
Abstract
For the regeneration of bone in tissue engineering applications, it is essential to provide cues that support neovascularization. This can be achieved by cell-based therapies using mature endothelial cells (ECs) or endothelial progenitor cells. In this context, ECs were used in various in vivo studies in combination with primary osteoblasts to enhance neovascularization of bone grafts. In a previous study, we have shown that cocultivation of human primary ECs and human primary osteoblasts (hOBs) leads to a cell contact–dependent up-regulation of alkaline phosphatase (ALP) expression in osteoblasts, indicating that cocultivated ECs may support osteogenic differentiation and osteoblastic cell functions. In the present study, we investigated this effect in more detail, revealing a time and cell number dependency of EC-mediated up-regulation of the early osteoblastic marker ALP, whereas osteocalcin, a late marker of osteogenesis, was down-regulated. The effect on ALP expression was bidirectional specific for both cell types. Functional inhibition of gap junctional communication between ECs and hOBs by 18α-glycyrrhetinic acid had only a weak suppressive effect on EC-mediated ALP up-regulation. In contrast, inhibition of p38 mitogen–activated protein kinase nearly completely prevented the EC-mediated stimulation of osteoblastic ALP expression. To investigate the molecular mechanism underlying the ALP up-regulation, we examined the effect of EC cocultivation on osteoblastic ALP promoter activity as well as mRNA stability. Cocultivation of ECs with hOBs significantly elevated the half-life of osteoblastic ALP mRNA without affecting its promoter activity. In summary, our data show that EC-mediated up-regulation of osteoblastic ALP expression is cell-type specific and is posttranscriptionally regulated via p38 mitogen–activated protein kinase–dependent mRNA turn-over.
Introduction
In previous studies, we 22 and others20,21,23 showed that cocultivation of ECs promoted a cell contact–dependent up-regulation in the expression of the osteoblastic differentiation marker alkaline phosphatase (ALP) in osteoblasts.
The present study aimed to analyze the underlying inter- and intracellular signaling mechanisms responsible for EC-mediated up-regulation of osteoblastic ALP expression. Our data indicate that this process is cell-type specific and is posttranscriptionally regulated via a p38 mitogen–activated protein kinase (MAPK)–mediated increase in ALP mRNA stability.
Materials and Methods
Cell culture
Human umbilical vein ECs (HUVECs) were purchased from Promocell (Heidelberg, Germany) and cultured in EC growth medium (ECGM) supplemented with 10% fetal calf serum (FCS) at 37°C, 5% CO2 in a humidified atmosphere. Only HUVECs from passage 2 to 5 were used for the experiments. MG-63 and Saos-2 osteosarcoma cells were cultured in Dulbecco's modified Eagle's medium, 10% FCS, 1% penicillin/streptomycin at 37°C, 5% CO2. MC3T3 cells were cultivated in minimum essential medium (MEM) Alpha Medium, 10% FCS, and 1% penicillin/streptomycin at 37°C, 5% CO2.
Human primary osteoblasts (hOBs) were isolated from fresh bone material as previously described. 24 Osteoblasts were cultured in medium 199 with Earle's salts, supplemented with 10% FCS, 1% L-glutamine, and 1% penicillin/streptomycin at 37°C, 5% CO2.
Human dermal fibroblasts were isolated from resections during plastic surgical procedures by collagenase digestion following standard protocols. Fibroblasts were cultured in Dulbecco's modified Eagle's medium containing 10% FCS and 1% penicillin/streptomycin at 37°C, 5% CO2.
Human mesenchymal stem cells (MSCs) were isolated and expanded as described before. 25 In brief, bone marrow aspirates were obtained from healthy adult donors by an iliac crest biopsy. Mononuclear cells were purified by density gradient centrifugation with Biocoll separating solution (Biochrom AG, Berlin, Germany). Subsequently, cells were filtered through 100-μm cell strainers (BD Labware, Franklin Lakes, NJ). Mononuclear cells were seeded in culture flasks at a density of 5 × 105 cells/cm2 in expansion medium (alpha-MEM, 10% FCS, 50 μg/mL gentamicin, and 5 ng/mL basic fibroblast growth factor) at 37°C, 5% CO2. The medium was changed twice weekly, washing out all nonadherent cells. Once adherent cells had grown to confluence, they were detached and reseeded at a density of approximately 2000 cells/cm2 and cultivated for two further passages.
Human primary chondrocytes were isolated from femoral heads. Femoral heads were obtained during a hip arthroplasty operation after a femoral neck fracture and kept buffered at 4°C. Within 8 h, cartilage was separated from bone, reduced to small pieces, and digested with collagenase CLS type II (Biochrom AG) at a concentration of 2 mg/mL (320 units/L). Ham's F-12 medium with 10% FCS (Invitrogen, Karlsruhe, Germany) and 1% penicillin/streptomycin (Invitrogen) was regularly used for all experiments. After an incubation period of 16 h, cells were filtered, washed, and used for experiments. All human primary cells were isolated with the informed consent of the patients according to hospital's ethics committee guidelines.
Cocultivation experiments with direct cell contact
The various cell types were cultivated in monoculture in 12-well cluster plates at 5 × 104 cells/well in ECGM with 10% FCS at 37°C, 5% CO2 for various time periods and assayed for ALP activity. For coculture experiments, hOBs were grown in coculture with HUVECs (2.5 × 104 hOBs + 2.5 × 104 HUVECs) or with human primary chondrocytes (2.5 × 104 hOBs + 2.5 × 104 chondrocytes) or with human primary fibroblasts (2.5 × 104 hOBs + 2.5 × 104 fibroblasts) in 12-well cluster plates in ECGM with 10% FCS at 37°C, 5% CO2 for various time periods. In some experiments, hOBs were replaced by human MSCs, MG-63 cells, Saos-2 cells, or MC3T3 cells and cocultivated with HUVECs at a 1:1 ratio for 48 h.
