
Abstract
Facial paralysis is a physically, psychologically, and socially disabling condition. Innovative treatment strategies based on regenerative medicine, in particular tissue engineering of skeletal muscle, are promising for treatment of patients with facial paralysis. The natural source for tissue-engineered muscle would be muscle stem cells, that is, human satellite cells (SC). In vivo, SC respond to hypoxic, ischemic muscle damage by activation, proliferation, differentiation to myotubes, and maturation to muscle fibers, while maintaining their reserve pool of SC. Therefore, our hypothesis is that hypoxia improves proliferation and differentiation of SC. During tissue engineering, a three-dimensional construct, or implanting SC in vivo, SC will encounter hypoxic environments. Thus, we set out to test our hypothesis on SC in vitro. During the first five passages, hypoxically cultured SC proliferated faster than their counterparts under normoxia. Moreover, also at higher passages, a switch from normoxia to hypoxia enhanced proliferation of SC. Hypoxia did not affect the expression of SC markers desmin and NCAM. However, the average surface expression per cell of NCAM was downregulated by hypoxia, and it also downregulated the gene expression of NCAM. The gene expression of the myogenic transcription factors PAX7, MYF5, and MYOD was upregulated by hypoxia. Moreover, gene expression of structural proteins α-sarcomeric actin, and myosins MYL1 and MYL3 was upregulated by hypoxia during differentiation. This indicates that hypoxia promotes a promyogenic shift in SC. Finally, Pax7 expression was not influenced by hypoxia and maintained in a subset of mononucleated cells, whereas these cells were devoid of structural muscle proteins. This suggests that during myogenesis in vitro, at least part of the SC adopt a quiescent, that is, reserve cells, phenotype. In conclusion, tissue engineering under hypoxic conditions would seem favorable in terms of myogenic proliferation, while maintaining the quiescent SC pool.
Introduction
To accomplish this, a three-dimensional construct should be engineered consisting of functional skeletal muscle fibers, vasculature, and innervation. Combining these premises to a clinically functional construct remains a major challenge. For vascularization, four main techniques are currently the subject of investigation: First, through in vivo implantation of a preformed three-dimensional muscle construct around a vascular pedicle. 6 Second, through implantation of myoblasts in a prevascularized fibrin gel. 7 Third, through stacking sheets of myoblasts on a vascular bed, the so-called cell sheet method.8,9 Finally, through coculturing myoblasts with endothelial cells. 10 To innervate a muscle construct, it is necessary to create a functional neuromuscular junction. This could be reached through coculturing myoblasts and neural tissue, 11 and by means of electrical stimulation. 12
Muscle tissue has its own endogenous repair and maintenance system that is based on myogenic progenitor cells, that is, satellite cells. The regenerative capacities and myogenic characteristics of these satellite cells has been the topic of many studies that aim at using satellite cells to tissue engineer muscle.13–15 In response to hypoxic ischemia in skeletal muscle, for example, after exercise or injury, satellite cells are activated. These activated satellite cells start to proliferate and differentiate, which contributes to regeneration of the affected muscle.16–18
In tissue-engineering functional skeletal muscle, angiogenesis and neurogenesis are imperative. However, vascularization of a tissue-engineered muscle construct may not be instantly functional after implantation in vivo. Thus, satellite cells in the center of the muscle construct will encounter hypoxia. Therefore, in this study, we focus on the influence of hypoxia on myogenesis in vitro.
In general, hypoxia is a crucial limiting factor in tissue engineering of constructs that exceed approximately 0.2 mm thickness. In these constructs, a steep oxygen concentration gradient occurs, with hypoxic levels inside that may result in cell death after implantation. 19 In vivo, skeletal muscle is shown to adapt to a prolonged hypoxic environment through upregulating myostatin, causing atrophy. 20 In mesenchymal stem cells, however, hypoxic preconditioning in vitro had profound benefits for cell survival after implantation. 21 Bovine satellite cells were shown to both proliferate and differentiate more efficiently under hypoxia. 22 However, species differences may exist, and the influence of hypoxia on human satellite cells has not yet been fully investigated. The importance of in vitro research shows from a recent clinical trial where myoblasts, that is, satellite cells, were transplanted into a hypoxic environment of the ischemic heart of patients suffering from heart failure. However, echocardiographic heart function did not improve and arrhythmia occurred, indicating cellular dysfunction. 23 In tissue engineering, as in cell therapy, satellite cells will encounter hypoxic environments. Therefore, for clinical application of human satellite cells, a better understanding of the response of human satellite cells to changes in oxygen level is required. In tissue-engineering facial muscles, although they are thinner than other skeletal muscles, their thickness exceeds 0.2 mm. Hence, satellite cells encounter lower oxygen concentrations. Therefore, before applying these muscle constructs in vivo, we need to study how satellite cells react to a hypoxic environment. Since satellite cells in vivo appear to respond to hypoxic muscle damage with activation, proliferation, differentiation, and maturation, while maintaining their reserve pool of satellite cells, our hypothesis is that in vitro hypoxia improves proliferation and differentiation of human satellite cells.
Materials and Methods
Satellite cell isolation
Muscle biopsies were obtained from eight healthy human donors, 30 to 66 years of age, undergoing reconstructive surgery. The age of the donors was 51.7±10.6 years, and there were three men and five women included. The study protocol was approved by our institutional ethics committee, and patients gave their informed consent. A muscle biopsy of either the latissimus dorsi (three donors) or the orbicularis oculi muscle (five donors) was collected in cold (4°C) phosphate buffered saline (PBS) supplemented with 10% penicillin/streptomycin 50ug/mL (Sigma-Aldrich). Specimens were stripped of any visible connective tissue and fat, weighted, and minced. The muscle specimen weight ranged between 200 and 800 mg. Enzymatic digestion was performed in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen/Gibco) containing 0.04 mg/mL (0.16 collagenase wünsch units/mL) liberase blendzyme 3 (Roche Applied Science) at 37°C for 1 h. The suspension was filtered through a 70 μm filter; 10% fetal bovine serum (FBS; Invitrogen/Gibco) was added to the flow trough and centrifuged at 300×g for 10 min at 4°C, after which the supernatant was discarded. Lysis-buffer was used to eliminate erythrocytes. After 2 min on ice, the suspension was centrifuged again at 300×g for 10 min at 4°C, after which the supernatant was discarded and the pellet was gently resuspended in proliferating medium (PM), containing DMEM, 20% FBS, and 1% penicillin/streptomycin.
