 5 and 7, respectively, as compared to control, whereas proliferation was increased by an average of 40% and 36.5% at days 5 and 7, respectively, by FGF-2 plus DEX, as compared to untreated cells (Fig. 2A). Treatment with FGF-2 plus DEX increased the proliferation of ATSCs by an average of 14.5% and 13.6% at days 5 and 7, respectively, as compared to FGF-2 treatment alone (Fig. 2A). To confirm these results, we carried out an analysis of cell number. After treatment with FGF-2 and DEX, the number of ATSCs increased by 81–103% as compared to control (Fig. 2B). These results demonstrated that combined treatment with low concentrations of FGF-2 and DEX strongly enhances the proliferation of ATSCs in vitro.

Induction of proliferation of ATSCs derived from three independent donors by FGF-2 and DEX. ATSCs were cultured with FGF-2 (1 ng/mL), DEX (10 nM), or FGF-2 plus DEX for 3, 5, and 7 days. (
Expression levels of MSC markers in ATSCs cultured with FGF-2 and DEX
To determine whether ATSCs that were expanded in the presence of FGF-2 and DEX maintained the expression of MSC cell surface markers, ATSCs were cultured with FGF-2 and DEX for 5 days, and the expression of CD29, CD44, CD90, CD105, and CD146 was examined by flow cytometry. More than 97% of cells that were expanded in the presence of FGF-2 and DEX were positive for CD29, CD44, CD90, and CD105, which was similar to untreated ATSCs (Table 1). However, CD146-positive cells were only between 4.5% and 9.3% (Table 1). This result is consistent with a previous study, demonstrating that CD146 expression was decreased during passage. 22 Therefore, ATSCs that were expanded in vitro in the presence of FGF-2 and DEX expressed MSC markers, including CD29, CD44, CD90, CD105, and CD146.
MSCs, mesenchymal stem cells; ATSCs, adipose tissue-derived mesenchymal stromal cells; FGF-2, fibroblast growth factor-2; DEX, dexamethasone.
Effect of FGF-2 and DEX on osteoblastic and adipogenic differentiation of ATSCs
To determine whether ATSCs cultured in vitro with FGF-2 and DEX maintained their multi-lineage differentiation potential, ATSCs were cultured in the absence (control cells) or presence of FGF-2 and DEX for 5 days. Expanded cells were collected, reseeded, and then cultured for an additional 14 days in osteogenic or adipogenic medium. ATSCs that were first expanded with FGF-2 and DEX were capable of differentiating into either osteoblastic (Fig. 3) or adipogenic cells (Fig. 4). Of note, ATSCs expanded in the presence of FGF-2 and DEX exhibited increased levels of osteoblastic and adipogenic differentiation as compared to control cells (Figs. 3 and 4). These results indicated that the expansion of ATSCs in the presence of FGF-2 and DEX maintains their osteogenic and adipogenic differentiation potential.

Osteoblastic differentiation of ATSCs expanded in the presence of FGF-2 and DEX. ATSCs were cultured with FGF-2 (1 ng/mL), DEX (10 nM), or FGF-2 plus DEX for 5 days. The expanded cells were reseeded and treated with osteogenic medium for an additional 14 days to induce osteoblastic differentiation. (

Adipogenic differentiation of ATSCs expanded in the presence of FGF-2 and DEX. ATSCs were cultured with FGF-2 (1 ng/mL), DEX (10 nM), or FGF-2 plus DEX for 5 days. The expanded cells were reseeded and treated with adipogenic medium for an additional 14 days. (
Tyrosine phosphorylation of Src kinase in ATSCs exposed to FGF-2 and DEX
To begin to elucidate the signaling pathways that were involved in the stimulatory effect of FGF-2 and DEX on the proliferation of ATSCs, cells were treated with FGF-2 and DEX for 1, 3, or 5 days, and the level of phosphorylation of key signaling molecules was examined by Western blot. Tyrosine phosphorylation of Src kinase was significantly increased by treatment with FGF-2 and DEX, whereas that of JNK, p38, and ERK was decreased (Fig. 5). These results suggested the potential involvement of Src kinase in the stimulation of proliferation and subsequent osteoblastic differentiation of ATSCs by FGF-2 and DEX.