For ALP and osteocalcin (OC) mRNA expression determinations, hOBs were grown either in monoculture in six-well plates at 8 × 105 cells/well in ECGM with 10% FCS at 37°C, 5% CO2 for various time periods, or in coculture with HUVECs (4 × 105 hOBs + 4 × 105 HUVECs/well).
Gap junctional communication was inhibited by adding 18α-glycyrrhetinic acid (18α-GA) (Sigma, Deisenhofen, Germany) to the cell culture medium at the indicated final concentrations. For protein kinase inhibition experiments, Gö 6983, Raf-1 kinase inhibitor I, Src kinase inhibitor I, PD 98059, SB 203580, and JNK inhibitor II (all from Calbiochem, Darmstadt, Germany) were dissolved in dimethyl sulfoxide and used at the indicated final concentrations.
Immunomagnetic separation
To separate HUVECs and osteoblasts after cocultivation, an immunomagnetic separation system (Invitrogen Dynal AS; Invitrogen) was used. In brief, cells were detached from the culture dishes by trypsin/ethylenediaminetetraacetic acid treatment. Enzymatic digestion was stopped by addition of 2 mL phosphate-buffered saline (PBS) and 5% FCS. Thereafter, cells were centrifuged (1000 rpm, 5 min) at room temperature and washed once with PBS and 0.1% bovine serum albumin. Cells were resuspended in 1 mL of PBS and 0.1% bovine serum albumin, mixed with 25 μL of magnetic beads (Dynabeads; Invitrogen) coated with an anti-CD31 antibody, and incubated on a rotator for 30 min at 4°C. The HUVECs binding to the CD31-coated Dynabeads were separated using a magnetic particle concentrator (Invitrogen Dynal AS; Invitrogen). The unbound cells (osteoblasts) were transferred to a new Eppendorf tube, and the separation process was repeated. Thereafter, osteoblasts were centrifuged (1000 rpm, 5 min) at room temperature and used for further experiments.
Immunocytochemistry
For immunostaining, cells were fixed with 2% paraformaldehyde for 2 min, washed with Tris-buffered saline (TBS)/Tween (0.05 M Tris-HCl, 0.15 M NaCl, and 0.05% Tween-20, pH 7.4) and incubated for 30 min with blocking solution (5% goat serum; Sigma) followed by incubation with a monoclonal anti-connexin43 (Cx43) primary antibody (Millipore, Schwalbach, Germany) at a dilution of 1:300 for 16 h at 4°C. In the negative controls, the incubation step with the primary anti-Cx43 antibody was omitted; instead, the cells were incubated with blocking solution for 16 h at 4°C. After washing with TBS/Tween, a goat anti-mouse secondary antibody (ready to use horseradish peroxidase–conjugated goat anti-mouse immunoglobulin; Dako, Hamburg, Germany) was applied and incubated for another 30 min at room temperature. Thereafter, cells were washed with TBS/Tween and exposed to DAB chromogen substrate (ready to use; Dako). The images of the stained cells were captured using a light microscope and its image analyzing software (Leica, Solms, Germany).
ALP detection
ALP activity in cellular extracts was determined as described earlier. 26 In brief, cells were washed once with PBS and then lysed by three freeze–thaw cycles in 500 μL of 25 mM Tris-HCl (pH 8.5) and 0.5% Triton X-100. Samples (20 μL) were mixed with 100 μL of CSPD substrate (Applied Biosystems, Forster City, CA) and incubated at room temperature for 30 min. Light output was measured by a plate luminometer (Spectrafluor Plus; Tecan, Crailsheim, Germany) in relative luminescence units.
Quantitative real-time reverse transcriptase polymerase chain reaction
TaqMan reverse transcriptase polymerase chain reaction was carried out as previously described. 27 Total RNA was prepared using RNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. Total RNA (3 μg) was treated with 3 units of deoxyribonuclease I (Invitrogen) to digest genomic DNA contamination. Random-primed cDNA synthesis was performed using 3 μg of deoxyribonuclease I–treated total RNA and 50 units of StrataScript reverse transcriptase according to the manufacturer's instructions (Stratagene, La Jolla, CA). TaqMan PCR assays were performed in 384-well optical plates on a LightCycler (Roche, Mannheim, Germany) using Absolute QPCR ROX Mix (Abgene, Hamburg, Germany) according to the manufacturer's instructions. Oligonucleotide primers and probes for human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (GAPDH forward: 5′-TGGGCTACACTGAGCACCAG-3′; GAPDH reverse: 5′-CAGCGTCAAAGGTGGAGGAG-3′; GAPDH probe: 5′-FAM-TCTCCTCTGACTTCAACAGCGACACCC-TAMRA-3′) and human OC (OC forward: 5′-TGAGCTCAATCCGGACTGTG-3′; OC reverse: 5′-GCCGTAGAAGCGCCGATAG-3′; OC probe: 5′-FAM-CGAGTTGGCTGACCACATCGGCTT-TAMRA-3′) were designed using Primer Express (Applied Biosystems) according to company guidelines. Oligonucleotide primers and the TaqMan probe for ALP were purchased from Applied Biosystems (Cat. No.: Hs01029144_m1). The thermal cycling conditions were 95°C for 15 min followed by 50 cycles at 95°C for 15 s and at 60°C for 1 min. Data were analyzed using the relative standard curve method, with each sample being normalized to GAPDH to correct for differences in RNA quality and quantity. Results from three experiments are expressed as mean arbitrary units ± standard deviation.
mRNA stability assay
hOBs were grown either in monoculture or in coculture with HUVECs at a ratio of 1:1 for 48 h before the addition of actinomycin-D (5 μg/mL final concentration). hOBs were then isolated at the indicated time points by negative anti-CD31 immunomagnetic separation. Total RNA was then extracted from hOBs and measured by real-time reverse transcriptase polymerase chain reaction for ALP and GAPDH expression. ALP mRNA expression was normalized to GAPDH levels. Data are presented as relative to control cells, at the time of addition of actinomycin-D.