Undigested tissue was subjected to another 2–3 cycles of digestion process. The different cell suspensions were pooled, and viable cells were counted using the trypan blue exclusion method with a Bürker counting chamber.
Satellite cell culture
Immediately after isolation, the cell suspension was divided into two fractions and cultured either under normoxia (21% O2) or hypoxia (2% O2) at 37°C. Cells were plated at 5.0×103cells/cm2 on culture flasks precoated with 1% gelatine. Medium was refreshed thrice per week.
When cells reached 70% confluence, they were enzymatically harvested using accutase (Invitrogen), counted with a Coulter counter, and replated at 5.0×103cells/cm2 on 1% gelatine precoated flasks.
Myogenic differentiation of satellite cells was induced at 100% confluence by switching to differentiation medium (DM), containing DMEM, 2% FBS, 1% penicillin/streptomycin, 1% insulin-transferrin-selenium-A (100×) (Invitrogen), and 0.4 μg/mL dexamethason (Sigma-Aldrich).
Passage number (Px) was defined as the xth sequential harvest of a subconfluent cell population. All differentiation experiments were performed using P6–P15.
The population doubling time (PDT) was calculated as N(t)=C(2)^t/d, N(t)=the number of cells at time t; d=doubling period, C=initial number of cells, t=time (days).
A CyQUANT cell proliferation assay (Invitrogen) was performed in accordance to the manufacturer's protocol to determine proliferation.
Immunofluorescent staining
Cells were cultured in PM on Thermanox® coverslips (NUNC Brand Products) coated with 1% gelatine in a 24-well plate. At 100% confluence, cells were washed twice with PBS and either cultured an additional 5 days in DM or fixed in 2% paraformaldehyde at room temperature for 10 min. Cells were dried under a ventilator and stored −20°C. After thawing, a permeabilization step was performed with 0.5% Triton X-100 (Sigma-Aldrich) in PBS at room temperature for 10 min. Nonspecific-binding sites were blocked with 10% goat serum in PBS for 30 min. Cells were incubated with the primary antibody in PBS and 2% goat serum at room temperature for 60 min. The primary antibody consists of either (1) a myogenic marker, rabbit-antihuman desmin (1:100) (Novus Biological), (2) a proliferation marker, mouse-antihuman Phosphohistone H3 (1:500) (clone 3H10; Millipore International Inc.), (3) a satellite cell marker, mouse-antihuman NCAM (1:200) (clone MEM-188; Biolegend), (4) a fibroblast marker, mouse antihuman MCA1399G (1:100) (AbD Serotec), (5) an adult muscle marker, mouse-antihuman myosin (MF20; 1:500), (6) a satellite cell marker, and mouse-antihuman Pax7 (1:10) (both Developmental Studies Hybridoma Bank). After three washes with 0.05% Tween in PBS, the cells were incubated with a secondary antibody-cocktail at room temperature for 30 min. The secondary antibody-cocktail was constituted of fluorescein isothiocyanate-conjungated goat-antirabbit IgG (Southern Biotech), Texas Red
Further, for flow cytometry, approximately 5×105 cells were resuspended in 100 mL fluorescence activated cell sorting (FACS) buffer consisting of PBS (four salts), 2 mM EDTA, and 0.5% (v/v) FBS. Cells were incubated with 5 μL mouse monoclonal fluorescein isothiocyanate-conjugated antibody antiNCAM (Biolegend) at 4°C for 30 min. After two washes with FACS buffer, cells were resuspended in 300 μL FACS buffer and examined by FACS analysis on an FACS Caliber system (BD Biosciences). The analysis was performed using WinList
Gene transcript analysis
At days 0 and 5, after switching to DM, total RNA was isolated from ∼200,000 cells using the Rneasy kit (Qiagen Inc.), in accordance to the manufacturer's protocol. Briefly, a lysate was made and diluted with an equal volume of ethanol (70%). RNA was collected on an RNA-binding filter by centrifugation. DNase treatment was performed by incubation with a DNase I solution at 37°C for 15 min. The RNA-binding filter was washed twice, and, subsequently, the RNA was eluted with 14 μL Elution Buffer. The RNA concentration and purity were determined by spectrophotometry (NanoDrop Technologies). For reverse transcription–polymerase chain reaction (RT-PCR) analysis, total RNA was reverse transcribed using the First Strand cDNA synthesis kit (Fermentas UAB). In summary, 1 μg of total RNA was diluted in a final reaction volume of 20 μL containing random hexamer primer (0.5 μg), RiboLock™ ribonuclease inhibitor (20 U), 1 mM dNTP mix, and incubated at 37°C for 1 h. The reverse transcription reaction was terminated by heating the mixture to 70°C for 10 min, after which the samples were placed on ice. RT-PCR was performed in a final reaction volume of 25 μL, consisting of 10× Taq polymerase buffer, 0.25 mM dNTP mix, 1.5 mM MgCl2, 1 μL primer-mix (Table 1), and 1U Taq DNA Polymerase. PCR reactions were performed at 94°C for 45 s, 57°C for 45 s, and 72°C for 1 min, for 35 cycles. Amplimers were separated in a 2% agarose gel and stained with 0.5 μg/mL ethidium bromide after electrophoresis.
ACTA1, α-sarcomeric actin; DES, desmin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Quantitative RT-PCR analysis was performed in a final reaction volume of 10 μL, consisting of 4.5 μL SYBR Green Supermix (Bio-Rad), 0.5 μL primer-mix (0.5 mM) (Table 2), and 5 μL cDNA (1 ng/μL). Reactions were performed at 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s, for 40 cycles. Analysis of the data was performed using science detection software 2.2.2.
