
Abstract
We have developed a simple and rapid method for isolation of human umbilical cord matrix stem cells (hUCMS). The umbilical cord contains a virtual inexhaustible source of adult stem cells. We have substantially modified, simplified, and improved previously reported hUCMS isolation procedures in terms of either used enzyme type, or digestion time, and substantially enhanced the final product yield and purity. The isolated hUCMS were positive for CD90, CD117, and SCF, and negative for CD31 and CD45 surface markers. mRNA and related proteins (i.e., Sox2, Oct4a, Nanog, ABCG2, and c-Myc) that coincide with an uncommitted cell status also were detected. hUCMS express genes and proteins for CD90 and Nestin that are associated with mesenchymal stem cells, as well as other genes that specifically relate to different embryonic germ layers, namely, Vimentin, Sox7, Sox17, FoxA2, E-cadherin, and N-cadherin. hUCMS showed multilineage cell differentiation potential into adipogenic, osteogenic, and neural cell phenotypes, under the influence of lineage-specific, differentiation culture media. Moreover, the basal expression of endocrine cell markers makes these cells seemingly suitable for endocrine cell phenotype differentiation. Noteworthy, Activin A induced hUCMS to acquire definitive endoderm cell markers.
Introduction
In 2003 Mitchell et al. had reported on a method for isolation of multipotent stem cells from the porcine umbilical cord Wharton jelly. 6 In particular, upon removal of the vascular network, the remainder tissue was minced into small fragments that were culture maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 20% fetal bovine serum (FBS) throughout 5–7 days, so as to promote spontaneous cell migration. Mixed cell populations, including elongated, mesenchymal-like, star-like, and round-shaped cells were obtained. The great potential of these cells was confirmed by experiments demonstrating their engraftment in the rat brain. 17 In 2004, hUCMS were first isolated by Chen and coworkers from the human umbilical cord according to a method where, upon removal of the vascular network, the Wharton jelly was scraped off, washed, and thereby exposed to enzymatic digestion with collagenase for 16 h at 37°C. 1 A second digestion, with 2.5% trypsin for 30 min at 37°C ensued. Finally, the obtained cells were culture maintained in DMEM-high glucose (HG) supplemented with 10% FBS, resulting in a fibroblast-like, homogeneous cell population. Weiss et al., 2 in 2006, introduced two major changes: (1) The open cord fragments were digested for only 1 h at 37°C using an enzymatic blend comprised of hyaluronidase, collagenase, and trypsin. The digest underwent mechanical manipulation to squeeze the cells out of the tissue. (2) A low serum culture medium was added, enriched in several other factors, including epidermal growth factor (EGF), platelet-derived growth factor BB (PDGF-BB), and dexamethasone. By this procedure, the authors obtained, in 5–7 days, a heterogeneous primary cell population, with mesenchymal-like cells associated with short and long processes as well as small round cells with a high nuclear/cytoplasm ratio. This protocol 2 was subsequently modified in 2009, 18 with minor changes, and it was exclusively applied to human umbilical cord.
Molecular bases of hUCMS immunosoppressive effects remain unknown, although some secretory products, such as prostaglandin E2 and transforming growth factor-β, have been deemed to play an important role.19,20
Despite the fact that hUCMS might represent an interesting and possibly useful cell population,9,21 in recent years no innovative protocols, aside of mechanical tissue disruption, or long-term enzymatic digestion has been reported thus far, with respect to their separation and purification.21,22–27
Aim of the present work was to establish a novel method for separation of hUCMS, with care being taken to cut off time-consuming steps, while improving quality and number of the isolated cells. hUCMS differentiation properties, toward multiple cell lineages, were investigated.