Western blot analysis
After serum starvation of cells for 24 h, 18α-GA (100 μM) or dimethyl sulfoxide (0.4%) was added to the cultures for 40 min. Thereafter, cells were stimulated by addition of FCS (final concentration 10%) or were left untreated. Cells were lysed on ice by the addition of lysis buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 0.5% NP-40, 10 μg/mL aprotinin, 5 μg/mL leupeptin, 50 μg/mL phenylmethane-sulfonylfluoride, 100 mM NaF, and 0.2 mM sodium orthovanadate). The particulate material was removed by centrifugation. The protein concentration of the supernatants was determined using the BCA protein assay kit (Thermo Scientific, Rockford, IL). About 10 μg of total protein was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to Hybond ECL nitrocellulose membranes (GE Healthcare, Munich, Germany), and probed with a polyclonal anti-active MAPK antibody (Promega, Heidelberg, Germany) for 2 h at room temperature. Detection was performed after incubation of the membrane with an anti-rabbit horseradish peroxidase–conjugated secondary antibody (Dianova, Hamburg, Germany) for 1 h at room temperature using an ECL Western blotting detection system (GE Healthcare), as described by the manufacturer.
Phospho-p38 MAPK enzyme-linked immunosorbent assay
hOBs were grown in monoculture or in coculture with HUVECs at a 1:1 ratio for the indicated time periods before hOBs were isolated by negative immunomagnetic cell separation. hOBs were washed twice with PBS and then lysed in lysis buffer (R&D Systems, Wiesbaden, Germany). Lysates were vortexed briefly and allowed to sit on ice for 15 min before the particulate material was removed by centrifugation at 2000 × g for 5 min. The protein concentration of the supernatants was determined using the BCA protein assay kit (Thermo Scientific). About 100 μL of the cell lysates was used in a commercial phospho-p38α enzyme-linked immunosorbent assay following the instructions of the manufacturer (R&D Systems). Phospho-p38α content is expressed in pg per mg of total protein.
Transient transfections and ALP promoter studies
Transient transfections of hOBs were carried out using the Nucleofector system (Amaxa, Köln, Germany). In brief, 8 × 105 cells were resuspended in 100 μL of nucleofector solution. About 2 μg of ALP promoter/luciferase plasmid, containing the −3850/−122 (relative to the first nucleotide of the adenosine thymidine guanosine (ATG) initiation codon, which was defined as +1) human ALP promoter linked to a luciferase reporter gene 28 was added, and cells were transfected using the nucleofector U-23 program. Cells from individual transfections were pooled and seeded immediately in prewarmed medium 199 with Earle's salts, supplemented with 10% FCS, 1% L-glutamine, and 1% penicillin/streptomycin in T75 cell culture flasks and incubated overnight at 37°C, 5% CO2. Thereafter, transfected hOBs were trypsinized, pooled, and reseeded in 12-well cluster plates, either in hOB monoculture (1 × 105 hOBs/well) or in coculture with HUVECs (0.5 × 105 hOBs + 0.5 × 105 HUVECs) in ECGM with 10% FCS. Cells were then incubated at 37°C, 5% CO2 for various time periods. Cells extracts were prepared using reporter lysis buffer (Promega). Luciferase activities in cell extracts were assayed using a microplate luminometer (MicroLumatPlus; Berthold, Bad Wildbad, Germany). Luciferase activities were normalized to hOB cell numbers and expressed as relative luciferase activity.
Results
Effect of HUVEC cocultivation on osteoblastic ALP and OC expression
hOBs were grown either in monoculture or in coculture with HUVECs at a 1:1 ratio for various time periods. Thereafter, hOBs were isolated by negative immunomagnetic cell separation and analyzed for osteoblastic ALP or OC mRNA expression and ALP activity. As shown in Figure 1, HUVEC cocultivation leads to a time-dependent increase in osteoblastic ALP expression on mRNA level (Fig. 1A) as well as on the level of ALP activity (Fig. 1B). The stimulatory effect of HUVECs on osteoblastic ALP mRNA expression could first be detected after a 24 h cocultivation period and was lasting for at least 72 h. On the level of ALP activity, a significant effect of HUVEC cocultivation was seen at the 48 h time point and was further increased by extension of the cocultivation period. Expression of OC, another important osteoblastic marker that is mainly expressed at later stages of osteogenesis,29,30 was also investigated in these cocultures (Fig. 1C). In this case, a significant time-dependent down-regulation of osteoblastic OC mRNA expression could be detected upon cocultivation with HUVECs, indicating that ALP and OC are reciprocally regulated by cocultivated HUVECs.