Statistics
All data are represented as means±SEM and were analyzed by Student's t test or analysis of variance (ANOVA) using Graph-Pad prism version 5 (GraphPad Software, Inc.).
Results
Proliferation of cells is initially increased under hypoxia
Cells isolated from enzymatically dissociated muscle tissue were divided into two fractions, one of which was cultured under normoxia (21% O2), whereas the other fraction was cultured under hypoxia (2% O2) during all passages. On average, 1.25±0.25×105 viable cells were isolated per gram of muscle tissue. We observed that the morphology of adherent cells was independent of the oxygen level. During low passages (P0–P5), cultures under both oxygen conditions contained both spindle-shaped and typical triangular “satellite-like” cells. During these low passages, the cells reached confluency twice as fast under hypoxia than under normoxia. The PDT at passage 0–5, determined by cell count, was 8.3 days for normoxic and 4.3 for hypoxic cultures (p=0.04, Fig. 1A).

Morphology and proliferation. During passages (P) 0–5, adhered cells had a heterogeneous morphology both at hypoxic and normoxic culture conditions. The PDT was lower for hypoxic cultures (*p=0.04)
At passages 6–8, in both oxygen conditions, colony forming units appeared and the cell population became homogeneous, that is, only the “triangular” shaped cell types had remained. This occurred one or two passages later in normoxic than in hypoxic cultures. The PDT of cells at high passage (P6–P15) decreased 3- to 7-fold and became similar for both oxygen conditions, namely 1.3 for normoxic and 1.2 for hypoxic cultures (Fig. 1B).
Quantification by TissueFAXS analysis after staining of phosphohistone H3, a marker for proliferation, confirmed the lack of significant difference in proliferation of high-passage cells under normoxia or hypoxia (Fig. 1C).
The CyQUANT assay also showed no significant difference in proliferation rate between cells under normoxia or hypoxia at high passage. However, in vivo, hypoxic insults actually represent a shift from normoxia to hypoxia, which cause satellite cells to respond either directly or indirectly, with proliferation. We investigated the influence of a decrease or increase of oxygen concentration on satellite cells that we had cultured under normoxic or hypoxic conditions, respectively. Remarkably, the transfer of cells from normoxic to hypoxic culture conditions, significantly increased their proliferation, with a maximum at 3 days (p=0.01, Fig. 2).
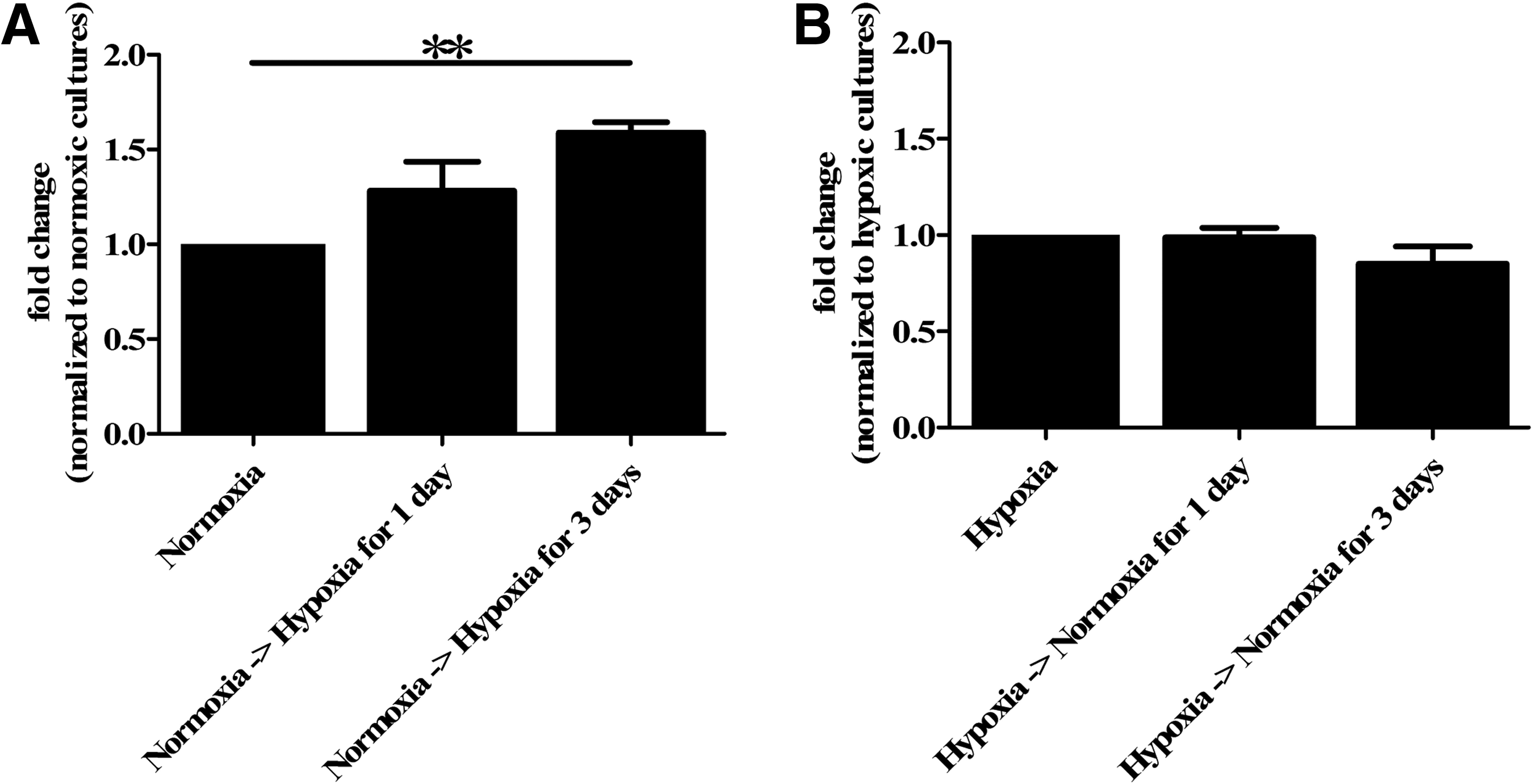
Proliferation after switching oxygen concentration. The CyQUANT assay showed an increased proliferation when cells at high passage (P6–P15) were transferred from normoxia to hypoxia. Hypoxic culture conditions significantly stimulated satellite cell proliferation rate by day 3 compared with satellite cells that remained in normoxic culture conditions (** p=0.01)
Desmin and NCAM expression by satellite cells is independent of the oxygen level
To further qualify and quantify satellite cells in our cultures, the expression of desmin, a myogenic cyotoskeletal protein, was determined by immunofluorescent staining. Of the cells cultured under normoxia, 59.8%±13.7% expressed desmin, and of the cells from the same donor, cultured under hypoxia, this was 41.1%±18.4% (Fig. 3B). This is lower, but does not differ significantly.