Reaserch Design and Methods
hUCMS procurement, isolation, and culture maintenance
Under official consent of the Hospital Board and patient's own informed consent, human umbilical cords, retrieved from caesarean deliveries, at the end of gestation were sent us from the Department of Obstetrics and Gynaecology of our School. Upon retrieval, the cord was soaked in phosphate-buffered saline (PBS) supplemented with antibiotics and transported to our laboratory on ice. Only the retrieved human cords associated with an overall ischemia time lower than 6 h were processed. Upon careful washing in PBS plus antibiotics, thereafter in Hanks' Balanced Salt Solution supplemented with 0.2 mg/mL Amphotericin B and Iodopovidone, a cord vein was cannulated to inject saline plus heparin (100 U/mL) to avoid clotting. The cord was then cut into about 10-cm pieces that were tied up at both ends by sutures. A digestion solution, made of 77 mM NaCl, 0.1 mg/mL bovine serum albumin (BSA; Biochrom; Biopsa), 1.5 mg/mL Hyaluronidase (Sigma), 0.5 mg/mL Liberase Purified Enzyme Blend (Liberase™ HI; Roche), in 0.02 M phosphate buffer at pH 7, containing 77 mM NaCl and 0.01% BSA, upon pre-warming at 37°C, was injected by a syringe directly into the Wharton Jelly. Usually, 20 mL of the enzymatic solution was injected into each cord's segment, in vertical position downsteram the suture. The injection induced full cord distension with no enzymatic leakage. The segments, upon injection, were then placed in 250 mL glass bottle, and soaked in 40 mL of the digestion solution in a shaking bath at 37°C for 30 min. At the end, the cords were jelly-free, whereas a few partially digested matrix residual tissue chunks were floating on the enzymatic solution. At the end of the digestion process, only the vascular and connective tissue skeleton was left. The preparation was then placed on ice, whereas 20 mL FBS (UE approved; Biochrom) was added. We used FBS to block the digestion and the serum was rapidly removed by centrifugation. Umbilical arteries and veins were easily removed, whereas the eventual partially digested jelly residues were easily scraped off the amnion. The tissue digest was spun at 1500 rpm for 5 min at 4°C, thereby resuspended in DMEM with antibiotics. To remove residual red blood cells (RBCs), the suspension was centrifuged on Lymphoprep™ gradients at 1850 rpm for 20 min at 4°C. hUCMS and small undigested matrix remnants floated at the Lymphoprep™/culture medium interface, with the RBC's settling down to the tube bottom. hUCMS and hyaluronic acid (HA) fragments were easily retrieved, and after washing in DMEM were plated. To allow free cells as well as matrix residues adhesion to culture flasks, the flask bottoms were pre-treated as follows: 5 mL HA in 0.3 M phosphate buffer, pH 5.35, at 1 mg/mL were placed into each flask at 37°C for 16 h. HA excess was then removed, whereas the flasks were rinsed with PBS, and stored at 4°C until use. The cells were seeded at a concentration of 6000–8000/cm2 per flask, in low serum culture medium, according to Weiss et al. 2 Upon 24h of the isolation, EGF (1 ng/mL; Peprotech, LiStarFish) and PDGF-BB (10 ng/mL; Peprotech) were added to the culture flasks. The cells were maintained at 37°C in humidified 95% air. Cell expansion throughout 80% confluence was achieved by treatment with 0.05% trypsin/EDTA (Gibco, Invitrogen) for 3 min at 37°C. Serum was subsequently supplemented to block trypsin activity.
Transcriptional expression analysis by reverse transcriptase–polymerase chain reaction and quantitative polymerase chain reaction
Total cellular RNA was extracted from the cultured cells using a commercial kit (SV Total RNA Isolation System; Promega) according to the manufacturer's instructions. After assaying RNA by agarose gel electrophoresis, cDNA was synthesized from 0.5–1 μg total RNA using an ImProm-II Reverse Transcriptase kit (Promega). The obtained cDNA was used as a template in a polymerase chain reaction (PCR) reaction, containing 200 nM of the primer pairs for amplification of the indicated gene product (see Supplementary Table S1. Supplementary Data available online at www.liebertonline.com/tea). Reverse transcriptase–PCR (RT-PCR) primers were designed using sequences from GenBank (www.ncbi.nlm.nih.gov/Genbank).
Quantitative PCR (qPCR) amplifications were performed using the Brilliant II SYBR Green qPCR Master Mix as directed by the manufacturer (Stratagene, M-Medical). The amplification conditions were optimized for the MxPro 3000 STRATAGENE and assays were run in triplicate under the following conditions: Taq polymerase activation, 95°C for 10 min followed by 40–45 cycles at 95°C for 30 s, at the indicated temperature of annealing for 45 s (see Supplementary Table S1 and Supplementary Data) and at 72°C for 45 s. PCR products were demonstrated to be a single PCR product by melting curve and electrophoresis analysis.
Immunocytochemistry
Immunocytochemistry was performed on cord 9 μm cryostat sections after fixation in 3.7% paraformaldehyde (PFA) overnight and on cells seeded on glass slides (diameter 12 mm), treated with 3.7% PFA for 10 min. In both instances, permeabilization (when necessary) with 0.01% triton x-100 in Dulbecco's(D)-PBS for 15 min and block with 1% BSA in D-PBS for 1 h were performed. Primary antibody was added overnight; thereafter, a secondary antibody was incubated for 1 h (see Supplementary Tables S2 and S3). Cell nuclei were counterstained with Hoechst 33342 (1:200; Sigma) and mounted with Mowiol®4-88 (Calbiochem), prepared according to the manufacturer's instructions, and 0.6% 1,4-diazabicyclo(2.2.2)octane (DABCO; Sigma), as anti-fading agent. Images were collected on Axio Observer.A1 fluorescence microscope, equipped with an AxioVision software driven camera.