Protein phosphorylation in ATSCs expanded in the presence of FGF-2 and DEX. ATSCs were treated with FGF-2 plus DEX for 1, 3, or 5 days, and the levels of phosphorylation of JNK, ERK, Src, and p38 were analyzed by Western blot. The phosphorylation of Src kinase was increased by FGF-2 and DEX.
Effect of Src kinase inhibitors on FGF-2 and DEX–induced proliferation of ATSCs
To further examine the involvement of Src or Src family kinases in the stimulation of proliferation of ATSCs, cells were cultured in the presence of FGF-2 and DEX, with or without (control cells) selective Src family kinase inhibitors (SU6656 and src kinase inhibitor I). The proliferation of ATSCs in the presence of SU6656 (Fig. 6A) and src kinase inhibitor I (Fig. 6B) was significantly decreased in a dose-dependent manner as compared to control cells. These results were consistent with the increased tyrosine phosphorylation of Src by FGF-2 and DEX, and strongly suggested that the stimulation of proliferation of ATSCs by FGF-2 and DEX involves Src kinase.
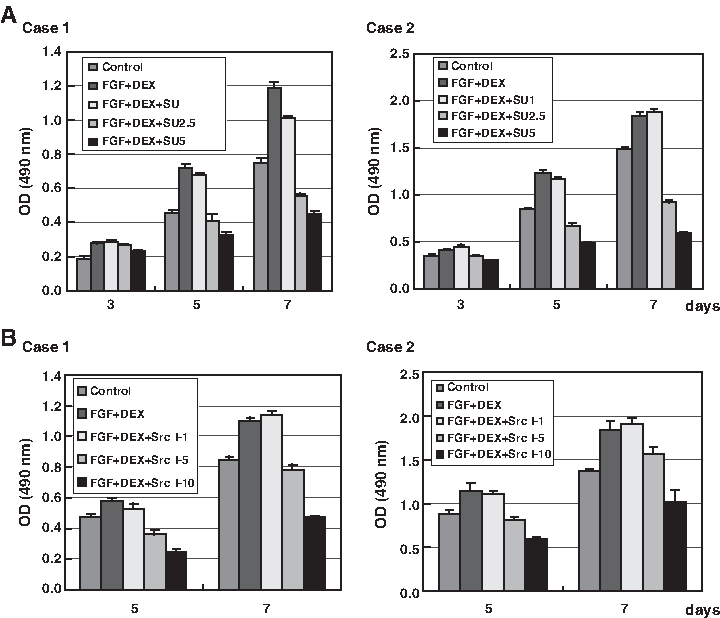
Effect of Src family kinase inhibitors (SU6656 and src kinase inhibitor I) on FGF-2 and DEX–induced proliferation of ATSCs. ATSCs were cultured with FGF-2 (1 ng/mL) plus DEX (10 nM) in the presence or absence of the indicated concentrations of SU6656 (
Effect of Src kinase inhibitors on FGF-2 and DEX–induced osteoblastic and adipogenic differentiation of ATSCs
To determine whether the inhibition of Src kinase affected osteoblastic and adipogenic differentiation, ATSCs were cultured with FGF-2 and DEX in the presence or absence (control cells) of SU6656, and then the expanded ATSCs were cultured for an additional 14 days in osteogenic or adipogenic medium. The osteoblastic differentiation of ATSCs that were pretreated with SU6656 was decreased as compared to control cells, based on AR-S staining. Similar results were observed in two independent donor-derived ATSCs (Fig. 7A). The level of adipogenic differentiation was also decreased by SU6656, as assessed by oil red O staining (Fig. 7B). These results suggested that Src kinase modulates the osteoblastic and adipogenic differentiation potential of ATSCs.