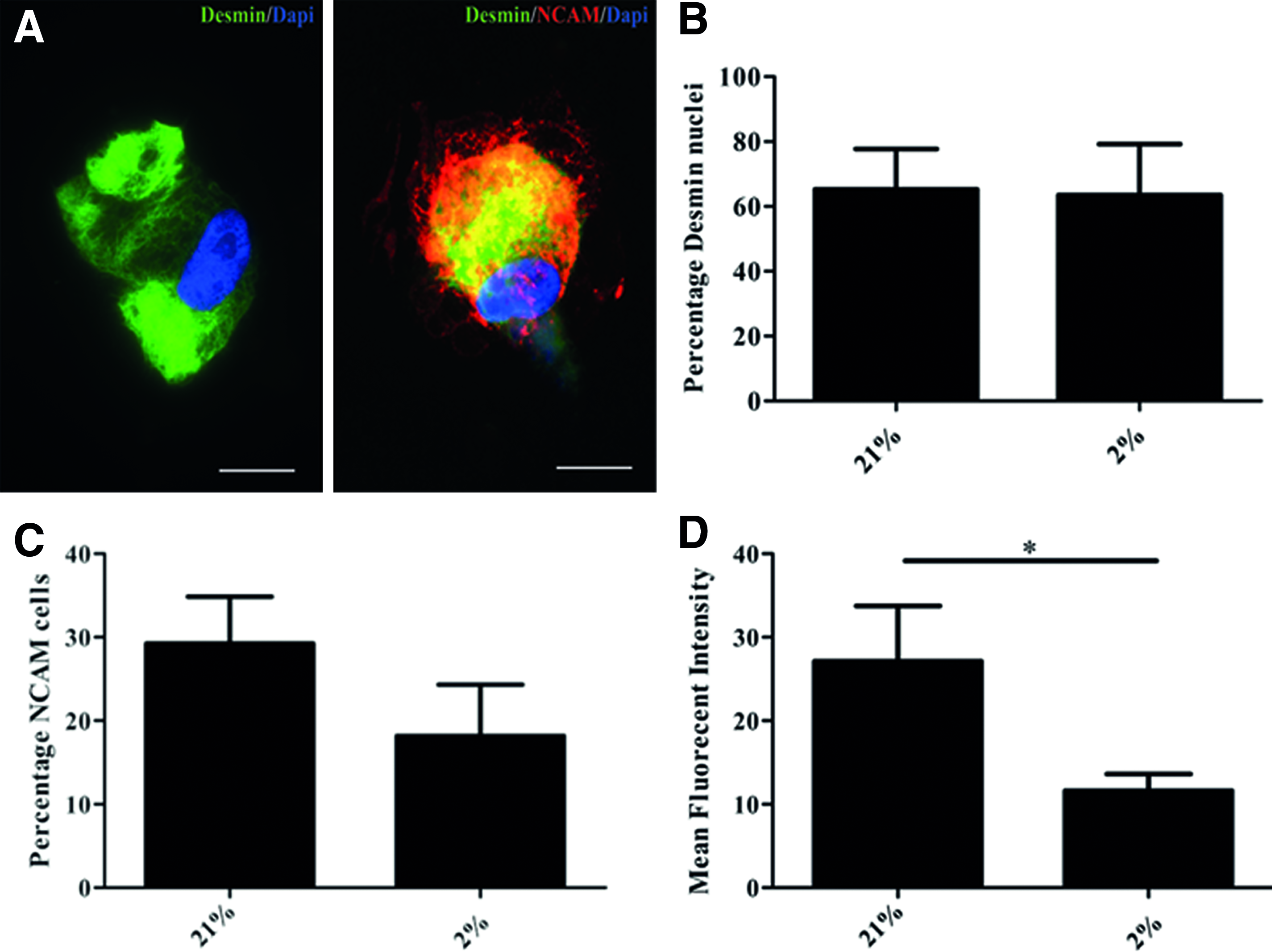
Protein expression analyses. Immunofluorescent image of a proliferating satellite cell at passage three showing positive staining for desmin (green) and a proliferating satellite cell at passage three showing double positive staining for desmin/NCAM (green/red)
The expression of NCAM, which is frequently considered a satellite cell marker, was measured by flow cytometry. Only approximately one third (28.4%±10.3%) of the satellite cells cultured under normoxia expressed NCAM, whereas no more than one fifth (18.2%±6.5%) of the cells cultured under hypoxia expressed NCAM. The mean fluorescent intensity of NCAM positive cells in normoxic cultures (27.1±6.6) was significantly higher than in hypoxic cultures (11.6±2.0, p=0.04, Fig. 3C, D).
Together, these findings support that desmin expression by satellite cells is unaffected by the oxygen level these cells are cultured in. Though the percentage of NCAM expressing cells is not affected, the NCAM expression per cell is decreased when satellite cells are cultured under hypoxia.
High satellite cell purity in high passage cultures is not influenced by hypoxia
To determine fibroblast contamination in our cell population, we performed a double staining of cells in low passage and in high passage with desmin and a fibroblast marker MCA1399G, and quantified with the TissueFAXS. In low-passage cultures, we found 13.5%±9.3% desmin positive cells and 75.6%±5.8% fibroblasts (Fig. 4A–C). In high-passage cultures, we found 91.3%±4.1% desmin positive cells and 1.9%±0.7% fibroblasts (Fig. 4B, C). We did not find an effect of hypoxia on fibroblast contamination. Together, these results show that in low-passage cultures, fibroblast contamination is high, but more importantly, in high-passage cultures, fibroblast contamination is very low, and cultures mainly consist of satellite cells.