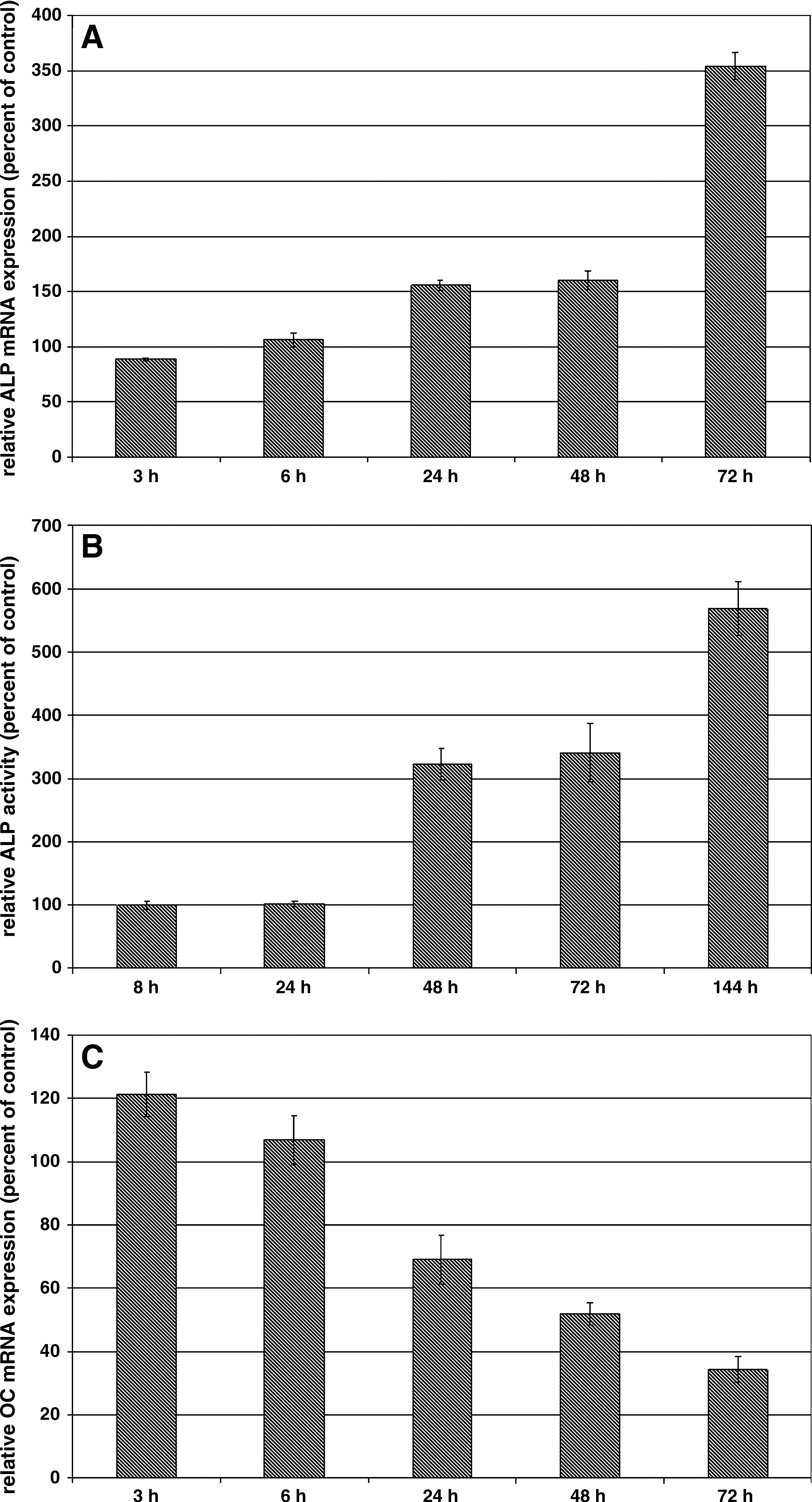
Effect of HUVEC cocultivation on osteoblastic ALP mRNA expression (
We next investigated the effect of HUVEC seeding density on osteoblastic ALP expression (Fig. 2). For this purpose, 4 × 104 hOBs were either cultured alone or cocultured with increasing amounts of HUVECs for 48 h. A significant inductive effect of HUVEC cocultivation on osteoblastic ALP activity could already be seen at a hOB/HUVEC seeding ratio of 1:0.25 (addition of 1 × 104 HUVECs), and this effect was further elevated with increasing numbers of coseeded HUVECs.

Effect of HUVEC cell numbers on osteoblastic ALP activity. hOBs were seeded at 40,000 cells/well in 12-well cluster plates and cultured alone (hOB control) or cocultured with the indicated amounts of HUVECs for 48 h. Thereafter, cell lysates were assayed for ALP activity. ALP activity is expressed in relative luminescence units (RLU). Shown are means ± SD from triplicate determinations.
Specificity of hOB/HUVEC interaction
To investigate the specificity of the HUVEC–hOB interaction in terms of osteoblastic ALP expression, hOBs were replaced by MSCs, Saos-2, MC3T3, or MG-63 cells in the cocultivation assays (Fig. 3A). A significant increase in ALP activity by cocultivated HUVECs could be detected in hOBs as well as in human MSCs, which can be considered as osteoblastic precursor cells. Interestingly, immortalized human osteoblastic cell lines such as Saos-2 and MG-63 or immortalized murine preosteoblasts such as MC3T3 were not responsive toward HUVEC cocultivation. Likewise, an up-regulation of osteoblastic ALP expression could only be seen after cocultivation with HUVEC, but not in cocultures with human primary chondrocytes or fibroblasts (Fig. 3B). These results indicate that there exists bidirectional specificity in the interaction between HUVECs and hOBs or preosteoblastic MSCs.