Total protein extraction
To extract proteins, the cell pellet was resuspended in RIPA lysis buffer: 10 mM Tris base pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.5% Igepal, 10 μL/mL phosphatase inhibitor cocktail (Sigma), and 10 μL/mL protease inhibitor cocktail (Sigma), and maintained for 20 min on ice. The suspension was then centrifuged for 15 min at 14,000 rpm at 4°C, with the supernatant thereafter used for western blotting, upon protein concentration assay. Proteins were assayed by the Bradford method (Bio-Rad Laboratories) referring to a standard BSA curve.
Western blotting
Protein samples (40 μg) were analyzed on 10% or 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membrane (BioRad Laboratories). The employed antibodies are listed in the Supplementary Data section of this article (Supplementary Tables S4 and S5). Immuno-detection was performed with Immun-Star HRP Chemiluminescent kit (Bio-Rad Laboratories) following vendor's recommendations.
Flow cytometry
Flow cytometry was performed on suspended pre-fixed cells (4% buffered formaldehyde, 20 min dark room temperature) by incubation overnight at +4°C with the following set of Abs: mouse anti-CD90 (1:50; Chemicon), mouse anti-CD117 (1:100; Santa Cruz Biotech), and mouse anti-SCF (1:50; Santa Cruz Biotech). Secondary Ab was FITC-conjugated goat anti-mouse IgG (1:200; Molecular Probes). Moreover, the following FITC-conjugated primary Abs were used: anti-CD31 and anti-CD45. Cells were examined by a flow cytometer (Coulter Corporation), equipped with an argon laser, excitation at 488 nm: green fluorescence was detected using a 525 nm band pass filter. At least 5000 cells were counted per condition.
Adipogenic differentiation
Adipogenic differentiation was induced in the confluent cultured hUCMS, at I through IV passage, by treatment with 0.5 mM 1-methyl-3-isobutylxanthine, 10−6M dexamethasone, 10 μg/mL insulin, and 200 μM indomethacin in DMEM in high glucose (Invitrogen), with 10% FBS. 14 At 3 weeks of culture, confluent cell monolayers (CM) incubated in 24-well plates were fixed with 10% buffered formaldehyde, and treated with fresh Oil Red O for 10 min at room temperature. Lipid droplets were detected and photographed. At the same time, total RNA was extracted from confluent hUCMS, cultured, and treated in 75 cm2 flask. After retro-transcription reaction, cDNA was assayed by qPCR for peroxisome proliferator-activated receptor γ (Pparγ) and fatty acid-binding protein 4 (Fabp4) gene expression.
Osteogenic differentiation
Osteogenic differentiation was induced by incubating the hUCMS, at I through IV passages, with 10−8 dexamethasone, 0.2 mM ascorbate phosphate, and 10 mM β-glycerolphosphate in α-MEM (Invitrogen) and in the presence of 10% FBS. 4 After 3 weeks, confluent hUCMS cultured and treated in 24-well plates were fixed with 10% buffered formaldehyde, and incubated with freshly made 2% Alizarine Red stain, pH 4.2, for 3 min; calcium deposition was observed and photographed. Total RNA was extracted from confluent hUCMS, cultured, and treated in 75 cm2 flasks, retro-transcripted, and assayed by qPCR for osteopontin (a protein located in the bone matrix) gene expression.
Neural cell differentiation
Neural differentiation was pre-induced by overnight treatment with basic fibroblast growth factor (Peprotech, LiStarFish) (10 ng/mL) in DMEM (Biospa) and 20% FBS, followed by 5 h induction with 2% DMSO and 200 μM butylated hydroxyanisole in DMEM plus 2% FBS. 28 An eventual hUCMS differentiation into neural cells was assayed at different passages, as compared to untreated controls. We performed morphological evaluation, including immunofluorescence staining for specific neural cell markers (Tubβ3, TH, Map2ab, NeuN, and Nestin), and qPCR for Tubβ3 gene expression, upon transdifferentation.
Differentiation toward definitive endoderm
This differentiation protocol was carried out in RPMI (Biospa) supplemented with 100 ng/mL recombinant human Activin A (Peprotech, Li StarFish), 0.5% FBS, 1×L-glutamine, 100 U/mL Penicillin, and 100 U/mL Streptomycin 29 for 5 days. We performed morphological evaluation, protein assay, and qPCR analysis for the indicated markers, at I and III culture passages, after differentiation, in comparison with untreated controls.
