
Abstract
Knee meniscus, a fibrocartilaginous tissue, is characterized by heterogeneity in extracellular matrix (ECM) and biomechanical properties, and critical for orthopedic stability, load transmission, shock absorption, and stress distribution within the knee joint. Most damage to the meniscus cannot be effectively healed by the body due to its partial avascular nature; thus, damage caused by injury or age impairs normal knee function, predisposing patients to osteoarthritis. Meniscus tissue engineering offers a possible solution to this problem by generating replacement tissue that may be implanted into the defect site to mimic the function of natural meniscal tissue. To address this need, a multiporous, multilamellar meniscus was formed using silk protein scaffolds and stem cells. The silk scaffolds were seeded with human bone marrow stem cells and differentiated over time in chondrogenic culture in the presence of transforming growth factor-beta 3 to generate meniscus-like tissue in vitro. High cellularity along with abundant ECM leading to enhanced biomechanics similar to native tissue was found. Higher levels of collagen type I and II, sulfated glycosaminoglycans along with enhanced collagen 1-α1, aggrecan, and SOX9 gene expression further confirmed differentiation and matured cell phenotype. The results of this study are a step forward toward biomechanically competent meniscus engineering, reconstituting both form and function of the native meniscus.
Introduction
Meniscal injuries are commonly associated with sports and age-related degeneration. Successful surgical restoration of the damaged meniscus has been a challenge due to its poor healing potential. The presence of a vascularized outer zone (10%–30% of the meniscal body), meniscal lesions in this zone heal spontaneously and can be sutured successfully with a high success rate.2–4 However, majority of meniscal tears are in the inner avascular zone lacking spontaneous healing, which ultimately leads to permanent degenerative changes, including osteoarthritis.1,2,4 Total meniscectomy is one of the treatments available for meniscal injury. However, this increases local stresses almost threefold on the underlying articular cartilage, leading to osteoarthritic changes in the knee.5,6 Further, the degree of osteoarthritis after meniscectomy was directly proportional to the amount of meniscus removed. 6 Thus, due to these degenerative outcomes, at present, the clinical goals are to preserve much of the intact meniscus and to remove only the damaged sections by arthroscopy, clinically termed partial meniscectomy. 7 Although partial meniscectomy is an improvement over complete excision of the tissue, stresses on the local articular cartilage remain, resulting in subsequent articular cartilage loss.6,8,9 Hence, a widely accepted solution to the present problem of meniscal injury has yet to be realized.
Patient-specific human stem cell three-dimensional (3D) scaffold–based meniscus engineering offers a possible solution to functional replacement tissue with a living, biodegradable, mechanically competent construct. Numerous strategies to repair and replace damaged meniscus have been adopted using different natural and synthetic matrices and cell types but with limited success due to graft failure mainly due to the lack of biomechanical matches or performance.3,10–21 Importantly, however, these studies suggest that if issues related to biomechanics are addressed, then cell- and scaffold-based interventions hold promise for effective meniscus repair and replacement.
To achieve functional tissue engineering and prevent graft failure, emerging efforts should be on both biochemical traits and mechanical properties.22,23 In our ongoing effort to engineer functional meniscus tissue matching native meniscus form and function, we reported a silk 3D scaffold meniscus model cultured with fibroblasts and chondrocytes based on the architectural model proposed by Petersen and Tillmann.24,25 In an effort to achieve native meniscus like biomechanics and to take this technology toward clinical implementation by making it more patient specific, in the present study we focus on engineering wedge-shaped multiporous silk meniscus tissues seeded with human bone marrow-derived mesenchymal stem cells (hMSCs) differentiated toward meniscus-like cells. Bombyx mori cocoon silk was chosen as the biopolymer due to its biocompatibility, versatile processability, mechanical features, and its history of use in biomedical applications, including sutures, ligament engineering, osteogenesis, chondrogenesis, and adipogenesis.26–32 Further, the unique mechanical properties of silk along with controlled degradability are useful features for neomeniscus tissue formation and integration.26,27,33–35
We hypothesized that with patient-specific stem cells, multiporous aligned scaffold layers will serve as 3D patterns to direct neotissue formation, resulting in more mature tissues with enhanced matrix content, organization, and biomechanics comparable to native meniscus tissue. hMSCs (human bone marrow stem cells) were used as the alternative cell source due to the availability, multilineage differentiation, and self-renewal potential.36,37 To evaluate this hypothesis, hMSCs were cultured over a period of 4 weeks in a chemically defined chondrogenic medium supplemented with transforming growth factor-beta 3 (TGF-β3) to study cell–scaffold interactions, accumulation, and distribution of extracellular matrix (ECM).
Materials and Methods
Preparation of silk fibroin solution
Silk fibroin solution (9, w/v%) was obtained from B. mori silkworm cocoons that were extracted in a 0.02 M Na2CO3 solution, dissolved in 9.3 M LiBr solution, and subsequently dialyzed against distilled water. 38
Meniscus scaffolds fabrication
To mimic the multiporous pore orientation of native meniscus, 3D aqueous-derived silk scaffolds were fabricated into individual layers with varying pore size and orientations. 25 The first two layers were fabricated according to our previously described salt leaching procedure, whereas the third layer was achieved using a freeze-drying method. 38 Briefly, for the salt leached method, 2 g of granular NaCl particles (350–400 and 500–600 μm for first and second layers, respectively) was added per 1 mL of 9 w/v% silk fibroin solution in meniscus-shaped polydimethylsiloxane (GE plastics) moulds at room temperature. Twenty-four hours later the molds were immersed in water to extract salt from the porous scaffolds over 2 days. Similarly, for the freeze-dried third scaffold layer, no salt was added and instead the silk solution was frozen at −80°C for overnight followed by lyophilization.
hMSC isolation and expansion
hMSCs were obtained from bone marrow aspirates (Cambrex Bio Science Walkersville, Inc.) from a 25-year-old healthy man and processed as previously reported.26,38 Briefly, cells from whole bone marrow aspirates were separated by density gradient centrifugation using a poly-sucrose gradient (1077 g/cm3, Histopaque; Sigma). Resuspended cells were plated at a density of 105 cells/cm2 tissue culture flasks in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U/mL), streptomycin (100 μg/mL), 0.1 mM nonessential amino acids, and basic fibroblast growth factor (1 ng/mL) (Invitrogen). This medium was termed growth medium. Cells were maintained in a humidified incubator at 37°C with 5% CO2. hMSCs were separated from hematopoietic stem cells on the basis of their adherence to tissue culture plastic; hematopoietic stem cells in suspension were removed after approximately 5 days of culture. hMSCs formed numerous colonies with time, and were subsequently expanded in the ratio of 1:3. For subsequent experiments, passage seven (P7) hMSCs were used. Late passage cells were deliberately used as a stringent test to check for their ability to differentiate into chondrogenic phenotype and ECM expression on the silk constructs.