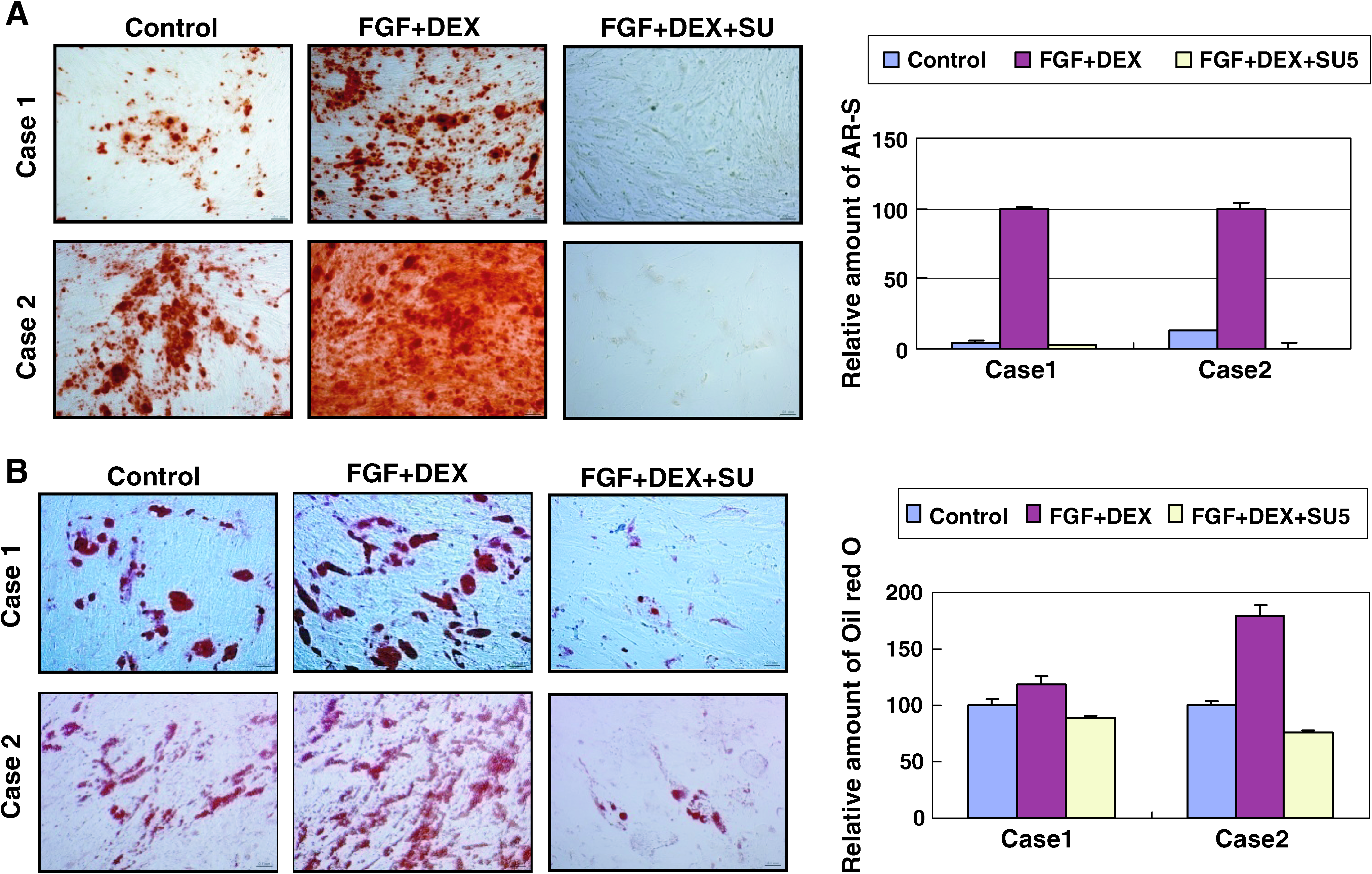
Effect of SU6656 on FGF-2 and DEX–induced osteoblastic and adipogenic differentiation of ATSCs. ATSCs were cultured with FGF-2 (1 ng/mL) plus DEX (10 nM) in the presence or absence of SU6656 (5 μM) for 5 days. Expanded cells were reseeded and treated with either osteogenic or adipogenic medium. (
Discussion
ATSCs have proven to be a useful source of cells for the treatment of defective tissues by tissue engineering. However, to be useful for clinical applications, particularly for the treatment of large lesions, it will be necessary to first expand ATSCs in vitro. Previously, we and others have shown that FGF-2 efficiently enhances the proliferation of BMSCs.9–11,23 A recent paper by Zaragosi et al. showed that FGF-2 plays a critical role in the self-renewal of ATSCs. 14 A low concentration of FGF-2 may not induce chromosomal abnormality in MSCs. 24 Therefore, FGF-2 appears to be an essential growth factor in the regulation of MSC proliferation and self-renewal. Here, we investigated ways to enhance the growth of ATSCs using a combination of FGF and steroids, and found that FGF-2 in combination with BET or DEX substantially enhances the growth of ATSCs in vitro (Fig. 1). The enhanced growth of ATSCs by FGF-2 and DEX was confirmed in ATSCs from three independent donors (Fig. 2). Previous reports by us and others using BMSCs and cementoblasts have shown that a combination of FGF-2 and DEX modulates the proliferation and biomineralization of BMSCs. However, the underlying molecular mechanism of this enhanced proliferation remains largely unknown.14,25,26
FGF-2 in combination with DEX enhanced the proliferation of ATSCs derived from three independent donors (Fig. 2). Because FGF-2–induced proliferation was enhanced by DEX, it is reasonable to hypothesize that FGF-2–induced signaling pathways are potentiated by DEX, thus resulting in enhanced ATSC proliferation. When we examined whether DEX enhanced the expression of FGF receptors (FGFRs) as a mechanism of amplification of FGF signaling, we found that the expression levels of FGFRI, II, and IV were unaffected by FGF-2 and DEX (Supplemental Fig. S1A, available online at www.liebertonline.com/ten). Although FGFRIII was induced by DEX, its expression was decreased by FGF-2 + DEX, suggesting that modulation of FGFRIII expression may not contribute to the enhanced proliferation of ATSCs. We also examined the phosphorylation levels of key signaling molecules involved in cellular proliferation, such as JNK, p38, ERK, and Src kinase. As shown in Figure 5, the levels of phosphorylation of p38, ERK, and JNK were decreased by treatment with FGF-2 and DEX, while the phosphorylation of Src kinase was substantially increased. These results imply that Src kinase plays a role in the stimulatory effect of FGF-2 and DEX on the proliferation of ATSCs. Subsequent analysis of cell proliferation in the presence of inhibitors of Src kinase confirmed that Src is involved in FGF-2 and DEX–induced stimulation of ATSC proliferation (Figs. 6 and 7).
