
Abstract
The objective of the present study was to evaluate the capacity of a tissue-engineered bone complex of recombinant human bone morphogenetic protein 2 (rhBMP-2)-mediated dental pulp stem cells (DPSCs) and nano-hydroxyapatite/collagen/poly(L-lactide) (nHAC/PLA) to reconstruct critical-size alveolar bone defects in New Zealand rabbit. Autologous DPSCs were isolated from rabbit dental pulp tissue and expanded ex vivo to enrich DPSCs numbers, and then their attachment and differentiation capability were evaluated when cultured on the culture plate or nHAC/PLA. The alveolar bone defects were treated with nHAC/PLA, nHAC/PLA+rhBMP-2, nHAC/PLA+DPSCs, nHAC/PLA+DPSCs+rhBMP-2, and autogenous bone (AB) obtained from iliac bone or were left untreated as a control. X-ray and a polychrome sequential fluorescent labeling were performed postoperatively and the animals were sacrificed 12 weeks after operation for histological observation and histomorphometric analysis. Our results showed that DPSCs expressed STRO-1 and vementin, and favored osteogenesis and adipogenesis in conditioned media. DPSCs attached and spread well, and retained their osteogenic phenotypes on nHAC/PLA. The rhBMP-2 could significantly increase protein content, alkaline phosphatase activity/protein, osteocalcin content, and mineral formation of DPSCs cultured on nHAC/PLA. The X-ray graph, the fluorescent, histological observation, and histomorphometric analysis showed that the nHAC/PLA+DPSCs+rhBMP-2 tissue-engineered bone complex had an earlier mineralization and more bone formation inside the scaffold than nHAC/PLA, nHAC/PLA+rhBMP-2, and nHAC/PLA+DPSCs, or even autologous bone. Implanted DPSCs' contribution to new bone was detected through transfected eGFP genes. Our findings indicated that stem cells existed in adult rabbit dental pulp tissue. The rhBMP-2 promoted osteogenic capability of DPSCs as a potential cell source for periodontal bone regeneration. The nHAC/PLA could serve as a good scaffold for autologous DPSC seeding, proliferation, and differentiation. The tissue-engineered bone complex with nHAC/PLA, rhBMP-2, and autologous DPSCs might be a better alternative to autologous bone for the clinical reconstruction of periodontal bone defects.
Introduction
As a trial to overcome these problems, tissue engineering has attracted much attention as a promising alternative to natural bone grafts. Tissue engineering induces bone tissue regeneration by the use of a composite graft that contains osteogenic cells and osteoinductive growth factors along with a synthetic osteoconductive matrix substitute. In periodontal tissue engineering, ideal scaffold and appropriate osteogenic cell are crucial for the successful engineering of alveolar bone tissues, as an ideal scaffold can provide a suitable environment for osteogenic cells to migrate, proliferate, differentiate, and promote new bone formation.
Recently, a developed ceramic/polymer composite material, nano-hydroxyapatite/collagen/poly(L-lactide) (nHAC/PLA) is attractive as a bone substitute because the novel biomimetic strategy used to generate it provides properties similar to natural bone. Natural bone is a structure composed of hydroxyapatite [Ca10(PO4)6(OH)2] crystals with low crystallinity and nanometer size deposited within an organic matrix (∼95% is type I collagen [COLI]). 10 It was reported that nano-hydroxyapatite and collagens assembled into mineralized fibril bundles, and then these bundles were uniformly distributed in a PLA matrix to form a porous scaffold. Thus, the three-dimensional porous scaffold materials mimicked the nano- to microscale hierarchical microstructure of natural, cancellous bone. 11 Cell culture and animal model tests showed that the composite material was highly osteoconductive, biocompatible, and bioresorbable.11–14 However, whether nHAC/PLA can be used in periodontal tissue engineering is still unclear.
Moreover, dental pulp is a niche housing neural-crest-derived stem cells. This niche is easily accessible and there is limited morbidity after collection.15–17 Previous studies have shown that dental pulp stem cells (DPSCs) are capable of differentiating into osteoblasts18,19 that secrete abundant extracellular matrix (ECM) and that can build a woven bone in vitro. 20 Further, DPSCs are capable of forming a complete and well-vascularised lamellar bone after grafting into immunosuppressed rats.21,22 The quality and quantity of regenerated bone formed by DPSCs was demonstrated in in vitro and in vivo experiments using stem cells and biomaterials.18,21–23 Thus, dental pulp housing neural-crest-derived stem cells sharing similar tissue origin with the mandibular bone cells may be considered as an interesting and potentially important source of autologous stem/progenitor cells that are ready for use for the repair/regeneration of periodontal bones. However, to our knowledge, the behavior of DPSCs on nHAC/PLA scaffold is still unclear.
Osteoinductive growth factors, such as bone morphogenetic protein (BMP), transforming growth factor b, and basic fibroblast growth factor, have been investigated to induce bone regeneration in the body. 24 Among them, BMP-2 has already been applied clinically for bone regeneration at the bone defect, because of their high osteoinduction activity. 25 The in vivo studies have also shown the recombinant human BMP-2 (rhBMP-2) to promote mineralized tissue formation effectively by DPSCs26–28 and bone cells. 29 Nonetheless, the studies on the capacity of the combination of nHAC/PLA, DPSCs, and rhBMP-2 to reconstruct critical-size alveolar bone defects are still limited.
To explore the feasibility of using stem cell-based bone regeneration to repair periodontal bone defects, we utilized New Zealand white rabbit as a preclinical animal model to test the regeneration of critical-size alveolar bone defects using nHAC/PLA+DPSCs+rhBMP-2 construct without genetic manipulation. Before transplantation, DPSCs were characterized and their in vitro differentiation potential was addressed by the production of osteoblasts. We evaluated bone formation of the composite graft implanted by X-ray scan, histological staining, fluorescence label, transfection of eGFP genes, and histomorphometric analysis.
Materials and Methods
Harvest and culture of dental pulp cells
The New Zealand rabbits (2.50–3.00 kg; Beijing, China) were obtained from laboratory animal center of the Academy of Military Medicine Sciences. All surgical procedures and care administered to the animals were approved by the University Animal Care Committee and performed according to institutional guidelines. The rabbits were killed by anesthesia. All teeth were dissected, and the surfaces of the teeth were cleaned with 75% alcohol. Apical areas were then removed to decrease the influence from periodontal and periapical tissue, and dental pulps were removed with a barbed broach. Dissected tissue was minced and incubated in 0.2% collagenase (Invitrogen) at 37°C for 4 h on a rotator set at 130 rpm. The released cells were collected into a centrifuge tube and pelleted. The pellet was resuspended in growth media including Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 IU/mL streptomycin (Invitrogen). Thereafter, the resuspended cells were seeded in 25-mL flasks (Corning Costar) and incubated at 37°C in 5% CO2. Culture medium was changed at 3- to 4-day intervals. When 70% to 80% confluence was reached, the cells were passaged and the first-passage cells were used to obtain DPSCs by limiting dilutions.
Colony efficiency assays and proliferation potential
The first-passage dental pulp cells were plated at limiting confluence to result in isolated single cells. Cultures were maintained in growth medium until the formation of well-defined colonies. The single dental pulp cell-derived colonies were harvested using sterile cloning rings and expanded in cloning medium (15% FBS and 1% antibiotic/antimycotic in F-12/DMEM [1:1]). Expanded clones were subcloned by limiting dilution. After 3 weeks of culture, cells were stained with 0.1% (w/v) Toluidine blue (Sigma-Aldrich) in 1% paraformaldehyde. The number of clones (>50 cells) were counted. The third-passage DPSCs cloned were removed with 0.25% trypsin (Sigma-Aldrich) and cultured in growth media for the various experiments.
Characterization and osteogenic differentiation of DPSCs
The first-passage DPSCs seeded on chamber slides in six-well culture plates (Corning Costar) were cultured in growth media for 3 days, fixed in 4% paraformaldehyde for 15 min, and examined for vimentin, keratin, and STRO-1.
The third-passage DPSCs seeded on chamber slides in 6-well culture plates were cultured in osteogenic media (growth media containing 10 nM dexamethasone, 50 μg/mL ascorbic acid, and 100 mM β-glycerophosphate [Sigma-Aldrich]) for 28 days and examined for dentin sialoprotein (DSP), osteocalcin (OCN), bone sialoprotein (BSP), COLI, alkaline phosphatase (ALP) activity, and ECM calcification. Samples were preincubated in 10% normal goat serum for 30 min, and incubated with mouse anti-human vimentin antibody and mouse anti-human keratin antibody (Santa Cruz Biotechnology, Inc.) at a dilution of 1:100 for 2 h according to the manufacturer's protocol. Samples were subsequently incubated with anti-mouse IgG TRITC (Santa Cruz Biotechnology, Inc.) at a dilution of 1:50 for 45 min. Nonimmune serum served as negative control. Subsequently, samples were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) for 15 min, and observed by fluorescence microscopy. Samples were preincubated in 10% normal goat serum for 30 min, and incubated with mouse anti-human STRO-1, DSP, OCN, BSP, and COLI antibody (MAB1038; R&D Systems, Inc.) at a dilution of 1:100 for 2 h, the cells were then incubated with biotin-conjugated goat-anti-mouse antibodies (Santa Cruz Biotechnology, Inc.) for 1 h, and incubated with streptavidin-biotin complex/horseradish peroxidase (Santa Cruz Biotechnology, Inc.) for 1 h. Staining was observed using 3,3′-diaminobenzidine (0.1 mg/mL, 0.02% H2O2; Santa Cruz Biotechnology, Inc.). Nonimmune serum served as negative control. Subsequently, samples were counterstained with hematoxylin for 15 min, and observed by a Leica microscope.