Cell culture on silk scaffold layers
To produce cell-seeded meniscal equivalent constructs, scaffolds were cultured with hMSCs and differentiated to chondrogenic phenotype mimicking native meniscal tissues. 3 Due to the large sample size, number, and limited cell numbers, instead of full-sized crescent-shaped meniscus, the scaffolds were cut into smaller discs (5 mm in diameter and 2 mm in thickness). Before cell seeding, scaffolds were sterilized with 70% alcohol, washed in sterile phosphate-buffered saline (PBS), and conditioned with growth medium overnight. To monitor cell response on the different meniscus layers for ECM deposition and cell proliferation, hMSCs were seeded into each scaffold layer. Twenty microliters of aliquots containing 0.8×106 hMSCs was loaded onto each scaffold layer separately. After 2 h for cell attachment, seeded constructs were cultured in 1 mL of growth medium for 3 days to allow cells to attach and spread onto scaffold pores. After attachment, scaffolds with cells were transferred to chemically defined chondrogenic medium containing DMEM with 1×penicillin–streptomycin–fungizone (PSF), 0.1 μM dexamethasone, 50 μg/mL ascorbate 2-phosphate, 40 μg/mL L-proline, 100 μg/mL sodium pyruvate, 1×ITS+ (6.25 μg/mL insulin, 6.25 μg/mL transferrin, and 6.25 ng/mL selenous acid), 1.25 mg/mL bovine serum albumin (BSA), and 5.35 μg/mL linoleic acid with 10 ng/mL TGF-β3 (R&D systems) in nontissue culture-treated 12-well plates. This chemically defined medium was previously been reported to induce and maintain chondrogenesis of hMSCs and to promote deposition of fibrocartilaginous ECM. 39 Medium with supplements was changed every 3 days for 4 weeks.
Biochemical assays for DNA, glycosaminoglycans and collagen content
Silk meniscal scaffolds with and without cells were digested for 16 h with papain digestion cocktail (125 μg/mL papain, 5 mM L-cysteine, 100 mM Na2HPO4, and 5 mM EDTA, pH 6.2) at 60°C for DNA and glycosaminoglycans (GAG) analysis. DNA content was measured using a PicoGreen DNA assay kit as per manufacturer's protocol (Invitrogen). In brief, the papain-digested samples were centrifuged and 25 μL aliquot of supernatant from each sample was added into a 96-well plate with wells containing 75 μL of 1×Tris-EDTA (TE) buffer. Then, 100 μL of Quant-iT PicoGreen reagent (1:200 dilution) was added to each well followed by measurement using a fluorimeter with an excitation and emission wavelength of 480 and 528 nm, respectively. A standard curve was generated using lambda phage DNA. For total sulfated GAG (sGAG), 1,9-dimethylmethylene blue (DMMB) assay was used. 40 Individual sample aliquots were mixed with DMMB reagent and absorbance was measured at 525 nm. For sGAG secreted into media, spent culture medium was stored at −20°C and later assessed for sGAG content using similar protocols. GAG was estimated using a standard curve generated using shark chondroitin sulfate (Sigma). For total collagen estimation, the samples were digested in pepsin cocktail (1 mg/mL pepsin, pH 3.0) at 4°C for 48 h. The collagen content was measured using a modified Hride Tullberg-Reinert method. 41 In brief, individual digested samples were dried at 37°C in 96-well plates for 24 h and reacted with Sirius red dye solution for 1 h with mild shaking. The dye solution (pH, 3.5) was prepared with Sirius red dissolved in picric acid–saturated solution (1.3%; Sigma) to a final concentration of 1 mg/mL. The samples were washed five times with 0.01 N HCL followed by resolving dye–sample complex using 0.1 N NaOH and recording absorbance at 550 nm. Total collagen was estimated using a standard curve using bovine collagen (Sigma). Scaffolds without cells were controls and values were subtracted in all assays to negate interference. To avoid variations from scaffold sizes and cell numbers, GAG and collagen contents were normalized against total scaffold weight and cell numbers, represented by the total DNA content measured by PicoGreen DNA assay kit (Invitrogen).
Histology and immunocytochemistry of constructs
For histology and immunocytochemistry, individual scaffold layers with differentiated stem cells were washed in PBS followed by fixation in 10% neutral buffered formalin for 24 h before histological analysis. Samples were dehydrated through a series of graded ethanol, embedded in paraffin, and sectioned at 5 μm thickness. For histological evaluation, sections were deparaffinized, rehydrated through a series of graded ethanol, and stained. Serial sections were stained with hematoxylin and eosin (H&E) as well as with Safranin-O and Alcian blue for sulfated proteoglycans in the matrix. Similarly, constructs were immunostained with monoclonal antibodies against collagen type I and II (Abcam). Immunohistochemical sections were deparaffinized, hydrated, and permeabilized. The sections were then incubated for 30 min with 1% BSA at 37°C followed by primary antibody for 2 h. The sections were washed and incubated with HRP-labeled secondary antibodies (Santa Cruz Biotechnology) followed by standard development in diaminobenzidine (Vector Laboratories). The sections were counterstained with hematoxylin.
Real-time polymerase chain reaction
For total RNA extraction, hMSCs cultured on silk 3D scaffolds in chondrogenic conditions for 14 and 28 days were placed on ice, and were cut into small pieces using sharp dissection scissors. The cut pieces were transferred into 2-mL plastic tubes containing 1.5 mL of Trizol solution (Invitrogen). After incubation for 15 min, the treated scaffolds were centrifuged at 12,000 g for 10 min/4°C. The supernatant was transferred to a new tube and 200 μL of chloroform was added. After further incubation for 5 min at room temperature, the solution was gently mixed for 15 s, followed by incubation for 5 min at room temperature. The tubes were further centrifuged for 15 min at 12,000 g/4°C. The upper aqueous layer was transferred to an RNeasy Plus mini-spin column (Qiagen). The RNA was washed and eluted according to the manufacturer's protocol. RNA samples were reverse-transcribed into cDNA using high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer's protocol in an M×3000 quantitative real-time polymerase chain reaction (PCR) system (Stratagene). All data analysis employed the M×3500 software (Stratagene) based on fluorescence intensity after normalization with an internal reference dye and baseline correction. Differences of gene expression were analyzed using the comparative Ct method (Ct [delta][delta] Ct comparison). Ct values for samples were normalized to an endogenous housekeeping gene. PCR conditions were 2 min at 50°C, 10 min at 95°C, and then 50 cycles at 95°C for 15 s, and 1 min at 60°C. The data were normalized to the expression of the housekeeping gene, glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) within the linear range of amplification and difference. The GAPDH probe was labeled at the 5′ end with fluorescent dye VIC and with the quencher dye TAMRA at the 3′ end. Highly purified human gene-specific primers for collagen-1-alpha-1 (COL1A1_NM_000088.3), aggrecan (ACAN_NM_013227.3), SOX9 (NM_000346.3), and housekeeping gene GAPDH (NM_002046.3) were ordered commercially (Applied Biosystems). Probes were purchased from Assay on Demand (Applied Biosciences).