Data analysis
Data were expressed as mean±standard deviation in at last three independent experiments and considered significant for p-values <0.01. All statistical analyses were done with Excel 2003 (Microsoft).
Results
So far, 20 human umbilical cords have been processed by our method. Mean cell count from each cord isolation, as assessed by Burker cytometer, after trypan blue treatment, was 15–30×104 cells/cm of cord.
Morphology of cells isolated from the Wharton jelly
At 24 h of the isolation (Fig. 1A) the culture was predominantly comprised of some adhering cells and a few partially digested matrix fragments. In the resulting suspension the Wharton jelly-derived mesenchymal stem cells were clearly detectable. These cells, in suspension, appeared round-shaped, and irregularly outlined under phase-contrast microscopyc examination, with an average size of 20 μm. At this time point, EGF (1 ng/mL) and PDGF-BB (10 ng/mL) were added to the culture to facilitate both, cell adhesion, and growth. After 5 days of the isolation, a selected mesenchymal stem cell population adhering to the plate bottom (Fig. 1A) was identified. The cells showed an elongated, triangle-like shape, with many of them showing mitotic figures. At 10 days of the isolation the cells reached full confluence and acquired a more pronounced fibroblast-like shape (Fig. 1A).

hUCMS morphology and stemness markers.
Immunophenotypic characterization of hUCMS
hUCMS at early stages were characterized by flow cytometry. A panel of five markers were tested. All hUCMS preparations were negative for the hematopoietic markers CD31 and CD45, but consistently positive for CD90, CD117, and SCF, that are typical of mesenchymal stem cells (Fig. 2A). Percent expression of these markers was quite similar in all performed isolations and in all the tested cell culture passages.
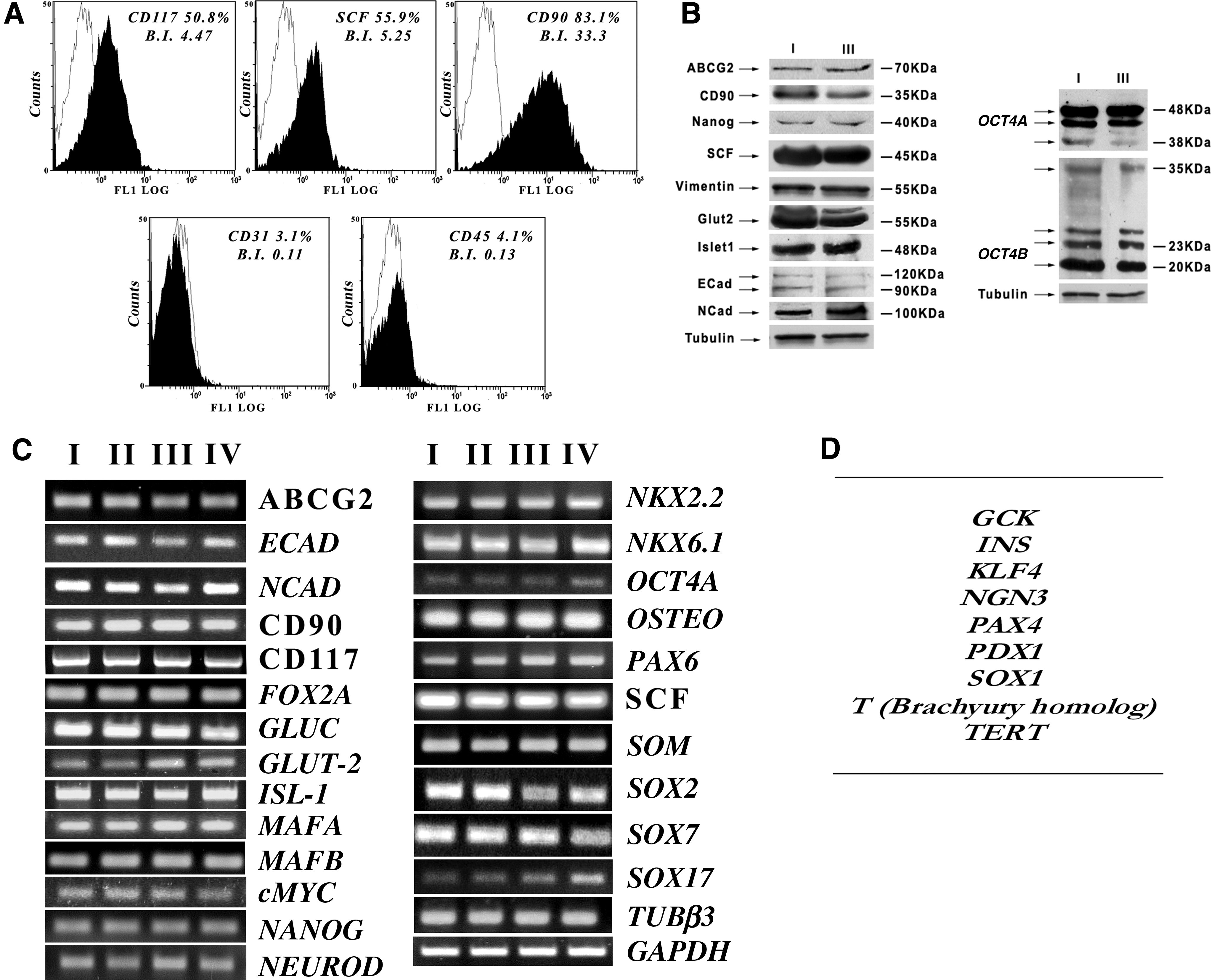
Characterization of hUCMS throughout the first four passages of in vitro culture expansion.