The ALP activity of DPSCs cultured for 28 days was assessed using Gomori calcium-cobalt staining. Briefly, the cells were washed with phosphate-buffered solution (PBS) for 5 min. An incubation solution containing 5 mL 2% barbital sodium, 5 mL 3% β-sodium glycerophosphate, 10 mL 2% calcium nitrate, 5 mL 2% magnesium sulfate (Sigma-Aldrich), and 25 mL distilled water was placed on each slide and incubated for 4 h at 37°C. Slides were then washed with distilled water and incubated in 2% calcium nitrate (Sigma-Aldrich) for 2 min. Slides were then incubated in 2% cobaltous nitrate (Sigma-Aldrich) for 2 min. Slides were then washed with distilled water and incubated in 1% ammonium sulfide (Sigma-Aldrich) for 1 min. Slides were then washed with running tap water and left to dry. The stained cells were photographed under a Leica microscope.
To estimate COLI, the differentiated cells cultured for 28 days were fixed with 10% formalin and stained histochemically using Van Gieson staining. Briefly, the cells were washed with PBS for 5 min and incubated in iron hematoxylin solution for 5 min. The cells were then washed with PBS for 5 min and returned blue with running tap water for 10 min. A trinitrophenol acid fuchsin solution was placed on the cells and incubated for 5 min. The cells were washed 95% ethanol and left to dry.
To detect ECM calcification with alizarin red staining, on day 28 of differentiation, the osteogenic media-cultured cells were fixed with 10% formalin. The cells were washed with PBS for 5 min. A 2% alizarin red solution (Sigma-Aldrich) was placed on each slide and incubated for 10 min at room temperature (RT). Slides were then washed with running tap water for 5 min and left to dry. The stained cells were photographed under a Leica microscope.
Adipocyte differentiation of DPSCs
Adipogenic differentiation was induced by culturing the third-passage DPSCs in growth media containing 100 nM human recombinant insulin, 1 μM dexamethasone, 0.5 mM isobuty 1-methylxanthine, and 200 μM indometacin (Sigma-Aldrich). Assessment of adipogenesis was performed with Oil red O (Sigma-Aldrich) staining, which demonstrates intracellular lipid accumulation. To perform this staining, the cells were trypsinized, collected, and plated onto chamber slides in multiple-well plates at 1×105 cells/cm2 for 24 h to allow attachment in growth media. These culture conditions achieve 100% confluence within the first 24 h. The media were then changed to adipogenic media. At the 14th day of differentiation, the cells on the chamber slides were fixed for 10 min in 10% formalin and washed with PBS for 5 min. The cells were then incubated in 2% Oil red O reagent for 30 min. The cells were then washed with PBS three times for 5 min each, followed by several changes of distilled water to remove excess staining. The cells were counterstained for 1 min with hematoxylin.
Transfection of eGFP genes
To trace directly the distribution and differentiation of DPSCs in vivo, the recombinant retroviral vector with green fluorescent protein (RV-GFP; Clontech) was used to label the third-passage DPSCs as described previously. 30 Recombinant RV-GFP expression vector was constructed and transfected into the packaging cell PT67. After G418 screening and amplification, cell clones producing high-level recombinant viruses were obtained and expanded in vitro. The virus supernatants from infected PT67 cell cultures were used to infect proliferating DPSCs directly.
Biometrics preparation and seeding of nHAC/PLA scaffolds
The nHAC/PLA material had some features of the natural bone composition and the structure: the porosity was 70%–90%; the pore size was (300–400) ± 150 μm. The nHAC/PLA materials (Beijing Allgens Medical Science & Technology Co., Ltd.) were cut into 10×4×3 mm blocks. The samples were rinsed with 100% alcohol to remove organic residues and with double-distilled water to remove inorganic residues (each solution for 10 min). Samples were then sterilizated by cobalt 60.
The GFP-labeled DPSCs were seeded onto nHAC/PLA. The constructs were incubated in growth media in 24-well plate for 2 h at 37°C, allowing the cells to adhere to nHAC/PLA and then 1 mL of additional growth media with/without 100 ng/mL rhBMP-2 was added into each well with nHAC/PLA or DPSCs+nHAC/PLA. The media were replaced on day 4 of incubation and the grafts were ready for in vitro studies and implantation in vivo.
Scanning electron microscopy
The GFP-labeled DPSCs were seeded separately onto chamber slides and sterile, resorbable nHAC/PLA scaffolds at 1×105 cells/cm2 per sample, and then the chamber slides, nHAC/PLA, and nHAC/PLA+DPSCs were cultured in growth media with 100 ng/mL rhBMP-2 for 2, 7, and 28 days. Culture medium was changed at 3- to 4-day intervals. Fixative was prepared from 2% paraformaldehyde and 2.5% glutaraldehyde (Sigma-Aldrich) in 0.1 M phosphate buffer. After fixation (30 min, 37°C), samples from chamber slides, nHAC/PLA, and nHAC/PLA+DPSCs were rinsed twice in PBS for 10 min and then washed five times (15 min each) in different ethanol concentrations (50%, 75%, 90%, and 95% v/v ethanol in distilled water and three times for 10 min each in analytical ethanol). After the ethanol washes, samples were rinsed in a series of different hexamethyldisilazane (HMDS) concentrations (33.3%, 50%, and 66.6% v/v) in analytic ethanol and three times in 100% HMDS (1 min each). Morphological characterization of cells and materials was done by means of scanning electron microscopy (SEM) using a Quanta 200 ESEM/SEM, FEI (Phillips) with beam energies of 6–25 kV and fitted with an energy dispersive spectroscopy apparatus. Samples were glued with conducing paste to appropriate mounting stabs, which were then coated with a several nanometer-thick layer of gold. The samples were examined under a Hitachi S-520 scanning electron microscope (Hitachi).
Protein content assay
The experiment was divided into two groups: nHAC/PLA+DPSCs and nHAC/PLA+DPSCs+rhBMP-2. The DPSCs were seeded onto nHAC/PLA at 1×106 cells/cm2 per graft. The protein content of nHAC/PLA+DPSCs cultured in growth medium with/without 100 ng/mL rhBMP-2 at 7, 14, 21, and 28 days of culture, as measured by total protein content, were assessed with a QuantiPro™ BCA Assay Kit (TaKaRa Bio, Inc.). Briefly, 0.5 mL 0.5% Triton X-100 was then added into each well with samples in the plates. The plates were shaken at 4°C overnight. Finally, the triton solutions with lysed cells were analyzed for protein content.
For the determination of the protein content, 100 μL of each sample (n=6) was added to the wells of a 96-well plate and then a 100 μL BCA solution was added. Then, the plate was continuously shaken for 2 h in the dark at RT. Finally, the protein content was measured with the QuantiPro™ BCA Assay Kit according to the guideline of the company. The protein content, expressed as mean±SD, was counted through a premade standard protein curve.
ALP activity assay
The same supernatants used to measure the protein content were also used to measure ALP activity using biochemistry automatic analyzer (Hitachi 7600). ALP activity was always expressed relative to the amount of total protein in the sample.
OCN content assay
The experiment was divided into two groups: nHAC/PLA+DPSCs and nHAC/PLA+DPSCs+rhBMP-2. The DPSCs were seeded onto nHAC/PLA at 1×106 cells/cm2 per graft. The nHAC/PLA+DPSCs were cultured in growth medium with/without 100 ng/mL rhBMP-2. After 28 days of culture, the media were collected from the wells, respectively. OCN content was assayed using a mouse-specific IRMA (Immutopics, Inc.). Briefly, the sample containing mouse OCN was incubated simultaneously with an antibody-coated bead and the 125I-labeled antibody. The OCN contained in the sample was immunologically bound by the immobilized and radiolabeled antibody to form a sandwich complex: bead/Anti-mouse, mouse, and 125I-anti-mouse OCN. At the end of the overnight incubation, the bead was washed to remove unbound labeled antibody and other components. The radioactivity bound to the bead was measured in a gamma counter. The radioactivity of the bound antibody complex was directly proportional to the bound antibody complex in the sample. As the amount of ECM proteins interfered with total cellular protein determination, the data were determined and expressed (as ng/mL) for each culture dish.
Mineral formation assay
Alizarin red staining was used to quantify calcium phosphate mineral formation in the two groups constructs mentioned above. Alizarin red was dissolved in distilled water at 2% (weight/volume) concentration and was adjusted to pH 4.2 with NaOH and passed through a 0.22 μm filter. The two group constructs in 24-well plates after 28 days of culture were rinsed with 0.01 M PBS three times, fixed in 75% ethanol, rinsed with distilled water, and stained at RT for 10 min with 1 mL of alizarin red solution per well. After staining, all constructs were washed with distilled water until supernatant was clear. For optical density measurements, each well was eluted for 30 min with 50 μL 10% cetylpyridinium chloride monohydrate. The optical density at 540 nm was determined using a microplate reader (Molecular Devices). Blank wells (without cells) were stained with dye and rinsed in the same manner. The blank-well optical density values were subtracted from the experimental well data points to control for stain retention by the walls of the well.
Preparation of fresh autogenous iliac bone graft
Fresh autogenous iliac bone was harvested at the time of surgery during the creation of alveolar bone defect. An incision of 3 cm was made and a corticocancellus bone block was harvested from the iliac bone (Fig. 1A). The periosteum and skin flap were replaced and sutured. The corticocancellous bone block was shaped with a dental drill and the bone graft was stored in a physiologic saline solution before it was grafted to alveolar bone defect.