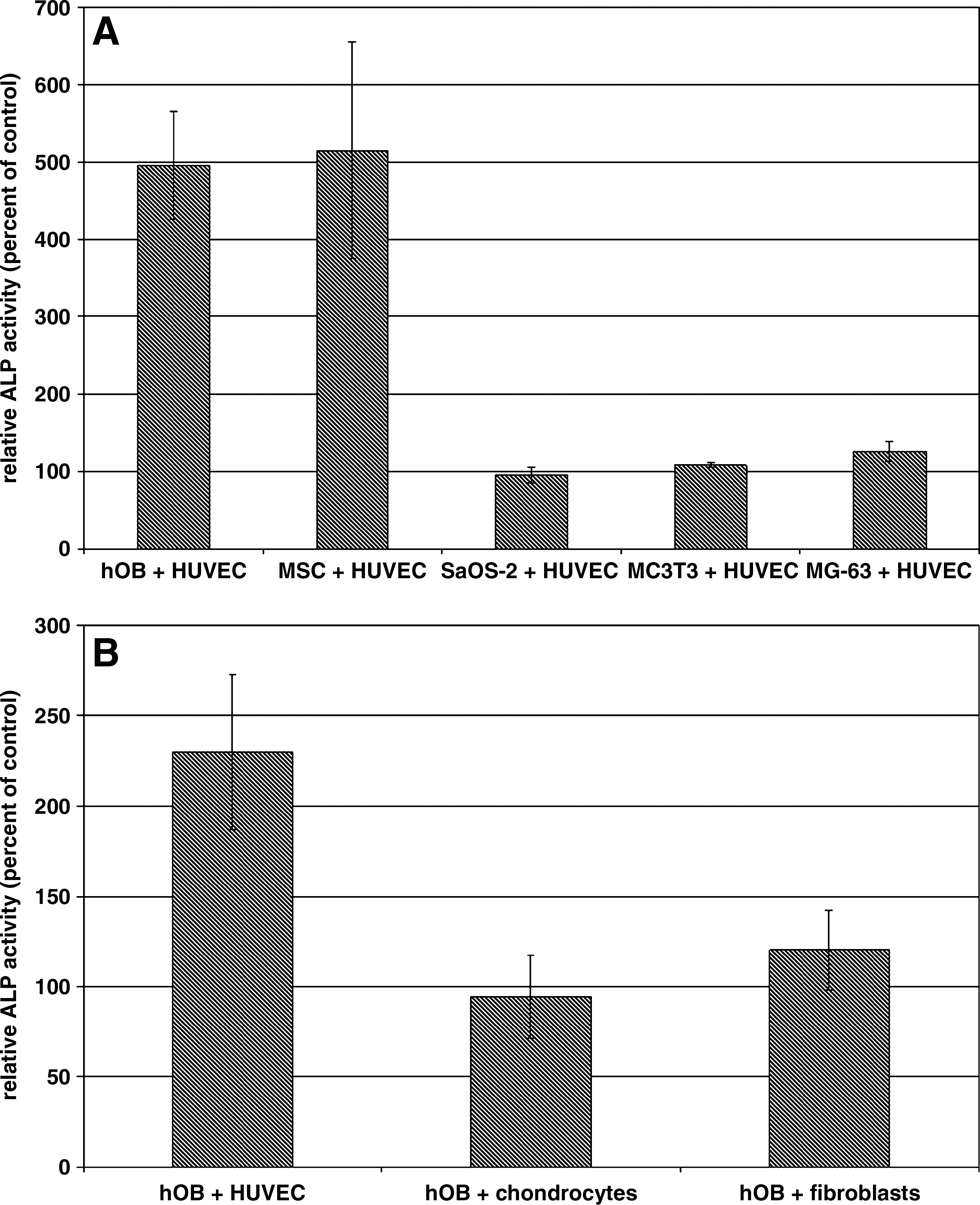
Specificity of hOB–HUVEC interaction. (
Inhibition experiments
We investigated the gap junctional communication between HUVECs and hOBs, because it was reported previously that HUVECs exert their effect on osteoblastic ALP expression only when both cell types were grown in direct cell contact.20–23 In osteoblasts, Cx43 is the predominant gap junctional protein that plays an important role in osteoblastic differentiation.31,32 Cx43 is also expressed in ECs.20,33 To investigate whether Cx43 is expressed in the cell populations used in our cocultivation experiments, we performed immunocytochemical stainings of Cx43 on hOBs and HUVECs grown in monocultures (Fig. 4A). Using a monoclonal anti-Cx43 antibody, Cx43 expression could be detected in hOB as well as in HUVEC cultures. The staining was specific for Cx43, since no cellular staining could be detected in negative controls where cells have been incubated in the absence of the primary antibody. From this experiment it became clear that both cell types synthesize the gap junctional protein Cx43. To investigate the involvement of gap junctions in the intercellular communication of hOBs and HUVECs, we performed functional gap junction inhibition experiments using 18α-GA, a specific inhibitor of gap junctions.34,35 As shown in Figure 4B, 18α-GA had only a marginal inhibitory effect on HUVEC-induced osteoblastic ALP expression, which did not follow a classical dose–response relationship; 18α-GA at a 10 μM concentration resulted in a 20.4 ± 16.2% inhibition of HUVEC-mediated induction of osteoblastic ALP expression. At the highest concentration of 100 μM, 18α-GA caused only a 4.6 ± 10.9% inhibition. From these experiments we conclude that heterotypic gap junctional communication between hOBs and HUVECs does not play an important role in HUVEC-mediated regulation of osteoblastic ALP expression. Since we have not seen a significant effect of 18α-GA on HUVEC-mediated induction of osteoblastic ALP expression, we investigated the effect of this gap junction inhibitor on serum-induced extracellular signal–regulated kinase (ERK) phosphorylation in hOBs. It was previously demonstrated that gap junctional communication in ROS17/2.8 osteosarcoma cells plays an important role in ERK1/2 phosphorylation and that 18α-GA treatment resulted in an attenuation in the activation of the ERK response to serum stimulation. 36 As shown in Figure 4C, FCS induced a time-dependent increase in ERK1/2 phosphorylation in hOBs. This effect was strongly suppressed when hOBs were grown in the presence of 18α-GA. This result confirms not only that hOBs respond to 18α-GA but also that this gap junctional inhibitor is effective in inhibiting gap junctional signaling in hOBs.

Expression of the gap junctional protein Cx43 and effect of functional inhibition of gap junctional communication between HUVECs and hOBs by 18α-GA. (
To investigate the intracellular signaling pathways underlying the ALP up-regulation, we performed inhibition experiments using various specific protein kinase inhibitors (Fig. 5A).