Detection of pluripotent/stem cell markers at mRNA, and protein level
hUCMS isolated by our method expressed, throughout the four examined passages, messengers typical of stem cell markers, namely, Oct4a, Nanog, Sox-2, ABCG2, and c-Myc, thus confirming their stemness identity (Fig. 2C). The relative gene-encoded proteins were assessed by immunofluorescence by comparing the cultured cells with those embodied in the cord matrix before isolation (Fig. 1B; see Supplementary Figs. S1–S3). In particular, Oct4 was detected at levels of both cytoplasm and nucleus as per recently published reports. 30 Moreover, the antibody used for this analysis enables detection of both principal splicing variants: Oct4A and Oct4B. Nanog and Sox2 also were detected in the cytoplasm and nucleus. ABCG2, a membrane glycoprotein found in multipotent adult stem cell lineages, associated with exclusion of many substances from the cytoplasm, including Hoechst, was significantly present in the hUCMS membrane. Here it formed clusters as previously described. 31 Noteworthy, all these markers are originally expressed by hUCMS within the Wharton Jelly as clearly detectable in the cord's cryostat sections (Fig. 1B).
Stem markers in hUCMS were also analyzed by western blotting on cells at passage I and III of in vitro expansion. In both passages, the presence of Oct4, Nanog, and ABCG2 was confirmed (Fig. 2B). In particular, Oct4 was present in both Oct4A and Oct4B isoforms. Oct4A, showed a double band signal, at approximately 48 kDa, possibly related to different, glycosylation-related, molecular arrangements within the same isoform. hUCMS also expressed Oct4B whose 190aa isoform might play a role in cell apoptosis rate. 30
hUCMS are positive for transcriptional factor c-Myc that is involved in regulating either progression of the cell cycle progression, or apoptosis or cell transformation (Fig. 2C): immunofluorescence analysis indeed showed its presence in the nucleus, in both culture, and cord sections (Fig. 1B). hUCMS also express messengers and proteins for CD90 and nestin that all identify mesenchymal stem cells (Figs. 1B and 2B, C). Expression of genes that belong to different embryonic leaflets (endoderm, mesoderm, and ectoderm) was evidenced through the various passages. In particular, Sox7 typical of the primitive endoderm, and Sox17 and Fox2A, typical of the definitive endoderm, were expressed (Fig. 2C). E-cadherin and N-cadherin typical of both epithelial and neural tissue, co-expressed along some stages of the epithelial–mesenchymal transition, were present at both messenger and protein levels (Fig. 2B, C and Supplementary Fig. S2). Surprisingly, MafB and MafA mRNAs, which usually appear during differentiation of the endocrine endoderm, also were expressed (Fig. 2C). Messengers of NeuroD and Glucagon associated with neuronal and endocrine cell lineages, respectively, were also present (Fig. 2C). Presence of Glut2 and Isl1 was confirmed at protein and messenger levels (Fig. 2B, C). NKx2.2, NKx6.1, and Pax6 were also expressed along the examined passages (Fig. 2C). RT-PCR analysis on the isolated hUCMS did not show, in our system, through all passages, detection of gene T (mouse Brachyury homolog), expressed in the mesoderm, or Sox1 from ectoderm, or Pdx1 from pancreatic endoderm, or Pax4 indispensable for β cell differentiation, or Tert encoding telomerase (Fig. 2D). Except for Pdx-1, which was weakly detectable in the cytoplasm of cultured cells, as well as cord section (see Supplementary Fig. S3), and the other, above-mentioned markers were undetectable, even under immunoflorescence examination (data non shown).