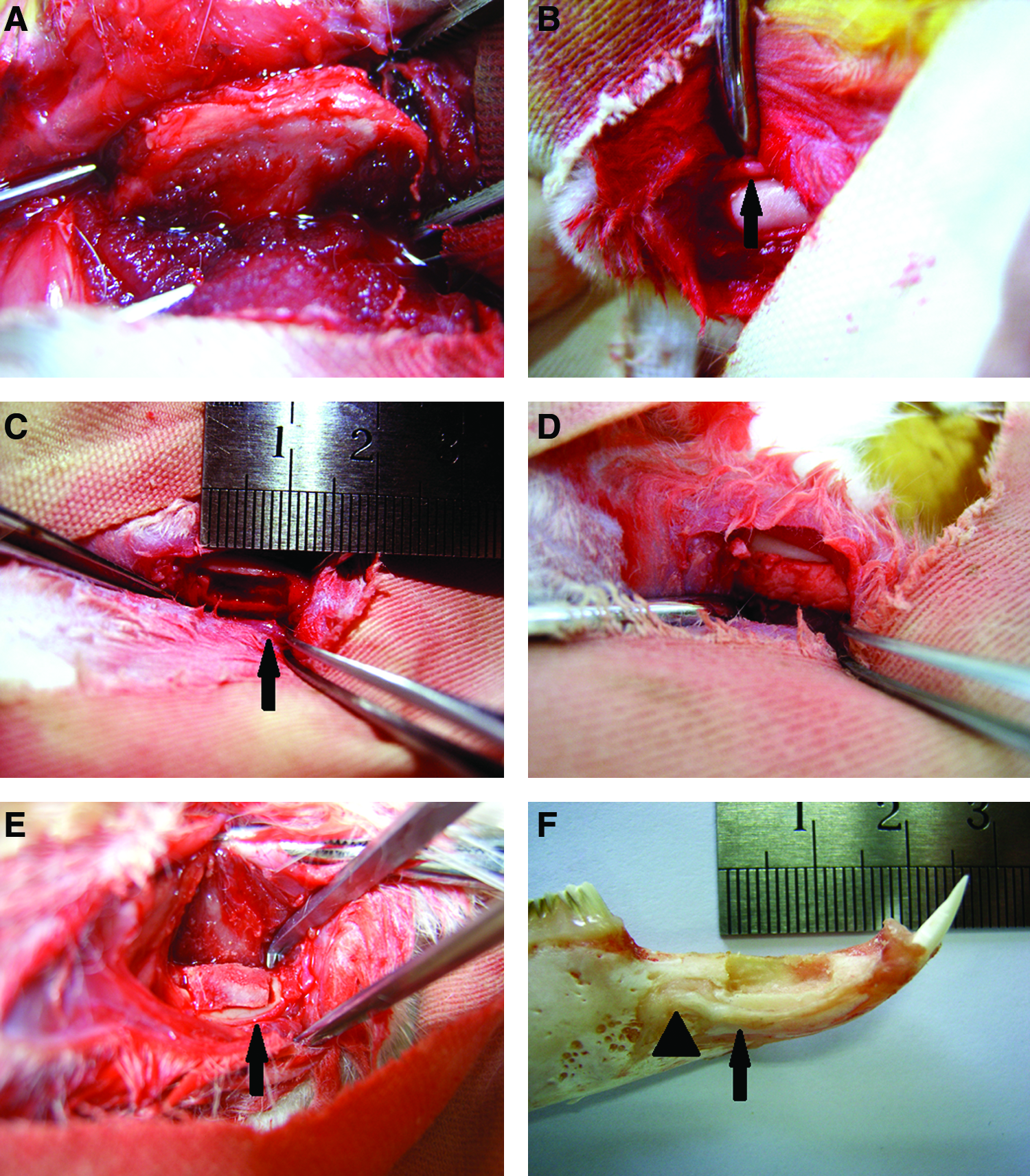
The schematic illustration of tissue-engineered bone complex and autologous iliac bone implantation into alveolar bone defects of rabbit.
Surgical procedure and tissue labeling method
The in vivo bone regeneration experiment was performed by use of a segmental critical-size alveolar bone defect model of skeletally mature female New Zealand white rabbits (Female rabbits, weight range of 2.50 to 3.00 kg, mean and standard deviation, 2.80±0.35; laboratory animal center of the Academy of Military Medicine Sciences, China). All surgical procedures and care administered to the animals were approved by the University Animal Care Committee and performed according to institutional guidelines. All rabbits were allowed to acclimate to the facility for a minimum of 1 week before their first operation. They were housed in separate cages in a climate controlled facility with free access to antibiotic-free food including commercial pellets, hay, and water. A 10 mm incision was made and the tissue overlying the diaphysis of the left alveolar bone of incisors of rabbits was dissected. A segmental defect (10×4×3 mm) was prepared in the alveolar of 36 rabbits with a surgical oscillating saw supplemented by copious sterile saline water irrigation (Fig. 1B–1F). A diameter of 5 mm corresponds to critical size in this experimental model. This size has been reported to prevent spontaneous healing during an animal's lifetime.31,32 The alveolar bone defects were treated with nHAC/PLA+DPSCs+rhBMP-2, nHAC/PLA+DPSCs, nHAC/PLA+rhBMP-2, or nHAC/PLA scaffold or were left untreated as a negative control; autogenous bone (AB) obtained from iliac bone was served as a positive control. The DPSCs were seeded onto nHAC/PLA at 1×108 cells/cm2 per graft. The nHAC/PLAs and constructs were then cultured in growth medium with/without 100 ng/mL rhBMP-2 for 7 days in vitro, and then transplanted into the alveolar bone defects of rabbits. After surgery, the soft tissue was approximated with interrupted 4-0 Vicryl (Ethicon, Inc.) and the skin was closed with 3-0 silk sutures. At 10 weeks postoperatively, the newly formed bones were labeled by an intraperitoneal injection of tetracycline (30 mg/kg of body weight; Sigma-Aldrich) dissolved in physiologic saline. After 10 days of injection of tetracycline, calcein (10 mg/kg of body weight; Sigma-Aldrich) dissolved in physiologic saline was administered intraperitoneally to the experimental rabbits. After 4 days, the rabbits were euthanized by an intravenous injection of pentobarbital sodium (Sigma-Aldrich) at 20 mg/kg body weight. The samples were removed surgically and fixed in formalin. X ray, confocal laser scanning microscopy (CLSM; GB-200; Olympus), toluidine blue staining, and goldner's trichrome staining were used to examine bone formation.
Assessment of bone regeneration
An X-ray confirmed the outlines of the defects 0 and 12 weeks after surgery. The implants were resected and fixed in 10% formalin for 3 days, and each resected bone was evaluated by light microscopy and CLSM. Specimens were trimmed using waterproof polishing paper without demineralization and cut into 5 μm sections, and stained with toluidine blue and goldner's trichrome for light microscopic observation. The specimens were observed by CLSM from the surface layer of the sections. In specimens without demineralization, calcein was displayed in green and tetracycline was displayed in yellow using two types of barrier filters, at BP 505–530 nm and LP 585 nm, respectively, with an argon laser excitation wavelength of 488 nm. Bone mineral apposition rate was determined by the tetracycline and calcein double labeling average interval/time period (10 days). The tracing of foreign DPSCs in the implanted constructs in vivo was determined using CLSM. Confocal images were recorded with a Carl Zeiss 510 META microscope (German).
For morphometric analysis, 5 sequential sections per implant were selected for evaluation under low magnification, allowing coverage of the entire implant. Using a Leica-Qwin 3.2 image analysis system (Leitz DMRD; Leica Microsystems Inc.), all slides were analyzed by two independent observers to identify the type of tissue (mature bone-like and osteoid-like). The extent of newly formed mature bone and osteoid was indicated by the percentage of total bone formation area within the section, and an average value was calculated for each implant. Data were then averaged across all implants within each group. Total scores per section were calculated and averaged for all sections to obtain an overall score for each implant. Data were then averaged across all implants within each group.
Statistical analysis
The data were assessed by the one-way ANOVA and Student's t-test, using computer-based SPSS 13.0 software. The results are expressed as mean±SD of 6 observations. For all analyses, p<0.05 level was used to indicate statistical significance.
Results
Culture and colony efficiency assays of DPSCs
To isolate dental pulp cells, single-cell suspension was obtained by enzymatic digestion and placed into the culture medium. After 1 day of culture, cells were adherent and often aggregated in groups. The cultures reached confluence after 5 days in culture. The isolated cells had typical fibroblastic morphology, spindle-shaped with extending cytoplasmic processes (Fig. 2A). To obtain DPSCs and determinate the proliferation and clonogenic potential of the cells, we performed a limiting dilution assay using first-passage pulp cells above mentioned. After 3 weeks of culture, a mean of 92% T 4% of wells, which were initially plated with one or two cells, contained colonies (formed 50 to 80 cells), with a doubling time of about 5 days (Fig. 2B, C). Colonies obtained reached confluence and were removed with 0.25% trypsin and cultured in growth media for the various experiments (Fig. 2D).

Isolation of DPSCs from dental pulp.
Characterization of DPSCs and expression of eGFP gene
The DPSCs had typical fibroblastic morphology, spindle-shaped with extending cytoplasmic processes. Immunofluorescence analysis showed positive staining for vimentin in DPSCs (Fig. 3A), negative staining for keratin (Fig. 3B). STRO-1-positive cells could be found in the cells population (Fig. 3C). The retroviral transfection was carried out on day 2 in culture, followed by a repeated transfection on day 3. Subsequently, transfected DPSCs were selected with G418 (100 μg/mL), and colonies expressing green fluorescence were expanded in growth medium. After several passages, the progenies of GFP-positive DPSCs continued to express eGFP (Fig. 3D).