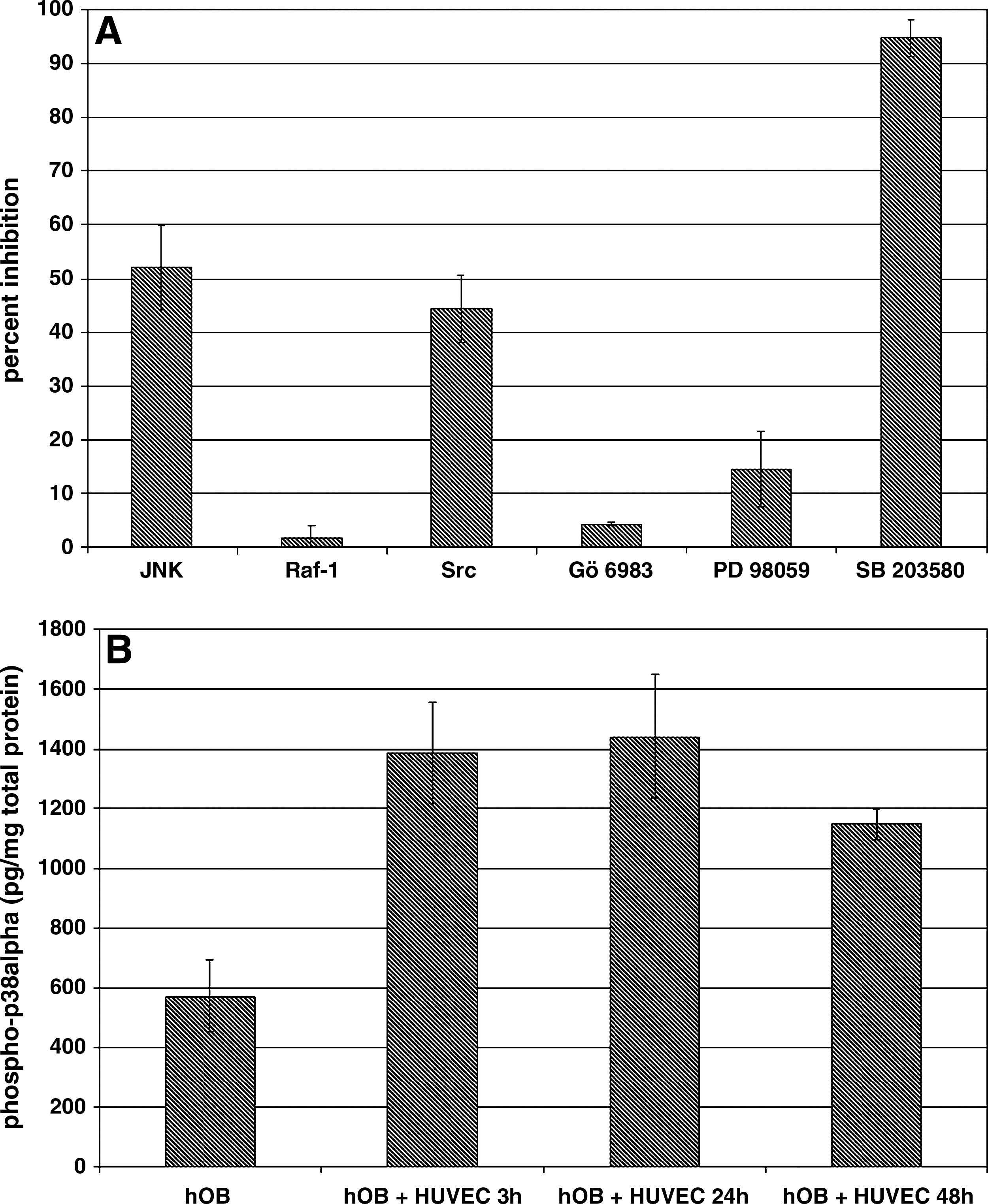
Effect of protein kinase inhibitors on HUVEC-mediated up-regulation of osteoblastic ALP activity and effect of HUVEC cocultivation on osteoblastic p38 mitogen–activated protein kinase (MAPK) phosphorylation. (
Treatment with JNK or Src kinase inhibitors resulted in a partial inhibition of HUVEC-mediated induction of osteoblastic ALP expression with 52 ± 7.8% inhibition for the JNK inhibitor and 44.4 ± 6.2% inhibition for the Src kinase inhibitor. In contrast, treatment with the p38 MAPK inhibitor SB 203580 resulted in a 94.6 ± 3.4% inhibition of HUVEC-mediated induction of osteoblastic ALP expression. Inhibition of the protein kinases ERK1/2 (PD 98059), c-raf (Raf-1), or PKC (Gö 6983) had only marginal effects on osteoblastic ALP expression. These data suggest that p38 MAPK is critically involved in HUVEC-mediated up-regulation of osteoblastic ALP expression, a notion that is further strengthened by the observation that HUVEC cocultivation leads to a time-dependent increase in p38 MAPK phosphorylation of hOBs (Fig. 5B).
Effect of HUVEC cocultivation on osteoblastic ALP promoter activity and mRNA stability
To further investigate the molecular mechanism underlying the ALP up-regulation, we examined the effect of HUVEC cocultivation on osteoblastic ALP promoter activity as well as mRNA stability (Fig. 6). hOBs were transiently transfected with the human −3850/−122 ALP promoter/luciferase plasmid and grown either in monoculture or in coculture with HUVECs for the indicated time periods. As shown in Figure 6A, HUVEC cocultivation did not affect ALP promoter activity. This result clearly indicates that the observed effect of HUVEC-mediated up-regulation in osteoblastic ALP mRNA and activity cannot be mediated by a transcriptional mechanism. Therefore, we investigated whether HUVEC cocultivation affects osteoblastic ALP mRNA stability. HUVEC cocultivation resulted in a significant increase in osteoblastic ALP mRNA stability (Fig. 6B), the half-life increasing from 6 h in hOB monocultures to 19.9 h in hOB/HUVEC cocultures.