Immunofluorescence confirmed the presence of hUCMS typical markers such as CD117, SCF, actin, vimentin, E-cadherin, N-cadherin, nestin, Tubβ3, and CK19 in both cultured cells and cord sections (see Supplementary Fig. S3). Finally, hUCMS showed basal-positive immunofluorescence patterns both in cell cultures and in cord sections for insulin, glucagon, and somatostatin (see Supplementary Fig. S3). Insulin was present at trace concentrations in these cells, as compared to patterns that usually are associated with human pancreatic islet cells (see Supplementary Fig. S4). Surprisingly, identification of all these factors in hUCMS likely was related to their multipotent, pleiotropic nature. The cells, in fact, express genes, at low levels, that are correlated with the undifferentiated state, but also genes that are linked to the three embryonic germ layers and trophoectoderm. 2 Recently, large numbers of growth factors, hormones, enzymes, and DNA-binding proteins have been described and identified as molecules that may act as both extracellular and intracellular signals. Some of these factors have been named “intracrines” and considered able to regulate either adult or pluripotent embryonic-like stem cells.32,33
Differentiation pathways
Stemness properties identify with the cell ability to differentiate toward several definitive cell phenotypes such as adipocytic, osteocytic, neural, and, as in our study, definitive endoderm that is a precursor for the endocrine tissue. On this purpose, the cells were exposed, at different passages, to differentiation protocols using ad hoc culture media.
Adipocytic
Cell cultures were examined under phase microscopy to assess extent of differentiation. The induction treatment, throughout 3 weeks, was associated with over 80% cell commitment toward adipocytic phenotype with evidence of fat vacuoles. These were confirmed by Oil Red O staining upon 3 weeks of culture maintenance. The red stained lipid vacuoli were undetectable in controls (Fig. 3A). qPCR showed that expression of both markers was noticeably increased. In particular, Fabp4 expression was 25 times higher than control (Fig. 3D), whereas Pparγ expression was detectable only in treated cells (see Supplementary Fig. S5).

hUCMS differentiation capacity.
Osteogenic
Differentiation capability of hUCMS into osteocytes was assessed after 3 weeks of culture induction by specific staining with Alizarine Red S. The treated cells picked up a typical dark red staining that is specific for extracellular stored calcium, whereas controls stained only faintly and not specifically (Fig. 3B). Genetic expression of osteopontin was lower in osteocytic differentiating cells than that in controls (Fig. 3D). Possibly, osteopontin messenger transcription could have been inhibited, at this juncture, by the protein itself.
Neuronal
As shown by immunofluorescence (Fig. 3C), the majority of neural cell phenotype markers was expressed by both treated and untreated cells. This is not surprising since these cells express differentiation markers that derive from all three the embryonic germ layers. Eventually, the special selected marker signals were differently cytolocalized in the treated versus untreated cells. In fact, Map2ab, Tubβ3, and nestin lose their perinuclear localization and were detectable distal to the nucleus, where axons and dendrites usually originate (Fig. 3C); TH and NeuN markers were detected in the nucleus of both treated and untreated cells (data not shown). Extent of cell differentiation was assessed by qPCR analyzing Tubβ3 versus Gapdh. The obtained data, so far, showed that treated cell monolayers were associated with a 50-fold increase in Tubβ3 mRNA expression as compared to untreated controls (Fig. 3D).
Induction of definitive endoderm
Differentiation induction toward definitive endoderm by Activin A and 0.5% FBS led hUCMS to strongly express genes that are typical of definitive endoderm in vertebrates, namely, Sox17 and FoxA2 in both tested passages (Fig. 3E). Likewise, Brachyury expression was negative in the treated as well as untreated cells, indicating that the selected conditions would not induce differentiation toward mesoderm. Moreover, no expression of Sox1 was evidenced, thereby confirming that ectoderm was not induced. Sox7 messenger, expressed by the extra-embryonic endoderm, was increased by Activin A (Fig. 3E). After induction, E-cadherin messenger expression was drastically diminished, whereas N-cadherin increased, possibly indicating that under these conditions, cell monolayers may undergo transition from epithelium to mesenchyma (Fig. 3E). hUCMS basally express MafB, NKx2.2, NKx6.1, and NeuroD, all progenitor endocrine cell markers that increased at the end of the treatment with Activin A (Fig. 3E). The treatment also increased basal MafA, a transcriptional factor that binds to Ripe3b, a conserved enhancer element that regulates pancreatic beta cell-specific expression of the insulin gene in the presence of Pdx1 and NeuroD. Moreover, Activin A activates Pdx1 messenger in treated versus control samples (see Supplementary Fig. S6) as previously reported.34,35 Both in basal and after treatment, hUCMS did not express Pax4, and Ngn3 messengers, as appropriate for their nature of endocrine, uncommitted progenitors cells. The fact that Activin A may lead the treated cells to maturity is supported by the induced, decreased expression of stemness-linked genes such as c-Myc and Nanog. On the contrary, Sox2 Oct4A, CD90, and ABCG2 were overexpressed (Fig. 3E).