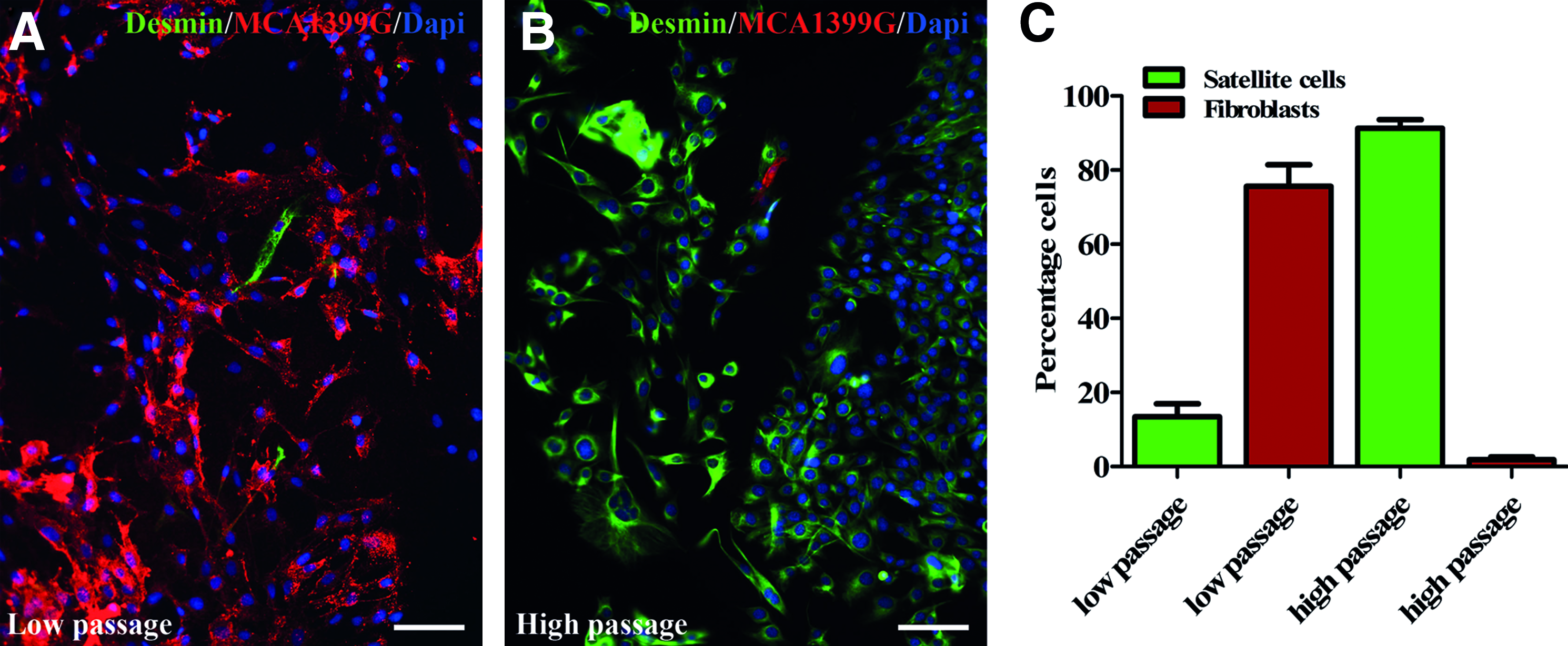
Satallite cell purity and fibroblast contamination. Immunofluorescent image of proliferation cell cultures at low passage (P3–P4)
Gene expression of early satellite cell markers seems to be promoted by hypoxia
In qualitative gene expression analysis, we found that myogenic regulator factors MYF5 and MYOD were upregulated in cells cultured under hypoxia, whereas desmin (DES) and α-sarcomeric actin (ACTA1), a marker indicative for structural myogenic differentiation, were downregulated in cells cultured under hypoxia. For satellite cell markers PAX7 and NCAM, there was no difference detected (Fig. 5A). With quantitative gene expression analysis, expression of MYOD and MYF5 was, respectively, 0.3- and 0.1-fold upregulated in cells cultured under hypoxia compared with those cultured under normoxia. The expression of DES and ACTA1 was downregulated, respectively, 0.5- and 1.5-fold. In contrast, PAX7 was upregultated 0.3-fold, whereas NCAM was downregulated 0.1-fold in satellite cells that were cultured under hypoxia (Fig. 5B). The differences at gene transcriptional level between cells cultured under normoxia or hypoxia are not significant; however, Figure 4 represents a trend that under hypoxic culture conditions, quiescent satellite cells are promoted.