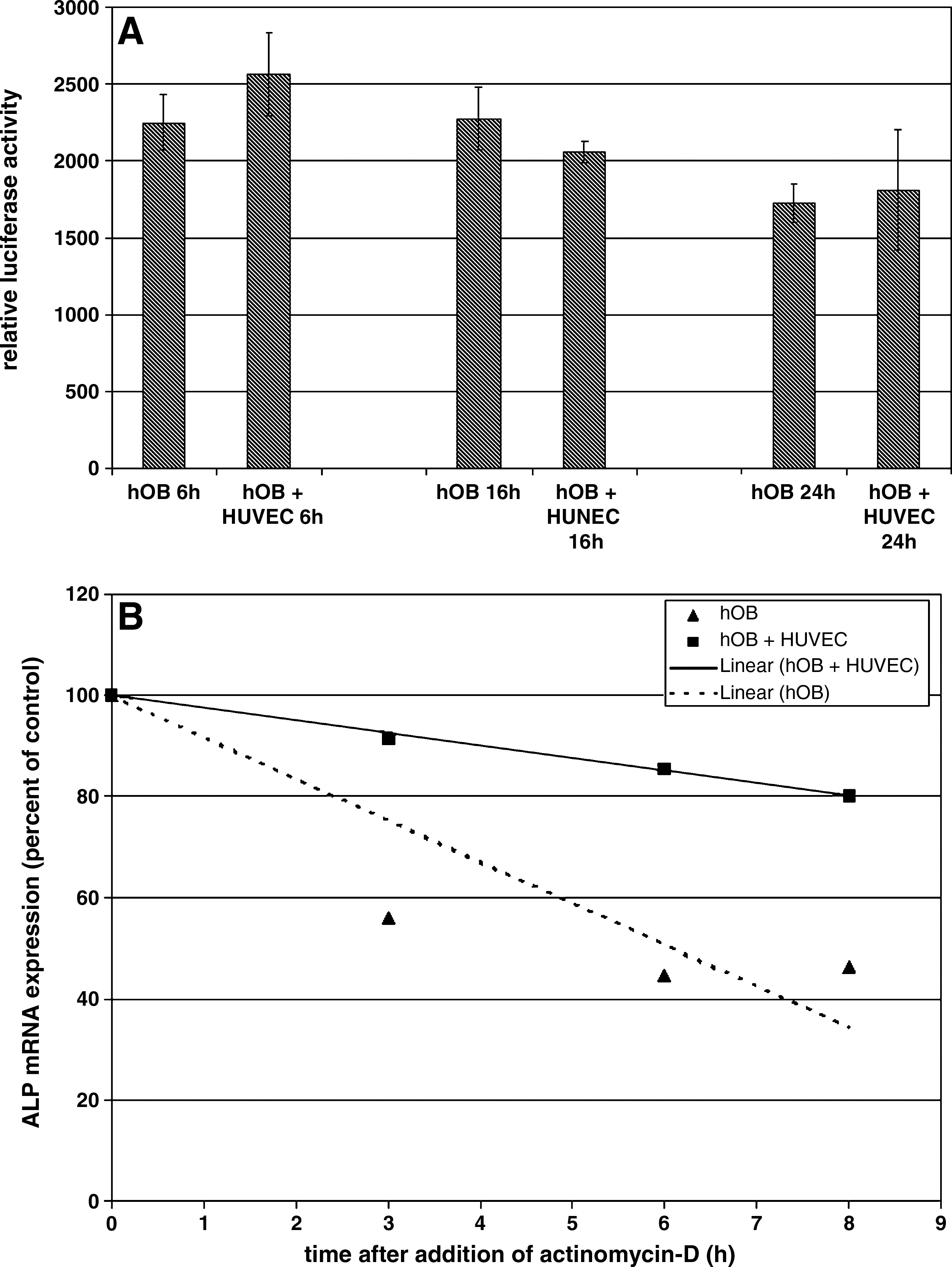
Effect of HUVEC cocultivation on osteoblastic ALP promoter activity (
In summary, our data indicate that ALP is up-regulated in hOBs by cocultivation with human primary ECs in a time-dependent and cell density–dependent manner. This effect is bidirectional specific and is posttranscriptionally regulated via a p38 MAPK–mediated increase in ALP mRNA stability.
Discussion
From the literature it is known that there exists an interactive relationship between ECs and osteoblasts during osteogenesis and angiogenesis.37,38 During endochondral ossification, hypertrophic cartilage is replaced by bone, and this process is highly dependent on neovascularization. Likewise, neoangiogenesis is a prerequisite for bone regeneration in the process of fracture repair. 5 It is well established that ECs and bone-forming osteoblasts can communicate via soluble paracrine acting mediators such as platelet-derived growth factor, vascular endothelial growth factor, or bone morphogenetic protein-2.39,40 However, direct heterotypic cell to cell interactions between ECs and osteoblasts may also influence osteoblastic differentiation and osteoblastic cell functions. For example our group 22 and others20,21 have shown that expression of the osteoblastic marker gene ALP is increased in primary osteoblasts or osteoprogenitor cells when these cells are cocultured in direct contact with ECs.
The present study aimed at the identification of the underlying molecular mechanisms of osteoblastic ALP regulation. In accordance with a previous report, 20 we have seen a time-dependent and cell density–dependent effect of HUVEC cocultivation on osteoblastic ALP expression. On mRNA level, the stimulating effect of HUVEC cocultivation could first be seen at the 24 h time point, whereas on the level of ALP activity this effect was somewhat delayed and could first be detected after 48 h of cocultivation. Concerning the seeding cell density, a hOB/HUVEC seeding ratio of 1:0.25 already provoked a robust increase in osteoblastic ALP expression, which was further elevated by increasing HUVEC cell numbers.