Confocal microscopy
hMSC attachment and spreading on individual B. mori silk scaffold layers after each time point was assessed using confocal microscopy. For microscopy, each individual scaffold layer was seeded with 0.8×106 hMSCs separately, and cultured for 1, 14, 21 and 28 days at 37°C, 5% CO2 in chondrogenic medium as mentioned above to allow cells to adhere and spread on the matrices. Upon harvesting day, scaffolds were washed three times with PBS (pH 7.4) followed by incubation in 3.7% formaldehyde in PBS for 10 min. The samples were further washed with PBS and pre-incubated with 1% BSA for 30 min. The constructs were then permeabilized using 0.1% Triton X-100 for 5 min. Incubation with rhodamine-phalloidin (Invitrogen) for 20 min at room temperature followed by PBS washing and counterstaining with Hoechst 33342 (Invitrogen) for 30 min was performed. Images from stained constructs were obtained using a confocal laser scanning microscope (Leica SP2 inverted microscope) equipped with argon (488 nm) and HeNe (534 nm) lasers; two-dimensional multichannel-image processing was performed using IMARIS software (Bitplane AG). Several microphotographs were taken of different samples and analyzed. Figures show one representative sample per group.
Scanning electron microscopy
Field emission scanning electron microscope (Zeiss Ultra55 or Supra55VP; Carl Zeiss AG) operating at 6 kV was used for imaging. Fractured-sections of the scaffolds were obtained in liquid nitrogen using a razor blade. The scaffold samples were sputter coated with platinum/palladium before analysis. Pore size and orientation were determined by measuring random 25 pores from scanning electron microscopic (SEM) images using ImageJ 1.40g program (Wayne Rasband).
Mechanical testing
Compressive mechanical testing of cell-seeded and nonseeded (control) hydrated silk scaffolds was on an Instron 3366 testing frame equipped with a 0.1 kN load cell. Tests for all scaffold types both seeded and nonseeded were carried out in 0.1 (M) PBS at 37°C in hydrated condition. Separate silk scaffold discs were punched out for compressive tests, with dimensions of 4 mm diameter and 3 mm height. For cell-seeded silk scaffolds, each layer was individually seeded with 1×106 hMSCs at day 1 and cultured for 28 days in chondrogenic medium with TGF-β3. Before compressive testing cell-ECM laden scaffolds were fixed with 3.7% formaldehyde for 15 min and washed with PBS. All tests were accessed with a conventional open-sided (nonconfined) configuration and were performed using a displacement control mode at a rate of 5 mm/min. After the compression tests, the compressive stress and strain were graphed based on the measured cross-sectional area and sample height (nominal ∼4–5 mm, measured automatically at 0.02 N tare load), respectively. The elastic modulus was calculated based on a linear regression fitting of a small strain section that precedes an identifiable plateau region.
Statistical analysis
All quantitative experiments are run at least in triplicate, and results are expressed as mean±standard deviation for n=4 unless specified. Statistical analysis of data was performed by one-way analysis of variance (ANOVA). Differences between groups of *p≤0.05 are considered statistically significant and **p≤0.01 as highly significant.
Results
Gross morphology and cell attachment on silk layers
Mimicking native meniscus pore heterogeneity in an in vitro wedge-shaped silk meniscus model, a three-layered silk scaffold system was fabricated using salt-leaching and freeze-drying (Fig. 1a, b). The three individual layers can be stacked on top of each other to form a single unit to demonstrate architectural control of the process similar to native meniscus tissue (Fig. 1). Figure 1b illustrates how the system was adopted into disc-shaped scaffolds for cell seeding and differentiation due to the limited availability of cells. The concept is to seed cells in each of these silk layers, culture over a period of time, allowing the cells to proliferate and deposit ECM, followed by stacking before use. Scaffolds can be stacked in a process analogous to our previous meniscus study using silk fibers to stitch all layers together or using small silk cylinders (riveting method) to hold the layers together. 25
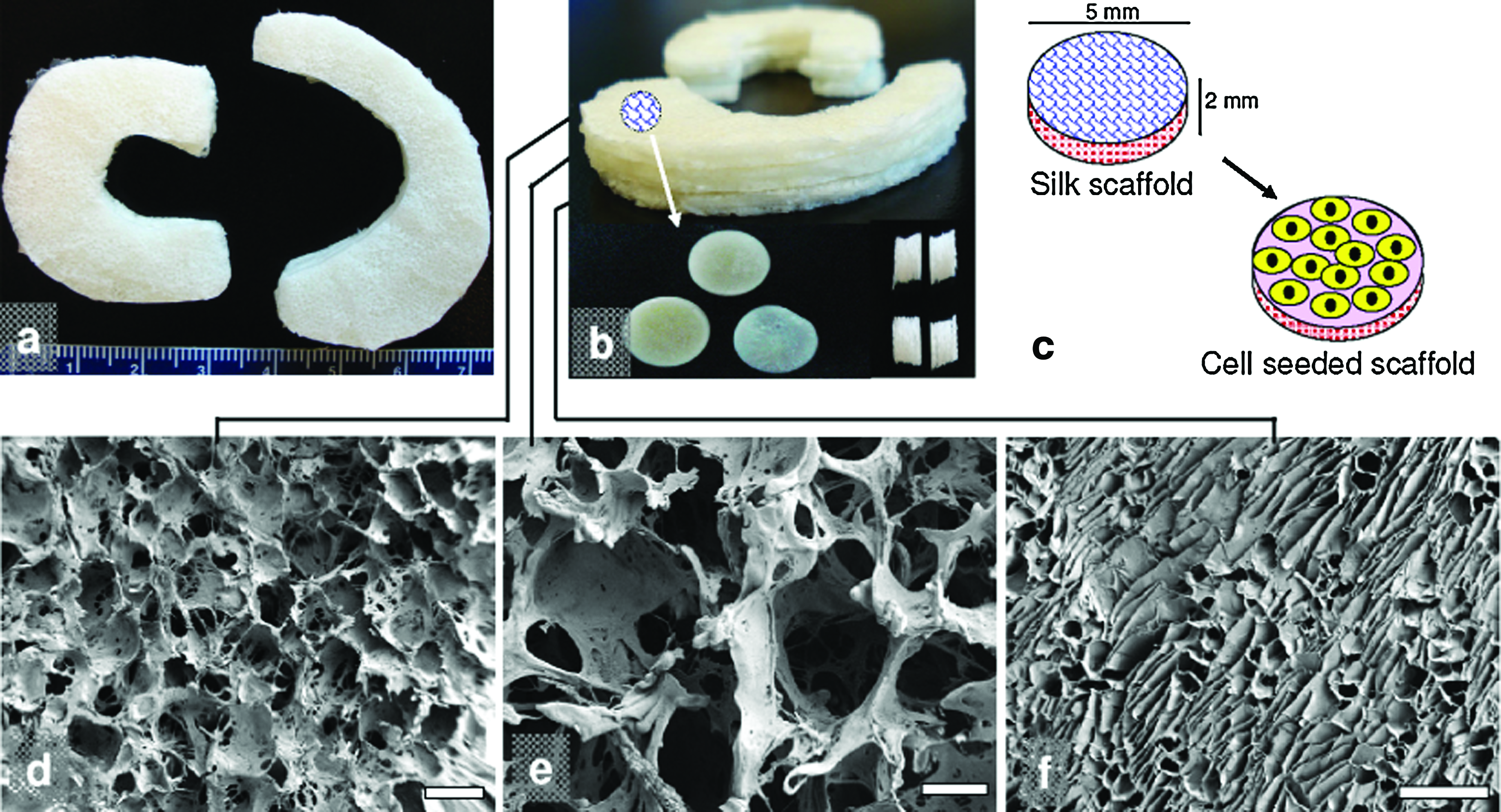
Regenerated silk scaffolds for functional meniscus engineering.