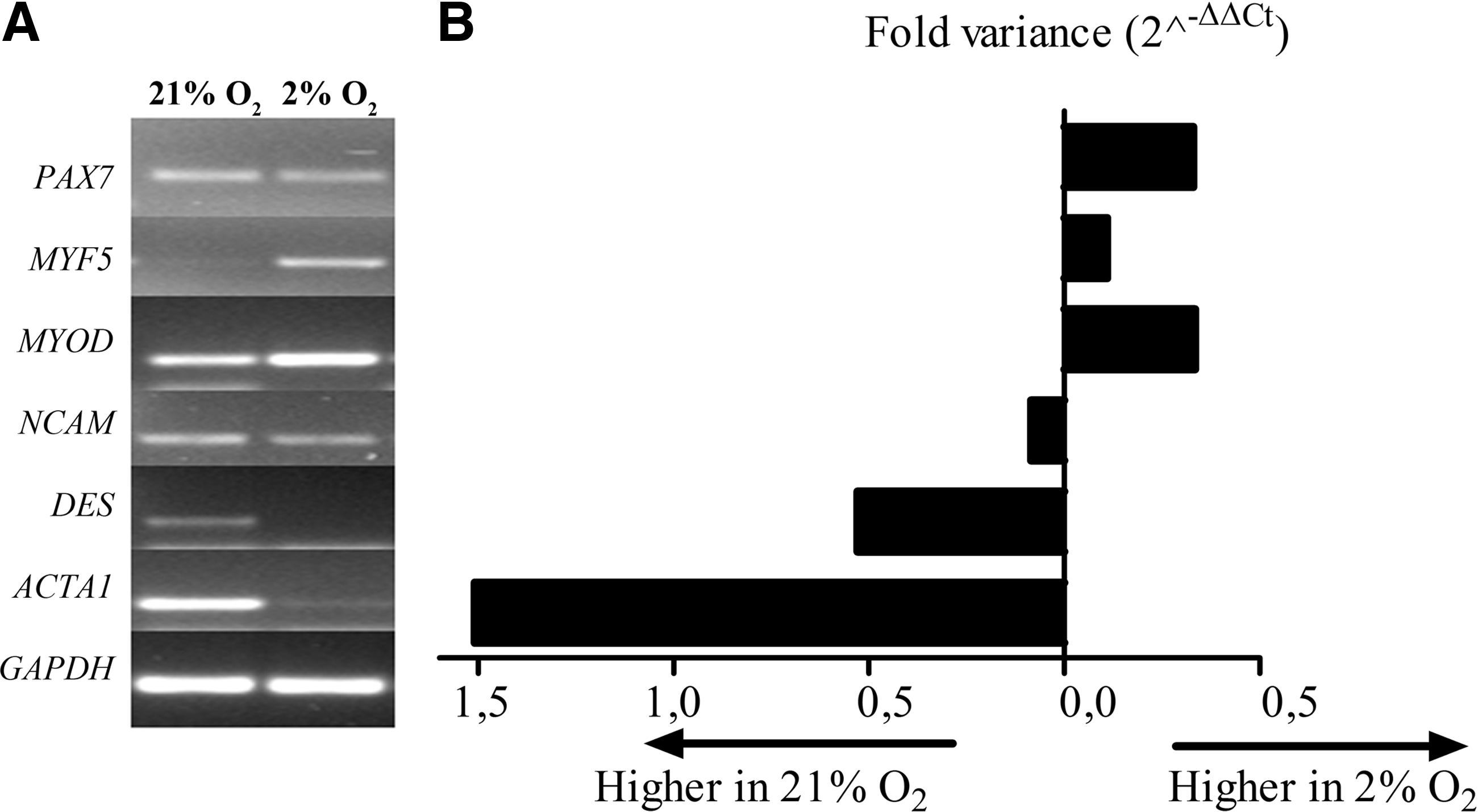
Gene expression analyses. Qualitative gene expression analyses showed the differential regulation of myogenic genes by proliferating satellite cells during passage 3–15
Myogenic differentiation of satellite cells unaffected by hypoxia
To determine whether satellite cells can form myotubes under hypoxic culture conditions, we exchanged the PM to DM when cells had reached 100% confluence in either oxygen concentrations.
We observed that cells in both culture conditions had started to fuse and form multinucleated cells that resembled myotubes within 3 days after myogenic induction, whereas at both days 3 and 5 mononucleated cells were still present (Fig. 6A–C). The multinucleated cells and the mononucleated cells expressed desmin (Fig. 6D–F). However, only the multinucleated cells expressed myosin (Fig. 6G, H), which confirmed their being myotubes. Further, after 5 days of differentiation, in high-passage cell cultures, cross-striations in the myotubes became visible after staining for myosin (Fig. 6I). These are the hallmarks of striated muscle.
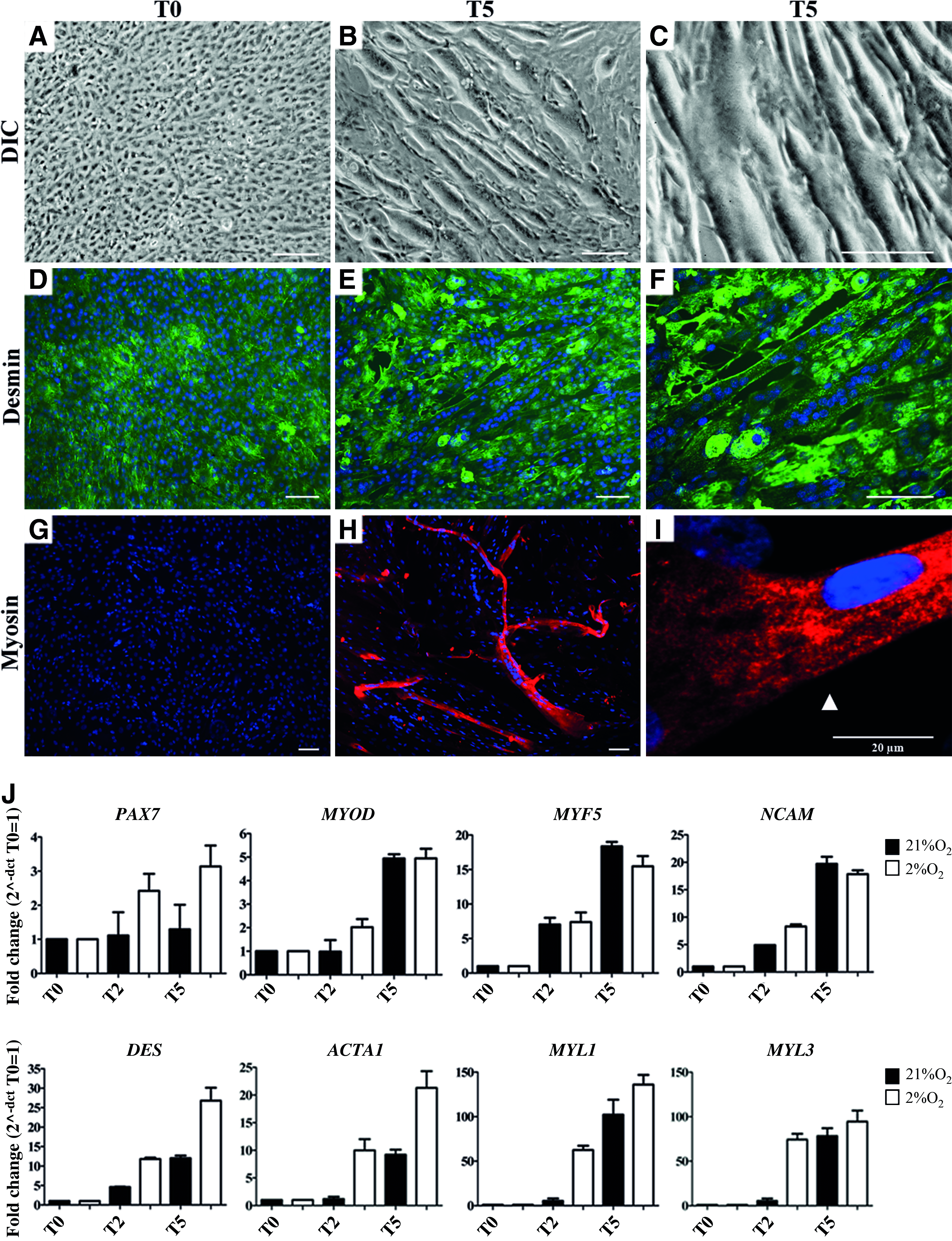
Differentiation of human satellite cells. In proliferating satellite cells at high passage (P6–P15), differentiation was initiated when satellite cells reached 100% confluence (T0) by switching to differentiation medium
During differentiation, the morphology of cells was not affected by the oxygen concentration: in normoxic culture conditions, there was differentiation of satellite cells and myotube formation, equally efficient in hypoxic culture conditions based on myotube size and increase in myoD positive nuclei (data not shown).
Quantitative gene expression analysis showed an upregulation of PAX7, the myogenic regulator factors MYF5 and MYOD, and NCAM during differentiation. Genes of myogenic differentiation such as DES, ACTA1, MYL1, and MYL3 showed a twenty- to hundred-fold upregulation concomitant with myotube formation. During differentiation, gene regulation is comparable between both oxygen conditions, though all genes are more upregulated by cells cultured under hypoxia (Fig. 6J).
The protein expression pattern of myosin matches the upregulation of MYL1 and MYL3, genes responsible for myosin expression.
Myogenic differentiation predisposes a fraction of satellite cells to quiescence
In DM, part of the satellite cells did not fuse into myotubes. To determine whether these cells remained activated satellite cells or whether they acquired a quiescent phenotype, an immunofluorescent staining for Pax7 was performed, which was quantified by TissueFAXS analysis. Data showed that during myotube formation, the percentage Pax7 positive cells did not change (Fig 7A, B). However, the Mean Fluorescent Intensity of Pax7 positive cells had increased (Fig. 7B, C). This increase in expression of Pax7 occurred only in the remaining mononucleated cells, but not in the myotubes (Fig. 7D). This observation concurs with our findings that gene expression of PAX7 is upregulated during myogenic differentiation.

Pax7 expression is maintained during myogenic differentiation. At day 0 (T0), myogenic differentiation was induced in confluent cultures of human satellite cells at high passage (P6–P15) and monitored on day 5 (T5). Immunofluorescence analyses by TissueFAXS showed that the fraction of Pax7 expressing cells (red) did not change during myotube formation
Discussion
The current study set out to investigate the influence of hypoxia on proliferation and differentiation of human satellite cells. One major finding is that hypoxia increases proliferation of low-passage human satellite cells in vitro. Second, satellite cells cultured for multiple passages under hypoxia maintain their myogenic properties and enhance their functional capacity of differentiation toward myotubes. Finally, another important finding is that during myogenic differentiation in vitro, part of the satellite cells acquired a quiescent phenotype. Taken together, these findings render human satellite cells highly suitable for tissue engineering of skeletal muscle for clinical applications.