Our experiments revealed a remarkable bidirectional cell specificity in terms of osteoblastic ALP regulation. For example, an increase in osteoblastic ALP expression could only be observed when hOBs were cocultivated with primary ECs but not when cocultured with human primary chondrocytes or fibroblasts, cell types that are located in proximity to osteoblasts in the natural bone environment. Likewise, upon cocultivation with HUVECs, ALP activity was only increased in primary osteoblasts and MSCs, but not in immortalized osteoblastic cell lines such as Saos-2, MG-63, or MC3T3.
This result is of particular interest because it indicates that immortalized cell lines do not necessarily behave as their physiological counterparts, a notion that is supported by numerous other examples from the literature.41–43
Concerning the intercellular communication between HUVECs and preosteoblastic cells, Villars and colleagues 20 have proposed a mechanism based on gap junctional communication. Using 18α-GA, a highly specific inhibitor for gap junctions, they have reported on a 76% to 82% inhibition in HUVEC-mediated up-regulation of osteoblastic ALP activity. However, in our hands this inhibitor was rather ineffective in inhibiting the HUVEC-mediated increase in osteoblastic ALP activity. For example, as shown in Figure 4B, at the 100 μM concentration, 18α-GA caused only a 4.6 ± 10.9% inhibition. The reason for this discrepancy between our data and the data published by Villars et al. 20 is not clear so far, but it is tempting to speculate that the conflicting results may be attributed to differences in the isolation procedure of the primary cells. Whereas the isolation procedure of Villars and colleagues favors the isolation of osteoprogenitor cells from bone marrow, our isolation protocol aimed at the isolation of differentiated primary osteoblasts from bone chips. However, by immunocytochemical studies, we have been able to show that both primary cell types used in our cocultivation experiments express the gap junctional protein Cx43. Therefore, it seems reasonable to speculate that there exists gap junctional coupling between hOBs and HUVECs via Cx43, a notion that is strongly supported by the data published by Villars and coworkers. 20 Moreover, by investigating the effect of 18α-GA treatment on serum-stimulated ERK1/2 phosphorylation in hOBs, we have seen that 18α-GA severely inhibited this cellular response. This result is of particular interest because it confirms that 18α-GA is biologically effective in inhibiting gap junctional signaling in hOBs without significantly inhibiting HUVEC-mediated up-regulation of osteoblastic ALP activity. Thus, the contribution of gap junctional communication in the modulation of osteoblastic ALP activity by HUVECs is still controversial and needs further clarification.
To determine the intracellular molecular mechanism by which cocultivated HUVECs increase osteoblastic ALP expression, we examined transcriptional activity of the ALP promoter as well as ALP mRNA stability. Primary osteoblasts were transfected with an ALP promoter/luciferase construct followed by cocultivation with HUVECs, and promoter activity was compared to transfected osteoblasts grown in monoculture. Cocultivation of HUVECs did not result in a significant increase in osteoblastic ALP promoter activity, indicating that the increase in ALP mRNA was not due to transcriptional activation of the ALP gene.
The stability of the ALP transcript was investigated using the transcriptional inhibitor actinomycin-D. For this purpose, osteoblasts were grown either in monoculture or in coculture with ECs for 48 h followed by the addition of actinomycin-D for various time periods. HUVEC cocultivation significantly modulated osteoblastic ALP mRNA stability by increasing the half-life from 6 h in hOB monocultures to 19.9 h in hOB/HUVEC cocultures. This result strongly indicates that the observed effect of cocultivated HUVECs on osteoblastic ALP expression is mediated by modulation of mRNA stability. From the literature it is known that ALP expression can be regulated by modulation of mRNA stability. For example, 1,25-dihyrdoxyvitamin D3 stimulates ALP expression in human osteosarcoma cells through mechanisms involving both transcriptional activation of the ALP gene, as well as stabilization of ALP mRNA. 44
In this context, it is interesting to note that p38 MAPK was identified to be an important component of intracellular signaling pathways involved in modulation of mRNA stability.45–47 In our experiments, treatment with the p38 MAPK inhibitor SB 203580 inhibited the up-regulation of osteoblastic ALP expression by cocultivated HUVECs by more than 90%. Moreover, we have seen that cocultivation with HUVECs leads to a significant time-dependent and long-lasting phosphorylation and hence activation of osteoblastic p38 MAPK. These results strongly indicate that p38 MAPK is critically involved in the regulation of osteoblastic ALP expression. However, we have also observed partial inhibition of HUVEC-mediated induction of osteoblastic ALP expression by treatment with JNK or Src inhibitors, suggesting that these protein kinases may also be involved to a certain extend in the regulation of ALP expression. In contrast, using inhibitors against protein kinase C, Raf-1, and ERK1/2, we have been able to show that the classical ERK pathway plays no role in the intracellular propagation of the HUVEC effect on osteoblastic ALP expression.
In the present study, we have focused our interest on the effect of HUVEC cocultivation on osteoblastic ALP expression, which can be considered as an early osteoblastic differentiation marker. However, there exist various other important osteogenic markers such as OC, which belongs to the group of late markers.29,30 Interestingly, an enhancing effect of HUVEC cocultivation was only detectable for ALP expression, whereas in contrast OC expression was diminished by cocultivated HUVECs, confirming results previously published by Guillotin and coworkers. 48 The reciprocal regulation of these two important osteogenic markers suggests that the formation of heterotypic cell contacts between ECs and osteoblasts or osteoprogenitor cells alone does not necessarily support osteoblastic differentiation in general.
Footnotes
Acknowledgments
We thank Beate vom Hoevel for excellent technical assistance. This work was supported by funding through the Deutsche Forschungsgemeinschaft (FI 790/2–1).
Disclosure Statement
No competing financial interests exist.