SEM images showed circular pores in the size range of 350–400 μm for the top and 500–600 μm for the middle layers (Fig. 1d, e). While the third (bottom) layer revealed a laminar morphology with laminar pores ranging between 60 and 80 μm (Fig. 1f). As evaluated from confocal images, hMSCs attached and proliferated on the individual silk meniscus scaffold layers (Fig. 2). At day 1, the early seeding stage, cells appeared clustered and attached onto the scaffold walls. Cell spreading was limited as empty scaffold pores were observed (Fig. 2a, e, i). In comparison, cells completely filled the voids of scaffold pores with well-developed and spread-out actin filaments as early as day 7. In all three individual silk meniscus layers, cells appeared dense and evenly distributed and attained confluence (Fig. 2). Cell proliferation on silk scaffolds was further supported from PicoGreen DNA assay results, as hMSCs proliferated with an increase of ∼39%, ∼25%, and ∼36% of their initial cell numbers at day 14 in the case of the top, middle, and bottom layers, respectively (Fig. 5d), whereas at day 28, hMSCs showed ∼108%, ∼111%, and ∼106% proliferation, respectively, for top-bottom layers upon culturing in chondrogenic media (Fig. 5d). At higher magnifications, cells appeared to form intricate meshwork with actin filaments within scaffold pores for the first two layers and dense and compact features within the third bottom scaffold layer having almost linear actin arrangement (Fig. 2d, h, l).
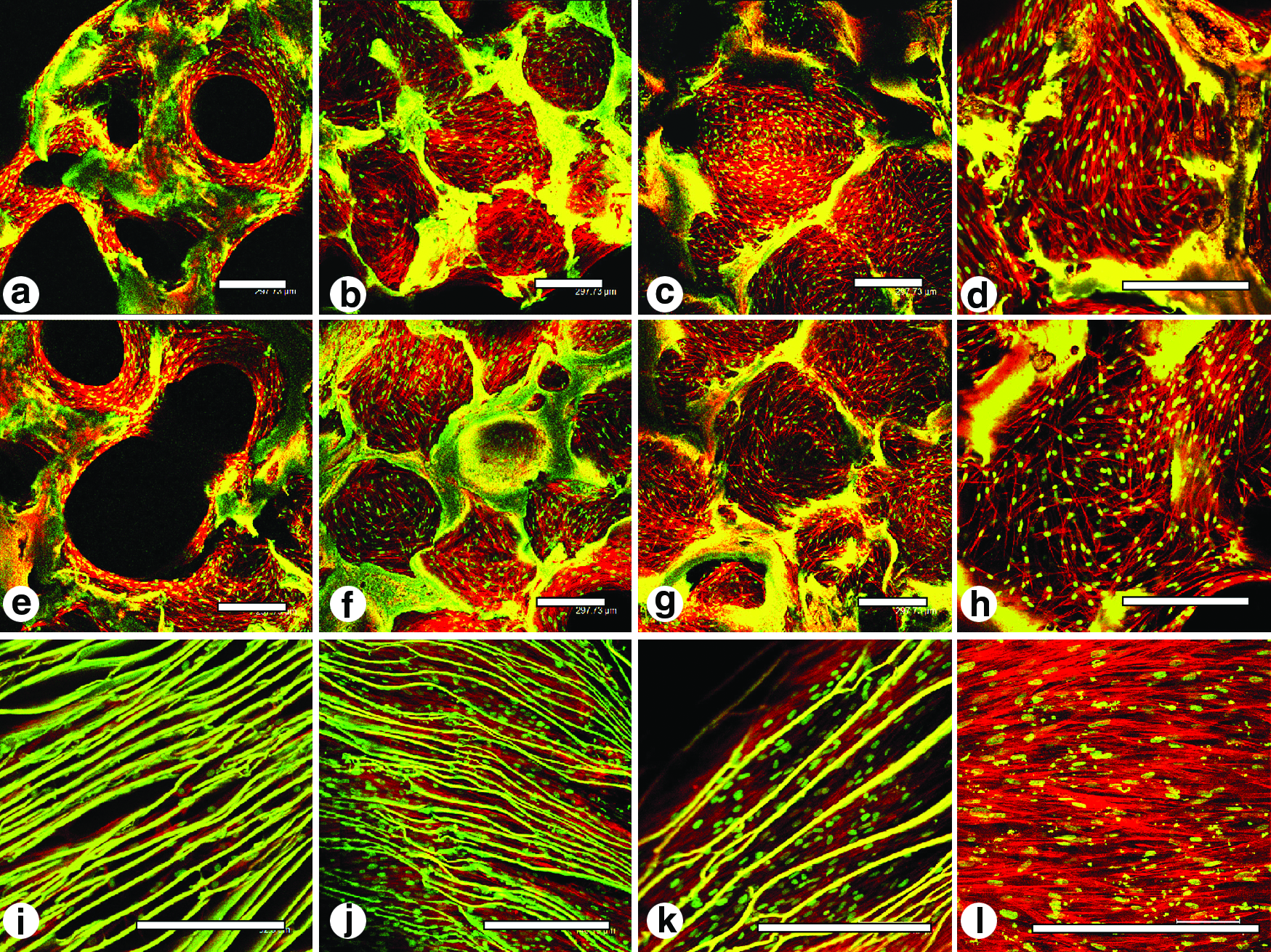
Confocal images of hMSCs showing attachment, growth, and proliferation on individual 3D meniscus silk scaffold layers in chondrogenic medium. Initial cell attachment on day 1
Histology and immunocytochemistry
Histology sections of individual silk meniscus scaffold layers revealed the accumulation of sGAG and collagen with time (day 28 vs. day 1) (Figs. 3 and 4). For Alcian blue, Saffranin O, and H&E, scaffolds showed intense staining on day 28 compared to day 1 for hMSCs grown in chondrogenic medium (Fig. 3a–c). For alcian blue staining, hMSCs showed intense blue color, a hallmark for abundant sGAG deposition, suggesting differentiation toward chondrogenic lineage. In comparison to the top and middle porous layers, the bottom laminar scaffold layer with differentiating hMSCs showed more compact and intense blue staining, suggesting a mature chondrocyte phenotype (Fig. 3b). For the top and middle layers, although alcian blue staining was comparable and as intense as the bottom third layer, the cells appeared less compact upon culturing in chondrogenic medium (Figs. 2–4). Similarly, intense Saffranin O stain further confirmed sGAG accumulation within all scaffold layers, indicating differentiation of hMSCs toward chondrocytes at day 28. Further H&E staining supported the confocal results and revealed cell attachment and distribution within each scaffold layer suggesting growth and proliferation (Fig. 3a). In all cases, cells appeared scattered throughout scaffold pores/layers on day 1, but attained confluence by completely filling void spaces by day 28.