Discussion
In light of their mesenchymal nature, low immunogenicity, and capability to induce immune tolerance, hUCMS might offer a possibly relevant source of adult stem cells to enter cell therapy protocols.9,10,21,22 However, hUCMS have not been harvested, so far, according to unequivocal as well as rapid methods. In fact, previously reported isolation protocols, according to the “plate and wait” procedure, simply were based on the spontaneous, physical migration of the mesenchymal cells from their native matrix. However, the procedure was too lengthy. Moreover, such a technique was essentially used in porcine animal models 6 with a few exceptions.26,27
Our proposed hUCMS cell isolation procedure consists of a new enzymatic digestion protocol where different proteases act synergistically to cleave the cells out of the original Wharton jelly matrix. This procedure not only is effective in terms of cell yield, but also it offers the opportunity to avoid time-consuming enzymatic digestion steps, the latter being associated with inevitable cellular membrane damage.
In particular, our method is based on direct delivery of an enzymatic solution into the Wharton jelly, similarly to methods for the pancreatic enzymatic digestion for islet cell isolation. 36 The umbilical cord is visibly distended upon the enzyme injection, which leads to detachment of the amnion from the underlying Wharton jelly matrix. The entire enzymatic digestion procedure does not take longer than 30 min, with the enzymatic activity being easily monitored and quickly stopped when and if necessary. The isolated cell yield (15–20×104 cells/cm) was higher than previously reported 2 : 10–50×103 cells/cm; the final yield of 5–6×104 cells/cord, considering that an average cord length ranges on 30–60 cm, amounts 1–2×103 cells/cm. 24 Cell growth seems to confirm that no cell membrane damage occurred during the process. Previously, we had attempted to isolate hUCMS from several human cords by a nonenzymatic procedure, just following tissue mechanical disruption, according to Weiss and coworkers, 6 supplementing DMEM with 20% FBS. However, contrary to the pig system, and unlike recent experiences with human cords,26,27 we did not observe any efficient cell migration from the chunked tissue. Hence, we maintained that the human cords need to be processed enzymatically, to maximize the separated cell yield.
In particular, we have selected hyaluronidase and human recombinant Liberase HI for our digestion enzymatic cocktail. Trypsin was eliminated from our mixture, unlike previous methods, 2 due to its potential induced damage to cell membranes. Hyaluronidase, on the contrary, looks appropriate since the Wharton jelly is mostly comprised of HA, whose digestion favors liberation of the hUCMS. Moreover, in the umbilical cord, HA is trapped in a collagen matrix whose digestion also facilitates cell liberation. A few reports detail on the collagenase type that should be employed for enzymatic digestion of the hUCMS. On the basis of our previous experience with isolation of pancreatic human islets from whole donor organs,37,38 we have excluded the use of type I and type V collagenase. These, in spite of their wide use, in fact are quite weak and associated with long digestion periods. Moreover, lot-to-lot variability adversely affects consistency of the results. 39 Other collagenase types, such as collagenase P, have been proven able to dissociate the cord tissue apart, although at cost of long digestion time (data non show). In our system, Liberase HI supplemented with hyaluronidase has proven to be the best enzymatic blend in terms of fast, gentle, and reproducible cord tissue digestion. Due to its specific chemistry and properties, Liberase HI is associated with elevated specific activity on the Wharton jelly, granting for an ideal digestion time, and preventing overdigestion if used at 0.5 mg/mL.
We decided to keep a few undigested/partially digested matrix fragments because we observed that they easily adhere to HA pretreated flasks. Upon adhesion, early in the culture flask, cells entrapped in the fragments adhere and replicate, thus resulting in further increase in the final cell yield.