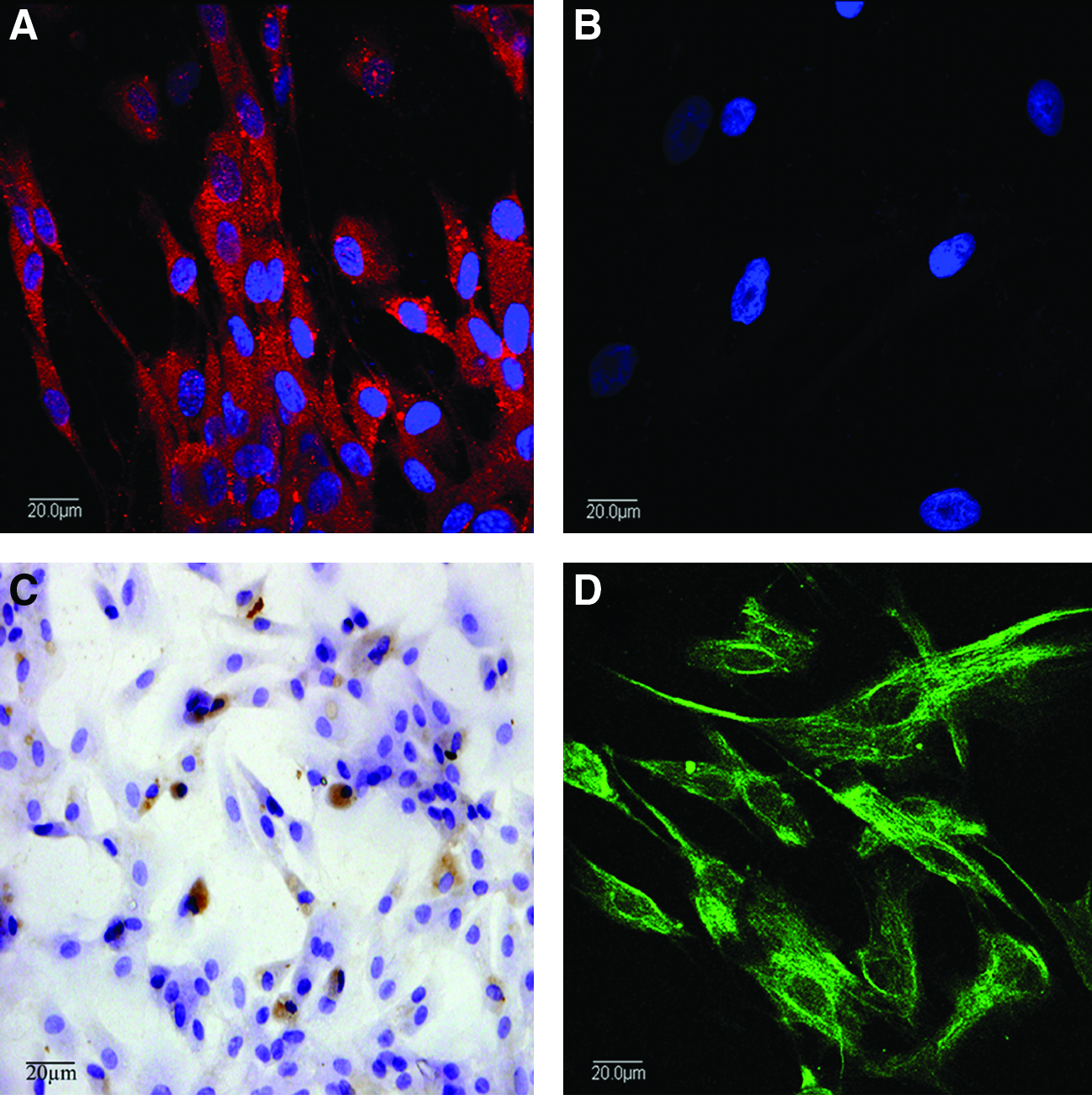
The characterization and expression of eGFP gene of DPSCs and schematic illustration of GFP-labeled nHAC/PLA+DPSCs implantation into an alveolar bone defect of rabbit. The DPSCs positively expressed vimentin
Osteogenic and adipogenic differentiation of DPSCs
When cultured in osteogenic media for 28 days, the DPSCs could undergo odontogenic differentiation and osteogenic differentiation. The cells expressed DSP, OCN, BSP, and COLI (Fig. 4A–D) by immunohistochemical staining. The ALP activity was intense in the cells using Gomori calcium-cobalt method (Fig. 4E). Sporadic nodule-shaped structures could be observed by alizarin red staining (Fig. 4F). Using this stain, nodules containing calcium mineral stain black. Large amounts of collagen fibers were stained in the cells using Van Gieson staining (Fig. 4G). The DPSCs did not spontaneously adipogenic differentiate during culture expansion. When cultured in lineage-specific differentiation culture medium for 14 days, the DPSCs within three passages could undergo adipogenic differentiation (Fig. 4H).
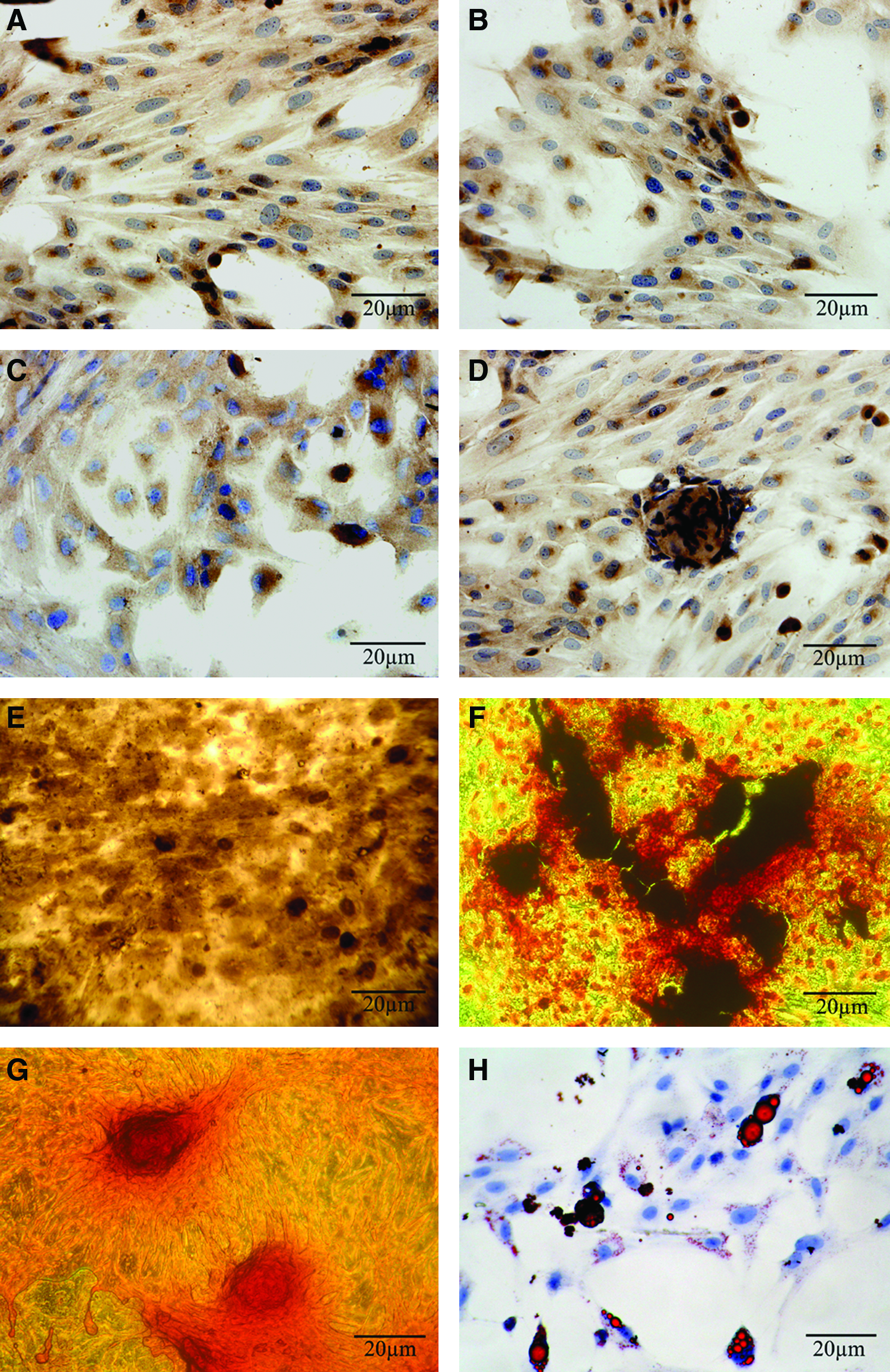
The characterization of DPSCs cultured in osteogenic and adipogenic media. The DPSCs positively expressed dentin sialoprotein
SEM analysis
Figure 5A and B shows the DPSCs cultured on the chamber slides in growth media with 100 ng/mL rhBMP-2 for 28 days by SEM. The cells became confluent, and formed a distinct multilayer. Some cells became round, and were covered with deposits.