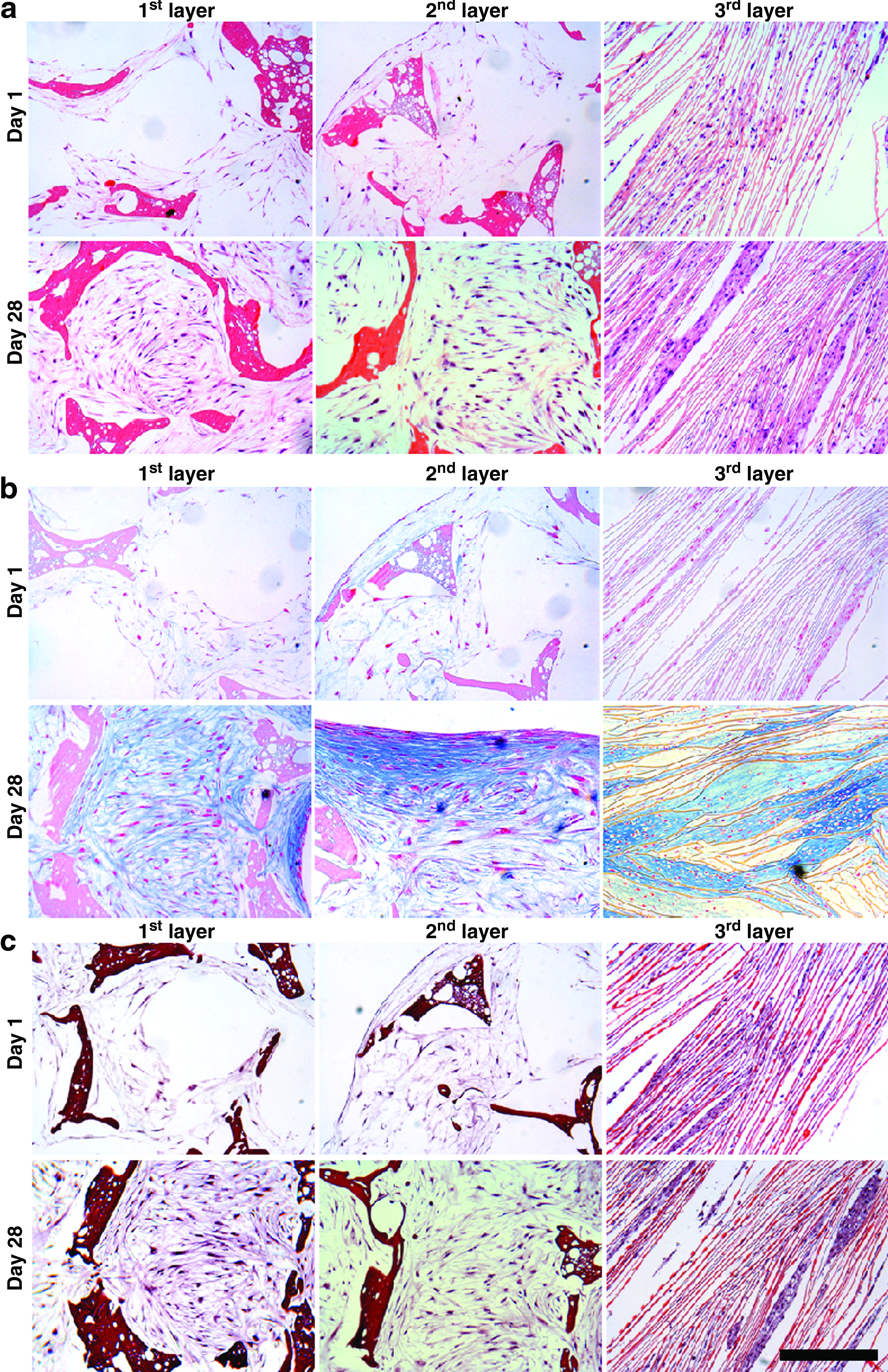
Histology sections showing hMSC cell growth and ECM deposition on individual 3D silk meniscus scaffold layers in chondrogenic medium. H&E staining
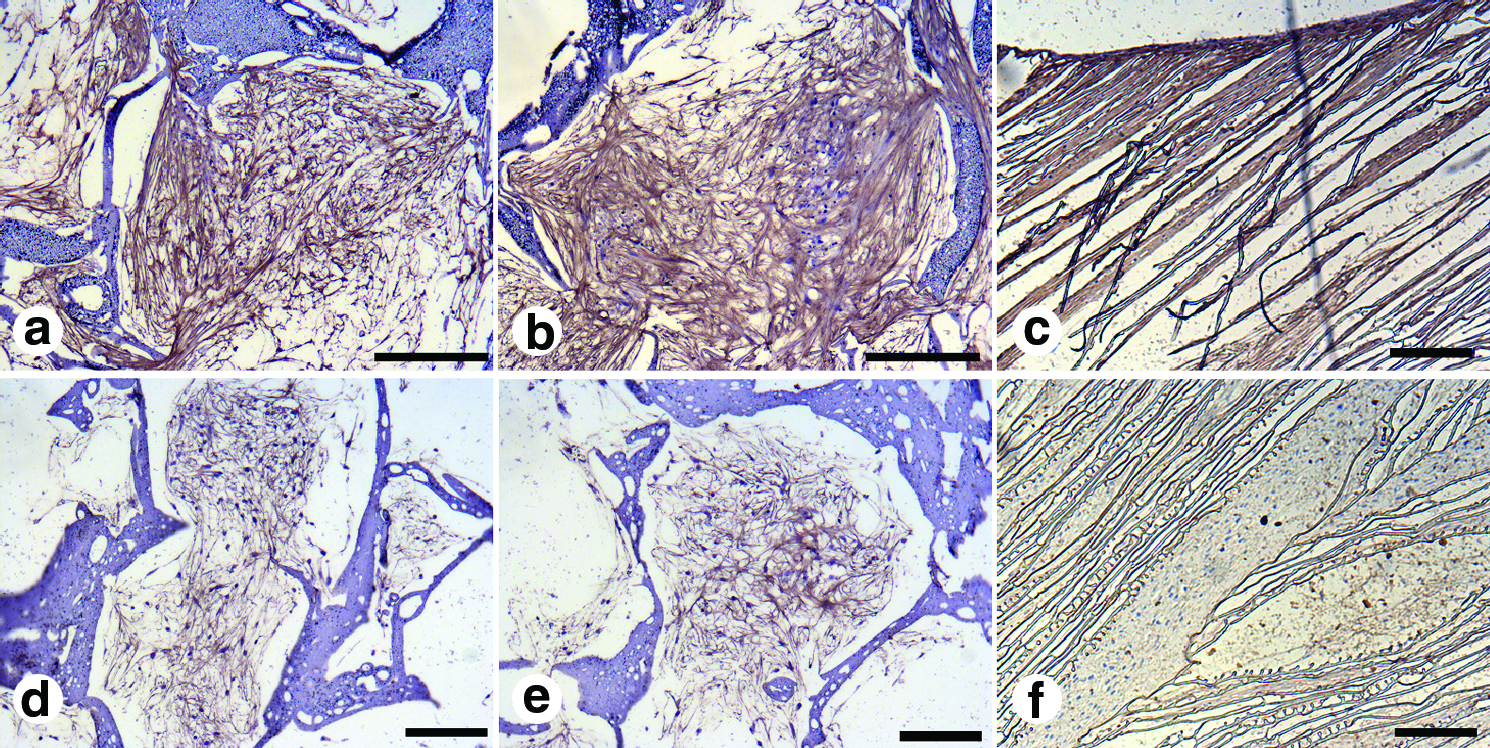
Silk scaffold histology sections showing immunostaining for collagen type I
Immunocytochemistry staining further revealed the presence of collagen type I and II deposition within silk scaffold layers at day 28. Differentiating hMSCs showed abundant collagen type I deposition as on day 28 compared to collagen type II in all silk layers (Fig. 4). Further collagen deposition appeared homogenously distributed and filled the scaffold pores.
Biochemical analysis
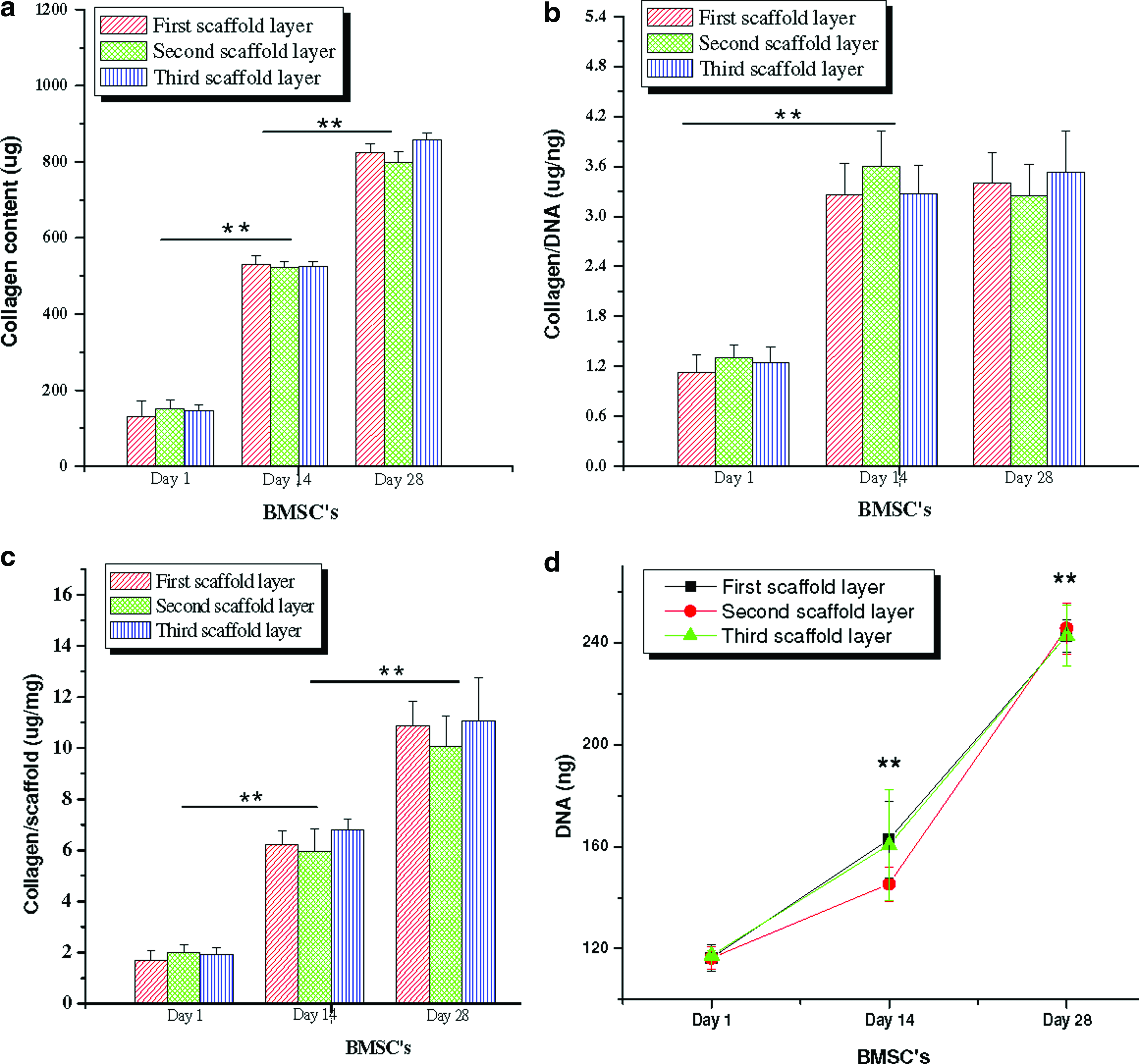
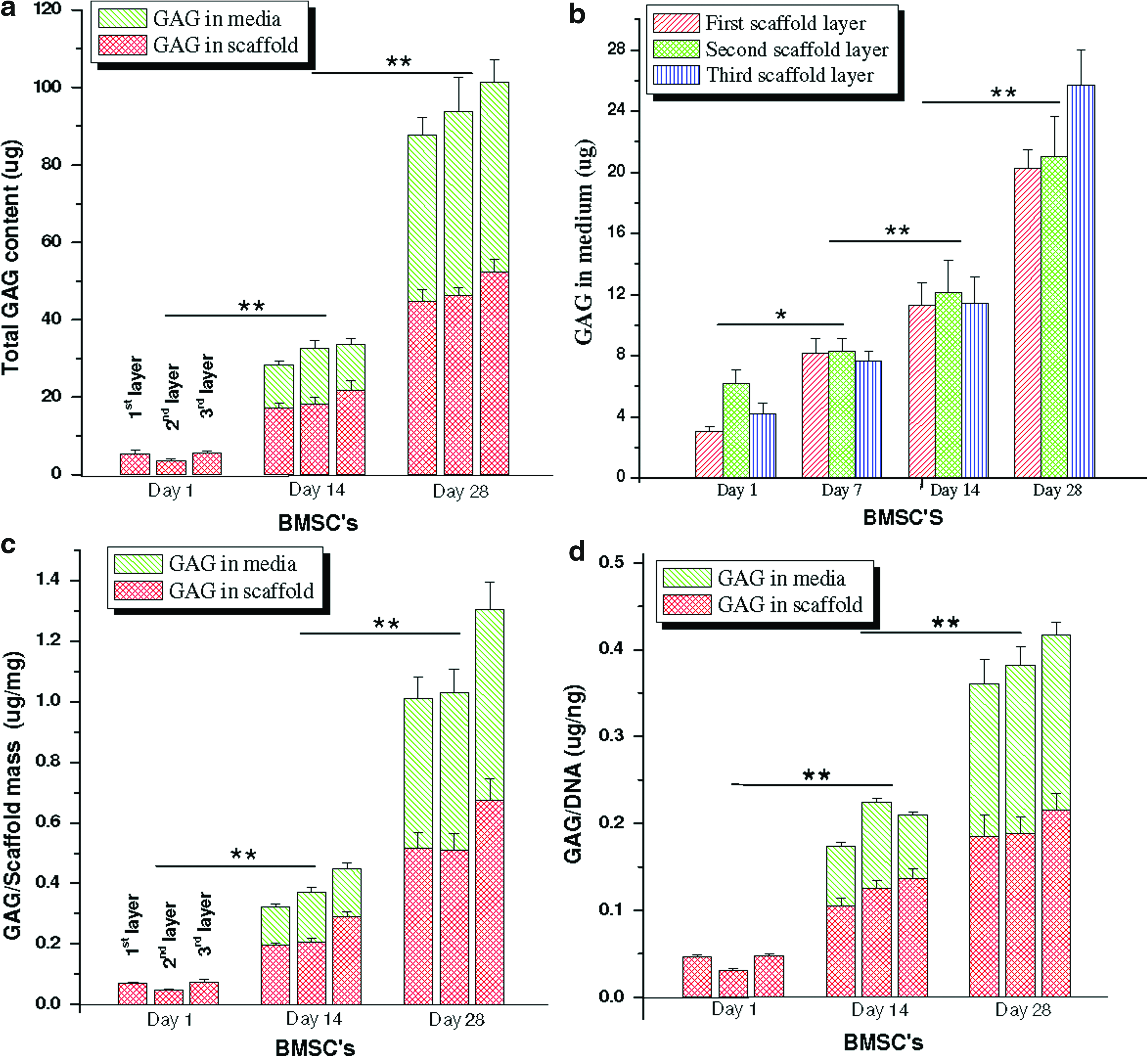
Biochemical analysis confirmed the differentiation of the hMSCs into a mature chondrocytic phenotype within scaffold meniscus layers. Both collagen and sGAG increased with time (p≤0.01) (day 28 vs. day 1) (Figs. 5 and 6). Total collagen increased ∼271% (520 μg) and ∼492% (820 μg) at day 14 and 28, respectively, in all three scaffold layers compared to day 1 (140 μg) (p≤0.01) (Fig. 5). Further, no