ATSCs that were expanded in the presence of FGF-2 and DEX were able to differentiate into either osteoblasts or adipocytes. Although we did not analyze the differentiation of the expanded ATSCs along other lineages, these results suggest that FGF-2 and DEX maintain or preserve the multi-lineage potential of ATSCs that are expanded in vitro. Significantly, ATSCs that were expanded in vitro with FGF-2 and DEX exhibited a higher tendency to differentiate into both osteoblasts and adipocytes. These results were in agreement with previous reports in which a combination of FGF-2 and DEX promoted the osteogenic commitment of BMSCs.18,19,27 In the current study, we also detected the expression of ALP in cells that were treated with FGF-2 and DEX, which indicates the commitment of ATSCs to an osteogenic lineage (Supplemental Fig. S1B). Treatment with FGF-2 plus DEX also promoted the commitment of ATSCs to the adipogenic lineage. The mRNA levels of PPARγ2, which plays an important role in adipocyte differentiation, were increased in cells cultured with FGF-2 and DEX for 5 days (Supplemental Fig. S1B). Thus, FGF-2 and DEX increased the osteogenic potential, as well as the adipogenic potential of ATSCs.
The inhibition of Src kinase resulted in the suppression of osteoblastic and adipogenic differentiation in ATSCs (Figs. 6 and 7). The tyrosine phosphorylation of Src kinase increased during FGF-2 and DEX–induced proliferation of ATSCs (Fig. 5), and treatment with inhibitors of Src family kinases suppressed the proliferation of ATSCs (Fig. 6). These results suggest that the osteogenic and adipogenic differentiation of ATSCs is modulated by the proliferative capacity of the cells. In fact, when the number of cells that were incubated in osteogenic medium was increased, we observed an enhancement of differentiation of ATSCs along the osteoblastic lineage (Supplemental Fig. S2, available online at www.liebertonline.com/ten). These results suggest that the inhibition of Src kinase causes the suppression of cell growth and subsequent differentiation of ATSCs along the osteoblastic or adipocytic lineage.
In summary, we have demonstrated that treatment of ATSCs with a combination of FGF-2 and DEX is an efficient method for expanding these cells in vitro and, importantly, can preserve the osteogenic and adipogenic potential of the cells. We also demonstrated that Src kinase is involved in the stimulation of proliferation of ATSCs.
Footnotes
Acknowledgments
This work was supported by a grant from the Ministry of Health and Welfare [0405-BO01-0204-0006] and by a Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MOST); Grant Numbers: M10646020001-06N4602-00110 and No. R01-2006-000-10756-0).
Disclosure Statement
No competing financial interests exist.