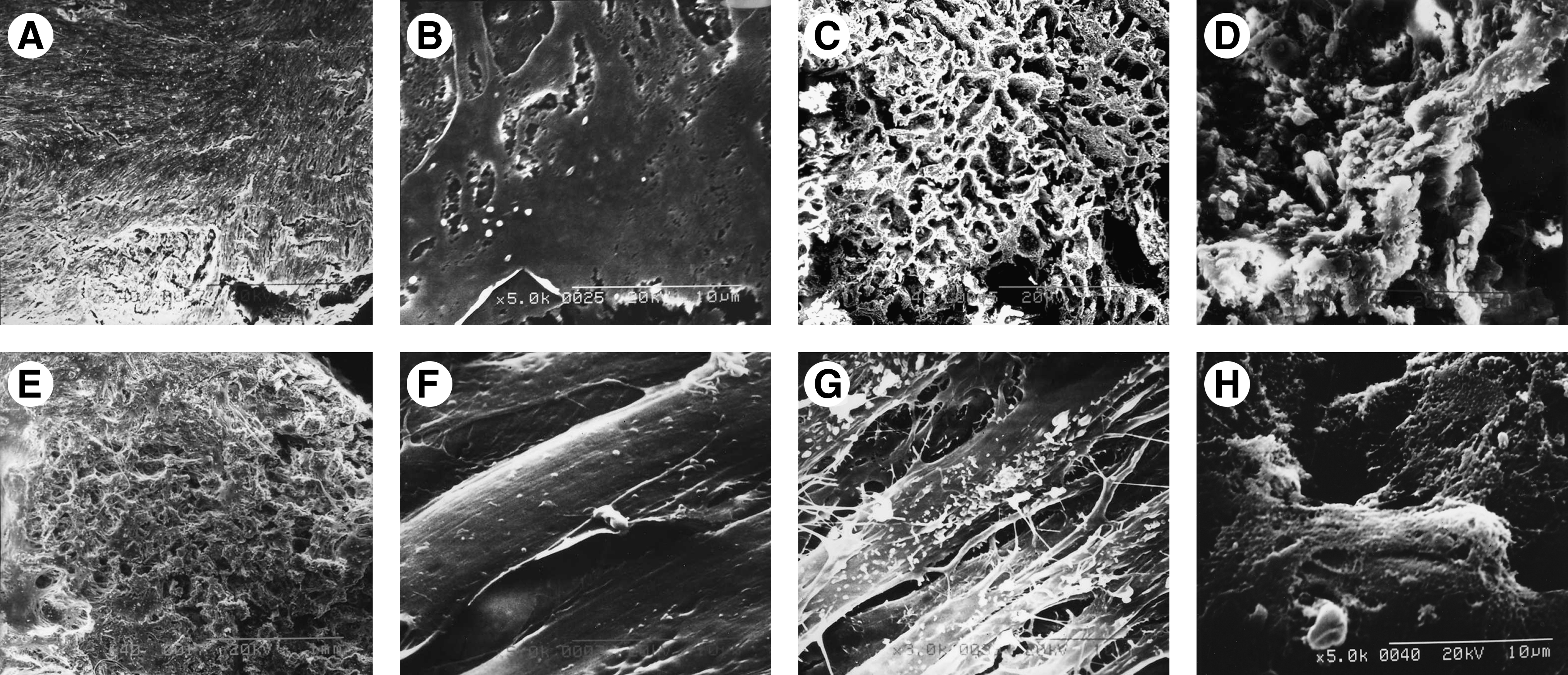
Scanning electron microscopy revealed GFP-labeled DPSCs grown on the chamber slide and on nHAC/PLA with rhBMP-2. Low magnification
The nHAC/PLA blocks were showed some features of natural bone in both main component and hierarchical microstructure by SEM (Fig. 5C, D). When the DPSCs were seeded on nHAC/PLA cultured in growth media with 100 ng/mL rhBMP-2 for 2 days, the cells adhered, extended, and connected with each other (Fig. 5E) and produced a few ECM on nHAC/PLA materials (Fig. 5F). After 7 days of culture, a large number of DPSCs could be seen adhered and significant proliferation to link flakiness on the surface and in the pore of the scaffold material; there were many filarious ECM on the surface of the cells (Fig. 5G). Some cells on nHAC/PLA were covered by deposits after 28 days of culture; network-like structures, formed by cell-secreted collagen, were also found (Fig. 5H).
Effect of rhBMP-2 on protein content, ALP activity, OCN content, and mineral formation of DPSCs cultured on nHAC/PLA
To investigate the effect of rhBMP-2 on protein synthesis of DPSCs seeded on nHAC/PLA, the constructs were cultured in growth medium with/without 100 ng/mL rhBMP-2 for 7, 14, 21, and 28 days. The results showed that protein synthesis of the both groups reached the highest at day 21, and rhBMP-2 significantly increased the protein synthesis of DPSCs seeded on nHAC/PLA at days 21 and 28 (Fig. 6A).

Effect of rhBMP-2 on the protein synthesis
To investigate the effect of rhBMP-2 on ALP activity of DPSCs seeded on nHAC/PLA, the constructs were cultured in growth medium with/without 100 ng/mL rhBMP-2 for 7, 14, 21, and 28 days. The ALP activity of the both groups reached the highest at day 21. The ALP activity of DPSCs cultured on nHAC/PLA with rhBMP-2 was significantly higher than that of DPSCs cultured on nHAC/PLA without rhBMP-2 at each culture time point (Fig. 6B).
To investigate the osteoblast function of DPSCs cultured on nHAC/PLA in growth medium with/without 100 ng/mL rhBMP-2, OCN, a marker of osteoblast function, was determined from culture supernatant at day 28. The rhBMP-2 significantly increased OCN content of DPSCs cultured on nHAC/PLA (Fig. 6C).
Alizarin red staining was used to quantify calcium phosphate mineral formation of DPSCs+nHAC/PLA constructs after 28 days of culture. The mineral formation in DPSCs+nHAC/PLA construct with rhBMP-2 was significantly higher than that in DPSCs+nHAC/PLA construct without rhBMP-2 (Fig. 6D).
Figure 6 shows that the rhBMP-2 could significantly increase protein content, ALP activity/protein, OCN content, and mineral formation of DPSCs cultured on nHAC/PLA.
The capacity of the combination of rhBMP-2, DPSCs, and nHAC/PLA to reconstruct critical-size alveolar bone defects in New Zealand rabbit
At 12 weeks, animals were killed, and the mandibles were harvested for X-ray, histological analyses, and CLSM. Figure 7 showed soft X-ray photographs of the alveolar bone defects at 0 and 12 weeks after surgery. Figure 7A showed an alveolar bone defect with nothing at 0 day ex vivo. It suggested that the incisor of the rabbit was not damaged. Figure 7B showed X-ray photograph of the alveolar bone defect with nothing at 0 day after surgery in vivo. When nHAC/PLA was implanted, the X-ray graph of the defect showed that nHAC/PLA had a weak diffraction (Fig. 7C). Figure 7D showed X-ray photograph of the alveolar bone defect with nothing at 0 day after surgery in vivo, but when AB implanted, the X-ray graph of the defect showed that AB had a strong diffraction (Fig. 7E). Compared to the X-ray graph of the defect with nothing just at 0 day after surgery (Fig. 7B, D), no bone formation was observed in the control group at 12 weeks after surgery, radiolucent lines were clearly visible between defect region and host bone (Fig. 7F). The radiopaque area of defect region gradually increased in nHAC/PLA, nHAC/PLA+rhBMP-2, nHAC/PLA+DPSCs, nHAC/PLA+DPSCs+rhBMP-2, and AB group, and bony unions were observed at the junction sites between each implant and host bone. Compared to control group (Fig. 7F), the X-ray graph at 12 weeks after implantation showed the increase of radiopaque area in the alveolar bone defect applied with nHAC/PLA and nHAC/PLA+rhBMP-2 (Fig. 7G, H), but the extent of radiopaque area for nHAC/PLA+DPSCs (Fig. 7I) was larger than that for nHAC/PLA or nHAC/PLA+rhBMP-2. Figure 7J and K separately showed the X-ray graphs of the defects with nHAC/PLA+DPSCs+rhBMP-2 and AB. No significant differences were detected between both groups according to the extents of radiopaque area of the defects, and they had the similar radiographic appearance as the rabbit mandible without surgery (Fig. 7L). Further, the density of X-ray graph of the defects with nHAC/PLA+DPSCs+rhBMP-2 or AB, compared to the right alveolar bone without surgery, respectively, had no significant difference either. However, the density of X-ray graph of the defects with nothing, nHAC/PLA, nHAC/PLA+rhBMP-2, or nHAC/PLA+DPSCs was significantly lower than that of the right alveolar bone.
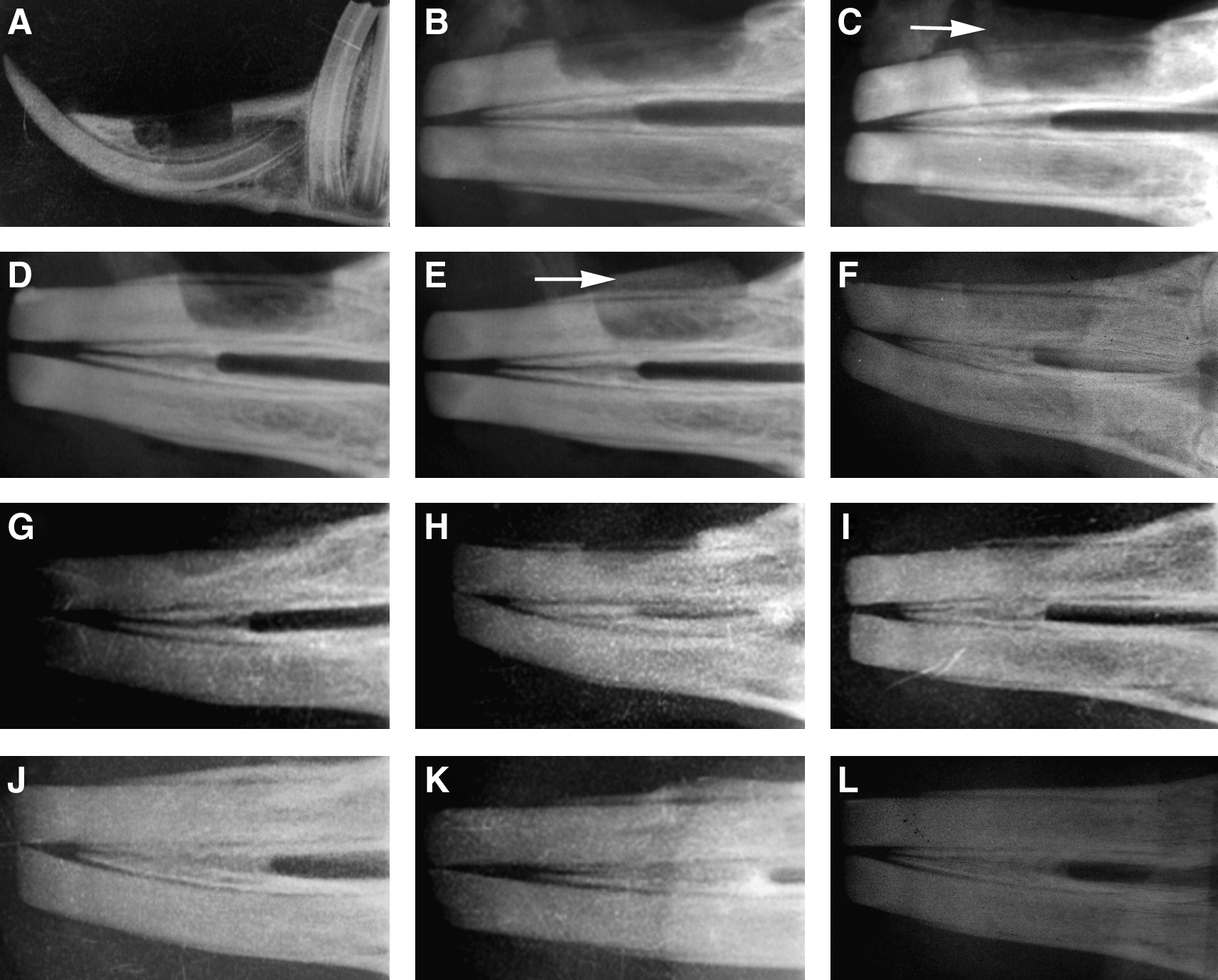
Radiographic appearance of alveolar bone defects at 0 and 12 weeks after application with the different grafts.