statistically significant difference in total collagen content was observed between the three individual scaffold layers at all three time points. Similarly, for total sGAG content (scaffold+media), the levels increased ∼50 (30 μg) and ∼176% (93 μg) in chondrogenic medium at the end of day 14 and 28, respectively, compared to day 1 (5 μg) for all silk scaffold layers irrespective of differences in morphology (p≤0.01) (Fig. 5). From the results it can be observed that substantial amounts (∼40%–50%) of total sGAG was secreted into the medium with time while culturing these differentiating cells in chondrogenic medium (Fig. 6). At day 14, estimated sGAG amount in the medium was approximately ∼13 μg, which increased to ∼47 μg at the end of day 28 (p≤0.01) (Fig. 6).
Biochemical assay results showing
Biochemical assay results showing
To minimize discrepancies from cell numbers due to initial cell seeding differences within individual scaffold layer groups, total collagen and sGAG content was normalized with total DNA. The normalized collagen content per unit DNA increased ∼1.75-fold (3.3 μg/ng) at day 14 in all layers compared to day 1 (1. μg/ng) (p≤0.01), although the relative amount was constant at day 28 (p≤0.01) (Fig. 5b). Individual scaffold layers showed no statistically different values for collagen per unit DNA in either of the days. Similarly, total sGAG per unit DNA (present in media and deposited in scaffolds) after 28 days of culture increased by ∼9.75-fold (0.39 μg/ng) in case of differentiating hMSCs compared to ∼3.2-fold (0.128 μg/ng) after day 14 (p≤0.01) (Fig. 6d). Compared to deposited sGAG within scaffolds, secreted GAG in the medium amounted to ∼30%–50% of the total content in all three silk layers (Fig. 6). The amount of sGAG amount secreted by hMSCs was estimated in the chondrogenic medium from individual silk scaffold layers for 28 days (Fig. 6b). Differentiating hMSCs slowly enhanced sGAG production over time, suggesting the onset of differentiation events, leading toward attainment of a mature chondrocyte phenotype. Maximum sGAG production was reached at day 28 amounting to ∼20–25 μg compared to ∼3–6, ∼7–8, and ∼12 μg on day 7, 14, and 21, respectively (Fig. 6a). This trend was observed for all three silk meniscus layers without any statistical differences between layers.