The proliferation rate of satellite cells during passages 0–5 was higher under hypoxia than normoxia, which may imply that satellite cells overgrow fibroblasts quicker under hypoxia. At a higher passage number, cultures consist of mainly satellite cells, and proliferation rate increases 3- to 7-fold, irrespective of oxygen level. These observations in vitro correlate to the response of satellite cells in vivo where hypoxia induces vigorous proliferation too. 18 Other studies also reported a beneficial effect of low oxygen levels on the proliferation of satellite cells.22,24,25 However, these investigators did not use human satellite cells, but a mouse cell line (C2C12), rat or bovine derived, or whole muscle fibers. Moreover, in these studies, only low-passage satellite cells were used, and they were subjected to hypoxia after initial normoxic culture. In those circumstances, we observed a similar, albeit, temporary increase in proliferation rate (Fig. 1A, Fig. 2). This is interesting in tissue engineering, because large numbers of cells are necessary and these cells should be able to withstand the hypoxic surroundings they will encounter to ensure survival. We have shown that satellite cells thrive on low levels of oxygen (2% O2).
A major finding of this study is that in cell populations derived of human skeletal muscle, Desminpos/NCAMpos and Desminpos/NCAMneg satellite cell populations are present. NCAM is an “established” satellite cell marker that is frequently used to purify cell population by cell sorting.26,27 However, in doing this, the Desminpos/NCAMneg population is discarded, which are satellite cells capable of differentiation. Moreover, a heterogeneous population of satellite cells may have a distinct benefit, because their capacity to differentiate and form functional myotubes, that is, generate force, requires supportive cells. 28
The finding that hypoxia decreases NCAM protein levels in satellite cells was corroborated by gene transcript analyses. As it appeared, satellite cells cultured under hypoxia harbor a more quiescent state in terms of gene transcripts, whereas normoxia promotes a more activated myoblast gene expression profile during proliferation (Fig. 5). Consistent with our findings is the upregulation of MYOD described in primary rat and bovine satellite cells cultured under hypoxia.22,29 However, these published results contrast with the murine myoblastic cell line C2C12. Under hypoxia, C2C12 cells show a delayed myogenic differentiation due to a downregulation of MYOD, which is one of the dominant myogenic transcription factors. This downregulation might be caused by an accelerated degradation of MYOD at the post-transcription level by C2C12. 30 However, this downregulation of MYOD is transient, which indicates that C2C12 myoblasts can adapt to hypoxic conditions too. 29 This indicates that C2C12 and human satellite cells respond differently to hypoxia. Further, human satellite cells also show donor variation at protein and gene expression levels. The donor variation could be a reflection of the functional heterogeneity within subpopulations of satellite cells.31–33 Also, due to their different embryonic origin, satellite cells derived from craniofacial muscle have properties that set them apart from satellite cells derived from limb muscles, 34 which might also be a cause for our donor variation. Further, there was some age variation in our research population. This is important, because when age increases, satellite cell population is decreased and their myogenic capacity is reduced.35–37 However, in this study, we could not relate the donor variation to muscle origin or donor age, because the variation was not significant and our population was too small.