Our isolated hUCMS express several markers, some typical of the undifferentiated embryonic stage (Oct4A, Sox2, and Nanog) at both messenger and protein levels. These markers are also detectable in the untreated cord tissue. As for Oct4, both isoforms A and B were identified. Form A typically correlates with stem cell self renewal capacity, 40 whereas form B seems to associate with stress resistance, thereby possibly preventing cell apoptosis. 41 Undetectability of Tert in our cell preparations could depend on the employed methods (qPCR vs. gene array), whereas its presence in adult stem cells is still debated, and only clearly confirmed in undifferentiated, pluripotent cell systems.42,43 Tert presence in adult mesenchymal stem cells looks unlikely. In fact, according to recent literature, Tert as well as other markers of cell pluripotency, while expressed during embryogenesis, tend to diminish through disappearance in postnatal and adult mesenchymal cells.44–47
In long-term cultured mesenchymal stem cells, tumor p16, p21, and p53 suppressor genes as well as DNA repair enzymes are inactivated, leading the cells to spontaneous changes and immortalization. 48 To exclude these events, we investigated on the presence or absence of p21 in both cord sections or cord-derived, isolated cells. The obtained data showed that p21 was not expressed in the cord sections but only on the isolated cells (see Supplementary Fig. S7). This finding was previously reported. 24 We speculate that absence of p21 in cord sections might correlate with the role played by cord mesenchymal cells during fetal growth: to adjust the umbilical cord size to the growing fetal size. Without p21, stem cells divide faster as indirectly proven by the fact that suppression of p53/p21 pathway increases generation of iPS. 49 Furthermore, p21 ko mice show high regenerative capacity of the damaged tissues. 50 On the contrary, presence of p21 in cord-derived isolated, cultured cells may correlate with need of inhibiting cell proliferation, so as to regulate mass size of stem cell compartment. 51
Our isolated cells do express markers that are typical of the three germ layers (Nestin, Sox17, and Vimentin). Overall, expression of these molecules leads to believe that they may associate with different cell phenotypes. In fact, we have preliminarily tested their ability to evolve toward adipocytic and osteocytic, with optimized differentiation results. Moreover, induction of neural differentiation leads to upregulation of proteins associated with this tissue, and already basally expressed by hUCMS.
In our work we have detected endoderm markers in basal hUCMS, as already described in the literature (Sox17, Sox7, FoxA2, and Nestin), but also others, so far undescribed, linked to the pancreatic endoderm (MafA, MafB, NKx2.2, NKx6.1, Pax6, NeuroD, and Glut2). Basal expression of nestin is very important, as proven by the fact that nestin-positive stem cells isolated from adult pancreatic islets have been shown to differentiate ex vivo into pancreatic endocrine/exocrine and hepatic cell phenotypes. 52 These markers may favor differentiation towards the endocrine cell phenotype upon exposure to 100 ng/mL Activin A in RPMI medium supplemented with 0.5% FBS for 5 days. In this experimental conditions, the cells acquired the typical features of the definitive endoderm and in particular upregulation of precursor endocrine genes (Sox17 and FoxA2); evolution of E-cadherin to N-cadherin; and raise in MafB and MafA, together with the upregulation of Oct4A, suggesting expression of endodermic markers during differentiation. 53 In our cell model, qPCR allowed for Pdx1 mRNA detection only after treatment with activin A. Such treatment also induced Fox2a and Sox17 messengers. At such differentation stage, toward the definitive endoderm, Pdx1 does not play a role as a glucose responsive regulator of insulin synthesis but eventually as developmental factor likely seen in the mouse model.34,54,55
Interestingly, modulation of the endocrine cell markers upon endocrine differentiation leads to believe that our cells may acquire the endocrine cell phenotype more easily as compared to human embryonic stem cells (hES). In fact, hES need an appropriate treatment with factors like Wnt 56 to reach a degree of differentiation that may approach our results. Moreover, upon differentiation into the definitive endoderm, we did not observe any expression of cell markers that are linked to mesoderm (Brachyury) or ectoderm (Sox1), this being, on the contrary, a pending problem with hES cells.
In conclusion, this work shows that our enzymatic digestion procedure enables bulk retrieval of mesenchymal stem cells from human umbilical cords that are associated with many of their typical properties. This is especially relevant in light of the actual trends of experimental medicine toward the new frontiers for cell and molecular therapy for several chronic disorders.
Only by replacing diseased/destroyed cells with viable and functional tissue could grant a cure for a specific disorder. While primary applications of this approach could be autoimmune or degenerative diseases such as type 1 diabetes mellitus or Parkinson or Alzheimer disease, other diversified disorders could potentially benefit from this new therapeutic strategy.
Footnotes
Acknowledgments
This work was supported in part by the “Consorzio Interuniversitario per i Trapianti d'Organo,” Roma, Italy. The authors thank Prof. Gian Carlo Di Renzo and Dr. Giuseppe Affronti (Department of Obstetrics and Gynecology, Hospital S. Maria della Misericordia, University of Perugia) and all their staff, especially Dott. Simona Freddio, for human umbilical cord retrieval. The authors are also grateful to Dr. Emanuela Falcinelli (Department of Internal Medicine, Section of Internal and Cardiovascular Medicine, University of Perugia) for her gracious assistance with flow cytometry.
Disclosure Statement
The authors have declared that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.