Histological observations with Goldner's trichrome staining demonstrated bone regeneration in the 10-mm segmental defect model of rabbit at 12 weeks after surgery (Fig. 8). Histological staining showed that no bone regeneration was detected at control group, whereas remarkable ingrowth of muscle fibers and soft connective tissue into the defect was observed (Fig. 8A). It was found that the alveolar bone defects in nHAC/PLA and nHAC/PLA+rhBMP-2 group were filled with abundant red-stained osteoid and a few green-stained newly formed bones; a great deal of osetoclasts were observed in newly formed osteoid (Fig. 8B, C). The defects in nHAC/PLA+DPSCs, however, were filled with a large amount of green-stained newly formed trabeculae and red-stained osteoid. Osteoblastic cells were lining the surface of newly formed bone (Fig. 8D). The maximal and robust bone formation was presented in the defects in nHAC/PLA+DPSCs+rhBMP-2. In some areas, nHAC/PLA+DPSCs+rhBMP-2 group showed that considerable numbers of osteoblasts and osteocytes in the newly formed thickened bone were seen (Fig. 8E), in low magnification, the quality of the bone was thicker and a majority of recovery was observed at 12 weeks after operation. The predominant orientation of the newly formed trabecular bone was parallel and regular. The local and abundant thickened new trabecular bone formed around the site of nHAC/PLA. When implanted composite decreased with the continuous biodegradation, it was replaced by the increased quantity of newly forming bone matrix. The new bone forming from the cortical bone extended to the central zone (Fig. 8G), but the defect site has not been fully closed yet as the natural rabbit alveolar bone without surgery (Fig. 8H). In AB group, there were areas of new bony regeneration as well as areas of autogenous grafted bone visible; newly formed bone was at different maturation stages exhibiting trabecular arrangement with nodular disposition involving predominantly adipose medullary tissue, sometimes characterized by a highly vascularized fibroadipose structure. The newly formed bone tissue was either a characteristic of mature bone, or highly cellularized involving randomly arranged osteocytes with osteoid seams lined by a continuous layer of osteoblasts surrounding the tissue in formation, regularly disposed amid osteoclasts. This organization suggests the continuing formation and remodeling of the original tissue, which characterizes fully active bone tissue. No inflammatory cells were observed in this group (Fig. 8F).

Histological sections stained by Goldner's trichrome of the alveolar bone defects at 12 weeks after implantation.
Figure 9 showed histological sections of alveolar bone defects and at 12 weeks after implantation by toluidine blue staining. Histological staining showed that no bone regeneration was detected at the alveolar bone control group, whereas remarkable ingrowth of muscle fibers and soft connective tissue into the defect was observed (Fig. 9A). Some cells had migrated into nHAC/PLA and nHAC/PLA+rhBMP-2 implants. They attached to the walls of the inner pores and spread in these scaffolds. Abundant dark blue-stained osteoid and a few light blue-stained mature bones had formed with some osteoblasts distributing along their edges in both groups (Fig. 9B, C). A large amount of light blue-stained trabeculae containing spindle mature osetocytes could be seen in nHAC/PLA+DPSCs group; the porous scaffolds were partly degraded and the residual materials were surrounded by areas of active bone formation (Fig. 9D). However, the implantation of nHAC/PLA+DPSCs+rhBMP-2 resulted in an extensive amount of light blue-stained trabeculae and partial bone union; the newly formed bones in treated defects were normal in histological appearance with osteocytes contained in lacunae (Fig. 9E). The gaps at the host bone–biomaterial interface were also observed in all treated defects. There were no visible fibrous connective tissues in the gaps, and new trabeculae had grown into the scaffold from the interface. The inflammatory infiltrate and tissue reaction had disappeared at 12 weeks. In AB group, there were areas of new bony regeneration as well as areas of autogenous grafted bone visible, newly formed bone exhibiting large medullar spaces, and irregular and thick bone trabeculae (Fig. 9F). Sometimes these trabeculae presented different maturation stages; active osteoblasts and osteoid tissue were observed on the surface of the preexisting or newly formed bone, indicating that the bone formation process was continued. No inflammatory cells were observed.
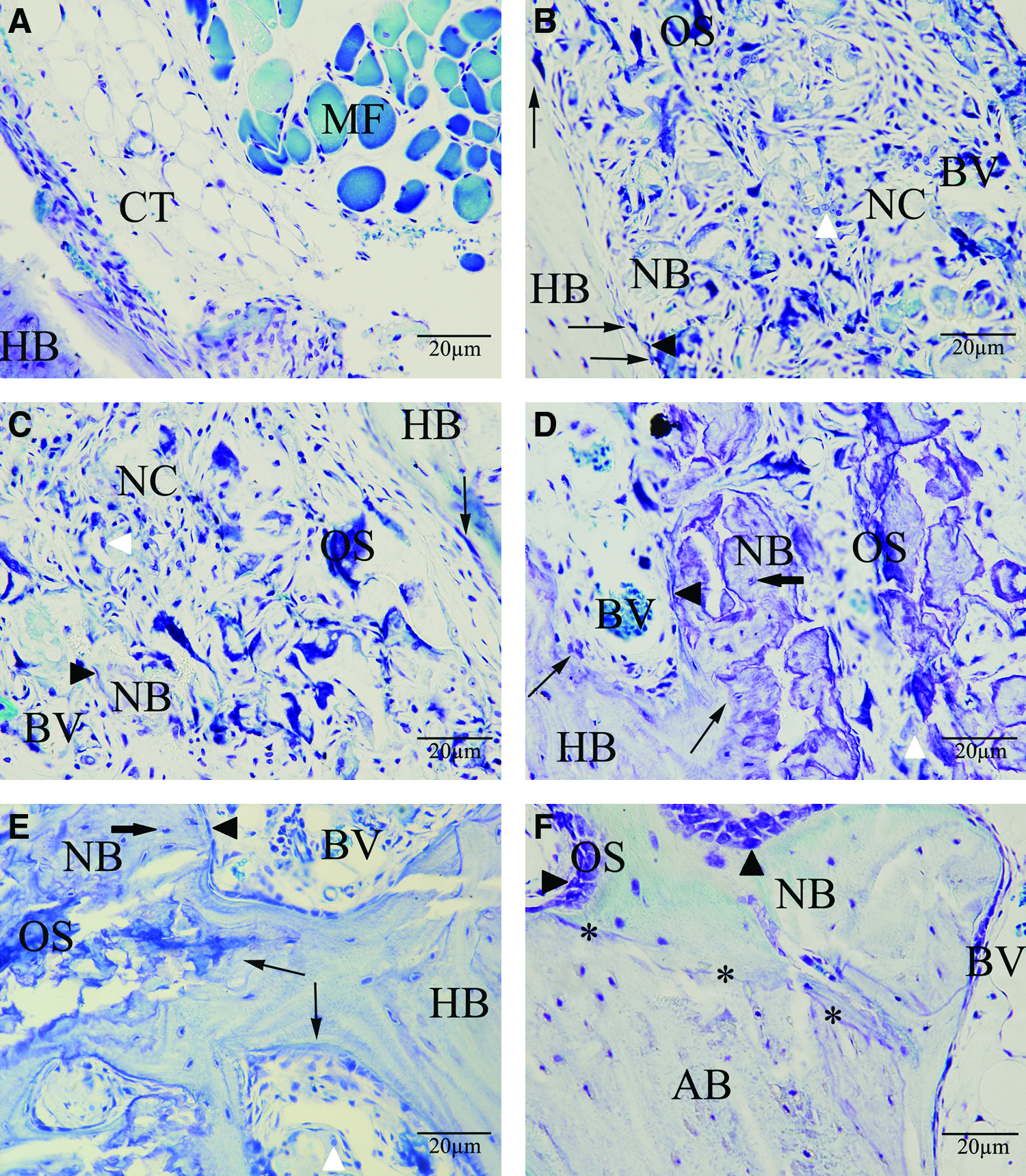
Histological sections stained by toluidine blue of the alveolar bone defects at 12 weeks after implantation.