During differentiation, mononucleated satellite cells formed multinucleated myotubes in both oxygen conditions. The myotubes expressed myosin, unlike mononucleated cells. Simultaneously, both MYL1 and MYL3, genes responsible for, respectively, myosin type-1 and myosin type-2 expression, were upregulated during differentiation. Both mono and multinucleated cells expressed desmin; therefore, the remaining mononucleated satellite cells most likely retained the capacity to form myotubes. Alternatively, even under myogenic conditions, desmin-expressing cells could (de)differentiate to quiescent satellite cells. This quiescent state was confirmed by the enhanced expression of Pax7 during myotube formation by mononucleated cells (Fig. 7). This also explains the upregulation of PAX7 observed at gene transcript level. We can, therefore, conclude that mononucleated cells maintain their satellite cell character or dedifferentiate toward their quiescent state (Fig. 8). We surmise that this in vitro synchronous bidirectional differentiation of satellite cells reiterates in vivo processes in which an appropriate niche of myogenic progenitor cells (quiescent satellite cells, or reserve cells 38 ) is recreated on regeneration of ischemic muscle tissue. Further, this is in concordance with the findings derived from mice and rat tissue, that satellite cells can both self-renew and differentiate after transplantation.39,40 Moreover, this process could be influenced by various signaling pathways promoting either myoblast and myotube formation or quiescent satellite cells.41–46 This offers great potential for future clinical applications of tissue-engineered muscle, where the presence of cells with a self-renewing capacity is essential. Moreover, we find that these processes are not influenced by hypoxia, which satellite cells will encounter on implantation.
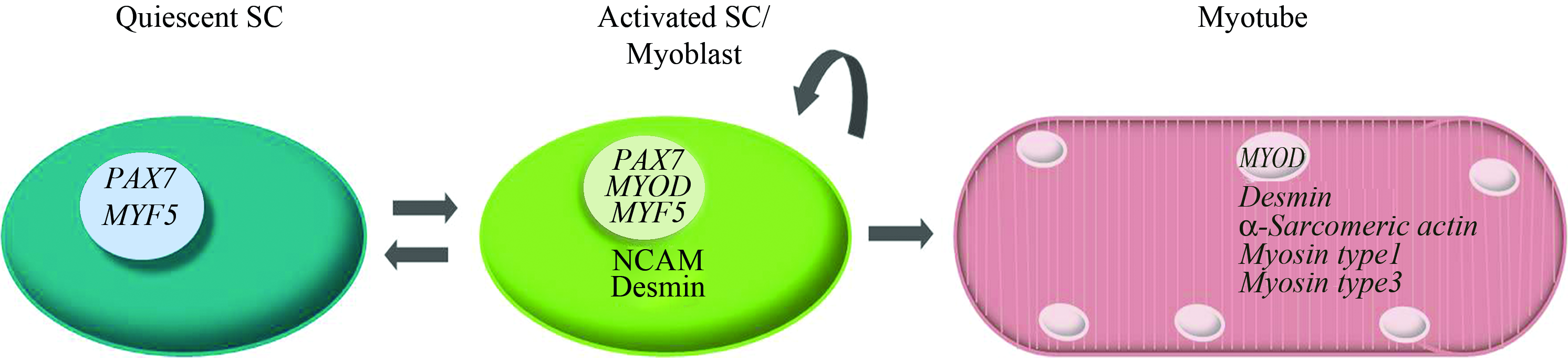
Transcriptional lineage of satellite cells. During differentiation of human satellite cells, myotubes are formed and gene and protein expression patterns shift toward mature muscle. However, some activated satellite cells differentiate into a quiescent state, with a cocurrent increase of Pax7 expression (Figs. 5 and 6). In vivo, this would represent a mechanism for satellite cells to maintain a reserve of myogenic progenitor cells (quiescent satellite cells, or reserve cells). The oxygen concentration during culture of human satellite cells is not a significant regulating factor in differentiation. However, low oxygen concentration does promote PAX7 expression, engaging satellite cells in myogenic lineage and promoting more efficient proliferation. Color images available online at www.liebertonline.com/tea.
For tissue engineering, where large cell numbers are required, hypoxia improves proliferation during low passage tremendously. We, therefore, recommend culturing human satellite cells under hypoxic conditions during the first passages.
Conclusion
Hypoxic culture conditions promote initial cell proliferation of human satellite cells. Moreover, although satellite cells differentiate into myotubes, they maintain a pool of quiescent satellite cells with qualities that are unaffected by hypoxia. Combined, this renders human satellite cells highly suitable for tissue engineering of skeletal muscle for future clinical application.
Footnotes
Acknowledgments
This study was funded by a research grant of the Graduate School W.J. Kolff Institute from the University Medical Center Groningen, University of Groningen, The Netherlands. The antibodies MF20 and Pax7 developed by resp. Fischman, D.A. and Kawakami, A. were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242. The TissueFAXS, “the equivalent to flow cytometry for multiparameter quantitative analyses in tissues,” was acquired with a NWO-ZonMW Medium Investment Grant (40-00506-98-9021).
Disclosure Statement
No competing financial interests exist.