The results of the histomorphometric analysis are summarized in Figure 11A quantified through goldner's trichrome staining. All slides were analyzed by two independent observers to identify the type of tissue (mature bone-like and osteoid-like). The extent of newly formed mature bone and osteoid was indicated by the percentage of total bone formation area within the section. The results showed that no bone regeneration was detected at the control group. The percentages of osteoid formation area for nHAC/PLA, nHAC/PLA+rhBMP-2, nHAC/PLA+DPSCs, nHAC/PLA+DPSCs+rhBMP-2, and AB group were, respectively, 18.59±1.88, 19.02±1.40, 11.73±1.83, 12.97±1.42, and 14.01±2.11. Histomorphometric analysis showed that nHAC/PLA+rhBMP-2 had the greatest percentage of osteoid formation area in five implanted groups; the values for nHAC/PLA and nHAC/PLA+rhBMP-2 were insignificant difference, but were significantly higher than those for other three implanted groups. Although this value for AB group was higher than that for nHAC/PLA+DPSCs or even nHAC/PLA+DPSCs+rhBMP-2, the difference was insignificant. The percentages of new mature bone formation area for nHAC/PLA, nHAC/PLA+rhBMP-2, nHAC/PLA+DPSCs, nHAC/PLA+DPSCs+rhBMP-2, and AB group were, respectively, 4.26±1.42, 4.31±0.74, 24.28±1.93, 48.19±2.66, and 44.27±3.25. Histomorphometric analysis proved that the value for nHAC/PLA was the lowest in five implanted groups, but was insignificant difference compared with nHAC/PLA+rhBMP-2. Further, these values for these two groups were significantly lower than that for nHAC/PLA+DPSCs. The maximal and robust new bone formation was presented in nHAC/PLA+DPSCs+rhBMP-2. However, there were no significant differences between nHAC/PLA+DPSCs+rhBMP-2 and AB group, but the values for the both groups were significantly higher than that for nHAC/PLA+DPSCs group. The percentages of total bone formation area for nHAC/PLA, nHAC/PLA+rhBMP-2, nHAC/PLA+DPSCs, nHAC/PLA+DPSCs+rhBMP-2, and AB group were, respectively, 22.86±0.55, 23.33±0.87, 35.95±2.53, 61.16±2.18, and 58.28±4.88. Histomorphometric analysis proved that the nHAC/PLA group had the lowest percentage of total bone formation area in five implanted groups, but there were no significant differences between nHAC/PLA and nHAC/PLA+rhBMP-2 group. The value for nHAC/PLA+DPSCs was significantly higher than that for nHAC/PLA or nHAC/PLA+rhBMP-2 group, but was significantly lower than that for nHAC/PLA+DPSCs+rhBMP-2 or AB group. The maximal percentage of total bone formation area was presented in nHAC/PLA+DPSCs+rhBMP-2. However, there were no significant differences between nHAC/PLA+DPSCs+rhBMP-2 and AB group.
In the specimens without demineralization, no calcein and tetracycline fluorescence was defected in control group (Fig. 10A); a little calcein and tetracycline fluorescence was defected in nHAC/PLA (Fig. 10B) and nHAC/PLA+rhBMP-2 (Fig. 10C). In contrast, abundant calcein and tetracycline fluorescence was displayed in nHAC/PLA+DPSCs (Fig. 10D), nHAC/PLA+DPSCs+rhBMP-2 (Fig. 10E), and AB group (Fig. 10F). To identify if the foreign GFP-labeled DPSCs engrafted at the transplanted bone defect sites had differentiated into osteoblasts, the GFP-labeled DPSCs were observed by fluorescence microscopy. Some GFP-positive cells were showed in green (Fig. 10D, E), which indicated that the GFP-labeled cells derived from autologous ex vivo-expanded DPSCs had differentiated into osteoblasts in vivo. Figure 11B showed bone mineral apposition rate during 10 days in vivo. The results revealed that bone mineral apposition rate for nHAC/PLA (1.31±0.078) and nHAC/PLA+rhBMP-2 (1.35±0.099) was insignificant difference, but was significantly lower than that for nHAC/PLA+DPSCs (1.77±0.11). The largest bone mineral apposition rate was presented in nHAC/PLA+DPSCs+rhBMP-2 (2.52±0.33), and it was an insignificant difference compared with the AB group (2.47±0.25), but these values for these two groups were significantly higher than that for nHAC/PLA+DPSCs.
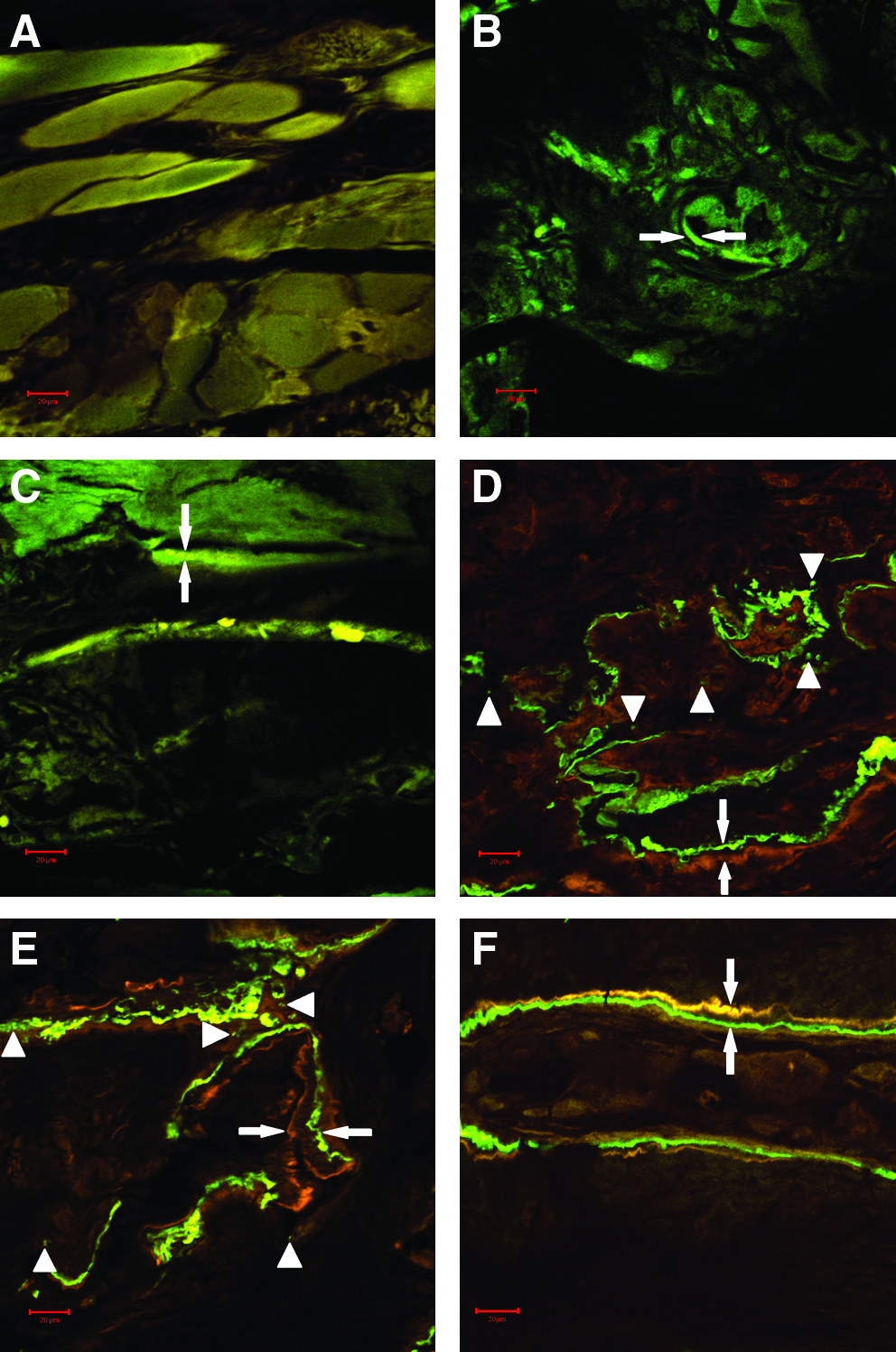
Tissue labeling and the tracing of foreign GFP-labeled DPSCs at 12 weeks after implantation.

Bar graph demonstrating the percentages of osteoid formation, mature bone formation, total bone formation
Discussion
Harvesting bone for autologous grafting is a daily problem encountered by craniofacial and oral surgeons. However, tissue engineering offers a promising new approach for periodontal bone repair, but there was still a lack of objective scientific data regarding the beneficial effects about using tissue-engineered bone complex for reconstruction of alveolar bone. In this study, we demonstrated that tissue-engineered bone with rhBMP-2 mediated autologous DPSCs and a biodegradable nHAC/PLA scaffold achieved an earlier mineralization and more bone formation when compared with nHAC/PLA, nHAC/PLA+rhBMP-2, and nHAC/PLA+DPSCs, or even autologous bone, which may facilitate to ensure the reconstruction of alveolar bone defect.
It is important to note that DPSCs have several advantages when compared with mesenchymal stem cells (MSC) derived from other sources; the method for their isolation is not invasive and they can be rapidly expanded in vitro for several potential clinical applications.16,21–23 We have been able to isolate the DPSCs from rabbit dental pulp through limiting dilution. The results have demonstrated that rabbit DPSCs are highly clonogenic, showing a high proliferation potential, and are able to self-maintain for long time. Previous experiments have demonstrated that stem cells, isolated from the pulp of human exfoliated deciduous teeth and expanded in vitro, showed 9% positivity for STRO-1, considered an early marker of MSCs.33–35 In our study, in addition to vimentin and keratin, we have challenged stem cells with the antibody STRO-1, and we have demonstrated that positivity for STRO-1 and vimentin and negativity for keratin were presented in DPSCs. For these reasons, we have associated STRO-1, vimentin, and keratin expression to isolate a population of MSCs. Our study has provided evidence that the DPSCs cultured in conditioned media represent an approachable niche of stem cells able to extensively proliferate and differentiate into several cytotypes, mainly osteoblasts, expressing osteogenic markers OCN, BSP, COLI, and ALP and forming mineralized nodules in vitro. Our single-colony-derived cells were still capable to differentiate in other cell lineages, such as odontoblasts and adipocytes, as shown in this study, confirming their pluripotent stem origin. Therefore, this cell population constitutes a large, ideal source of osteoblasts already suitable for bone regeneration, autotransplantation, and tissue-based clinical therapies in humans.
The nHAC/PLA we selected has good biocompatibility and osteoconductive capacity.11–14 SEM results show that the DPSCs attached and spread well, and retained their osteogenic phenotype on nHAC/PLA in the culture containing rhBMP-2. No major adverse biologic reactions were found between DPSCs and the composite graft compared to the chamber slide. These findings suggested that nHAC/PLA provided a suitable environment for DPSCs to migrate, proliferate, and differentiate, which indicated that the composite graft was a potential scaffold for bone tissue engineering.
Osteogenic differentiation of DPSCs seeded on nHAC/PLA is another key issue determining the success in bone and periodontal regeneration via a tissue-engineering approach. Of all classified cytokines, BMPs are recognized to possess the greatest in vivo bone stimulatory capacity and are the only growth factors known to stimulate MSC to differentiate along osteoblastic and chondrogenic lineages.36,37 As a heterogeneous cell population with multidifferentiation potential, the in vitro lineage-directed induction is necessary to direct committed differentiation of DPSCs seeded on nHAC/PLA before implantation. In this study, we demonstrated that rhBMP-2 could significantly increase protein content, ALP activity, OCN content, and mineral formation of DPSCs cultured on nHAC/PLA. It indicated that rhBMP-2 could provide an appropriate in vitro osteoinductive environment for DPSCs cultured on nHAC/PLA.
To evaluate the capacity of tissue-engineered bone complex to reconstruct critical-size alveolar bone defects, we implanted nHAC/PLA and nHAC/PLA/DPSCs incubated in growth media with/without 100 ng/mL rhBMP-2 in vivo for 7 days and AB into alveolar bone defect in rabbit. By definition, if the original defect is critical size, it will not spontaneously heal31,32; for this size defect, after 12 weeks of implantation, histological results showed that the control group had no bone formation. The nHAC/PLA group had abundant engineered osteoid and a few engineered mature bones formed. Large numbers of blood vessels, osteocalsts, and some osteoblasts were also observed in this group. These results indicated that bone regeneration was dependent on the matrix implanted. The nHAC/PLA were highly biocompatible and osteoconductive, and could be used as a potential scaffold for the alveolar bone regeneration.
Analogous to nHAC/PLA group, nHAC/PLA+rhBMP-2 group also had abundant engineered osteoid and a few engineered mature bones, large numbers of blood vessels, osteoclasts, and some osteoblasts. Histomorphometric analysis showed that the newly formed total or mature bone areas observed in the groups of nHAC/PLA and nHAC/PLA+rhBMP-2 was insignificant difference. The reason might be the lower rhBMP-2 concentration (100 ng/mL) in media. The nHAC/PLA might adsorb rhBMP-2 from the media, but the minute amounts of rhBMP-2 left in the scaffold at the time of implantation would hardly account for the extensive ossification capacity of the construct. 38
However, the X-ray graph, histological results, and polychrome fluorescent labeling observation demonstrated that the implantation of nHAC/PLA alone or nHAC/PLA+rhBMP-2 resulted in slow, incomplete healing, as the osteogenic response was too low, although nHAC/PLA was able to instruct the in vivo environment to form abundant engineered osteoid. The few cells that migrated to the defect site could not create enough ECM for adequate healing. To promote an enhanced osteogenic response, our findings demonstrated that cells and/or osteogenic growth factors must be included with the implanted scaffold. When we implanted nHAC/PLA with un-induced DPSCs in vitro in the alveolar bone defect, histological results showed that abundant engineered osteoid and more engineered mature bone were formed in the implant, and histomorphometric analysis and mineral apposition rate demonstrated that nHAC/PLA+DPSCs group had significantly higher and faster bone formation than that for nHAC/PLA or nHAC/PLA+rhBMP-2 groups. There may be three reasons for this: (1) The structure and the composition of nHAC/PLA really had effect on osteogenic differentiation of DPSCs. (2) The important in vivo bone defect environmental factors played a key role in osteogenic differentiation of DPSCs. (3) The transplanted DPSCs sharing similar tissue origin with the mandibular bone cells themselves could also secrete various cytokines that may stimulate the function of local progenitor cells. In a word, it is likely that the factors released and osteoprogenitor cells recruited from alveolar bone defect environment, and proteins secreted by the implanted DPSCs, and the structure and the component of nHAC/PLA may function together to drive more bone regeneration. Meanwhile, these results also indicated that embryologically derived from the neural crest cell DPSCs share similar tissue origin with the mandibular bone cells and, therefore, could serve as a potential cell source for the regeneration of alveolar bone defects.
Further, when we implanted nHAC/PLA with induced DPSCs in vitro by ostogenic growth factor, the nHAC/PLA+DPSCs+rhBMP-2 tissue-engineered bone complex had an earlier mineralization and more bone formation inside the scaffold than nHAC/PLA, nHAC/PLA+rhBMP-2, or nHAC/PLA+DPSCs. The results indicated that bone repair was significantly enhanced when DPSCs cultured on nHAC/PLA were induced by exogenous rhBMP-2 in vitro. Several studies have also demonstrated, even in apparently ideal graft conditions, that bone repair is significantly enhanced when BMP delivery is supplemented with osteogenic cell populations, suggesting that the local stem cell niche is a limiting factor,39–42 and the implanted scaffold with adult stem cells induced by osteogenic growth factors could create enough ECM for adequate healing.
Meanwhile, the X-ray graph of the defects also showed that bony unions were observed at the junction sites between the above mentioned implants and host bone except for control group. Histological results showed that the gaps at the host bone–biomaterial interface were also observed in all treated defects. There were no visible fibrous connective tissues in the gaps, and new trabeculae had grown into the scaffold from the interface. These results suggested that nHAC/PLA could very well integrate into host alveolar bone and could be used as a potential scaffold for the alveolar bone regeneration.
To discern nHAC/PLA+DPSCs+rhBMP-2 tissue-engineered bone complex tested is a valid alternative technique for the reconstruction of periodontal bone defects, a positive control gold-standard fresh autogenous iliac bone graft was also tested in this study. The X-ray graph, histological results, and polychrome fluorescent labeling observation demonstrated that the approach presented here showed similar, even better, results than the gold standard method. To our knowledge, the newly developed nHAC/PLA scaffold with rhBMP-2-mediated DPSCs still has not been studied in an animal alveolar bone defect model. However, in this study, the excellent repair results of 1.0-cm segmental defect in a rabbit alveolar bone have been demonstrated as implanted in this composite. The results indicate that the nHAC/PLA+DPSCs+rhBMP-2 composite is desirable for dental, craniofacial, and orthopedic repairs, especially where shaping and contouring for esthetics are needed.
Moreover, we also observed that alveolar bone defect with DPSCs+nHAC/PLA+rhBMP-2 has not been fully closed yet as the normal alveolar bone in rabbits without surgery by histological staining after 12 week of implantation. The normal anatomy of bone is a result of evolutionary functional optimization via structural adaptation. When we describe various tissue engineering strategies to regenerate bone, it will be productive to frequently return to and review this anatomy. We can consider the contributions of each of these natural components to the function of bone as we attempt to discover acceptable substitutes for those functions. Thereby, for effective tissue regeneration, it is important to develop optimized biologic composite grafts culture and implantation conditions based upon the influence of the native tissue microenvironments. The further study is to investigate the significance of multiple osteogenic factors combined based upon the influence of the native bone tissue microenvironments to obtain the most optimizing in vitro osteoinductive microenvironments, and help identify suitable carriers to induce transplanted DPSCs and better integrate them into the surrounding environment to improve tissue engineering-mediated alveolar bone tissue regeneration.
In this study, to investigate DPSC-mediated bone formation in vivo, we labeled DPSCs with GFP and used DPSCs to repair critical-size alveolar bone defects over the incisor in rabbit. At 12 weeks after transplantation, green fluorescence signals were detected by fluorescence microscopy within newly formed bone. The results confirmed that GFP-labeled DPSCs had differentiated directly into new bone. These findings suggested that DPSCs were implanted to segmental defect and contributed to new bone regeneration in the restoration of the rabbit alveolar bone defect.
Conclusion
Our in vitro and in vivo experiments have proved evidence: (1) embryologically derived from the neural crest cell, DPSCs sharing similar tissue origin with the mandibular bone cells, could serve as a potential cell source for the regeneration of alveolar bone defects. (2) The rhBMP-2 could promote osteogenic capability of DPSCs cultured on nHAC/PLA. (3) The nHAC/PLA supported DPSCs proliferation and differentiation, and could well integrate into host alveolar bone, and could be used as a potential scaffold for the alveolar bone regeneration. In conclusion, the nHAC/PLA+DPSCs+rhBMP-2 might be a better alternative to autologous bone for the clinical reconstruction of periodontal bone defects.
Footnotes
Acknowledgment
The authors thank the staff and faculty of Institute of Stomatology, Chinese People Liberation Army General Hospital.
Disclosure Statement
No competing financial interests exist.