Further, to monitor effect of individual scaffold mass and or volume and morphology on hMSCs differentiation and maturation, and on ECM deposition (if any), total collagen and sGAG were normalized per unit scaffold mass (Fig. 5c). Upon normalization, total collagen was independent of unit scaffold mass and morphology in individual layers, resulting in similar collagen deposition at days 1, 14, and 28 (Fig. 5c). However, there was a constant increase in total collagen content of ∼6 and ∼11 μg/mg scaffold at the end of day 14 and 28 compared to ∼2 μg/mg scaffold at day 1 (p≤0.01) (Fig. 5c). Similarly, upon normalization, no significant difference in sGAG amount was observed for differentiating hMSCs per unit scaffold within individual silk scaffold layers at days 1, 14, and 28. However, like total collagen, sGAG also increased with time both in the medium and within the scaffolds to ∼0.35–0.45 and ∼1–1.3 μg/mg scaffold at day 14 and 28, respectively, compared to ∼0.04–0.06 μg/mg scaffold at day 1 (Fig. 6c).
Real-time gene expression
To further support the biochemical results and confirm chondrogenic differentiation of hMSCs, real-time reverse transciptase–PCR was carried out for cartilage-specific genes. Upon culturing hMSCs in chondrogenic medium for 28 days, there was upregulation of all chondrogenic markers with time, suggesting in vitro chondrogenesis in the silk scaffolds irrespective of individual layer sand scaffold morphology. Further, expression of collagen 1-α-1, a major constituent of native meniscus cell phenotype, was observed to increase nearly fivefold at day 28 compared to day 14 (p≤0.01) (Fig. 7a). Similarly, expression of SOX9 and aggrecan (AGC) genes, considered a hallmark for mature chondrocyte phenotype, was enhanced with time, reaching a maximum at day 28 (p≤0.01) (Fig. 7b, c).
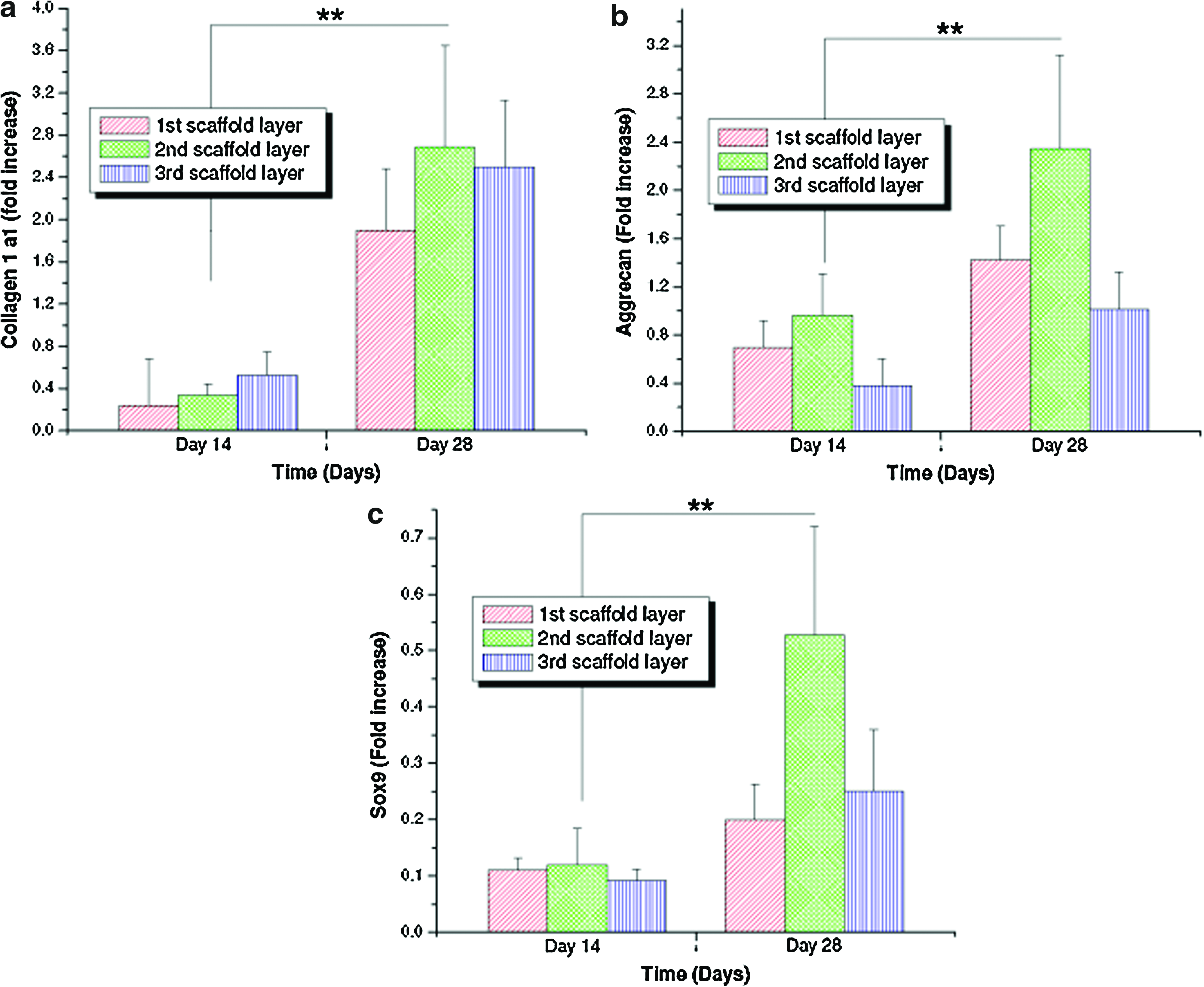
Real-time gene expression results showing fold increases of
Mechanical characterization
To evaluate biomechanical properties of the constructs, compressive testing was used to compare control and hMSC-seeded scaffolds. Compressive modulus for nonseeded (control) silk meniscus scaffolds was approximately 274, 297, and 150 kPa for the 350–400 μm (top), 500–600 μm (middle), and 60–80 μm (bottom) scaffold layers, respectively (Fig. 8). However, in the cell-seeded constructs, the compressive strength was higher with values of approximately 643, 542, and 424 kPa, respectively, for the top-bottom scaffold layers. Compressive modulus values were doubled due to possible combinatorial effects of bulk silk properties along with deposited ECM (mostly collagen and sGAG) from the differented hMSCs.
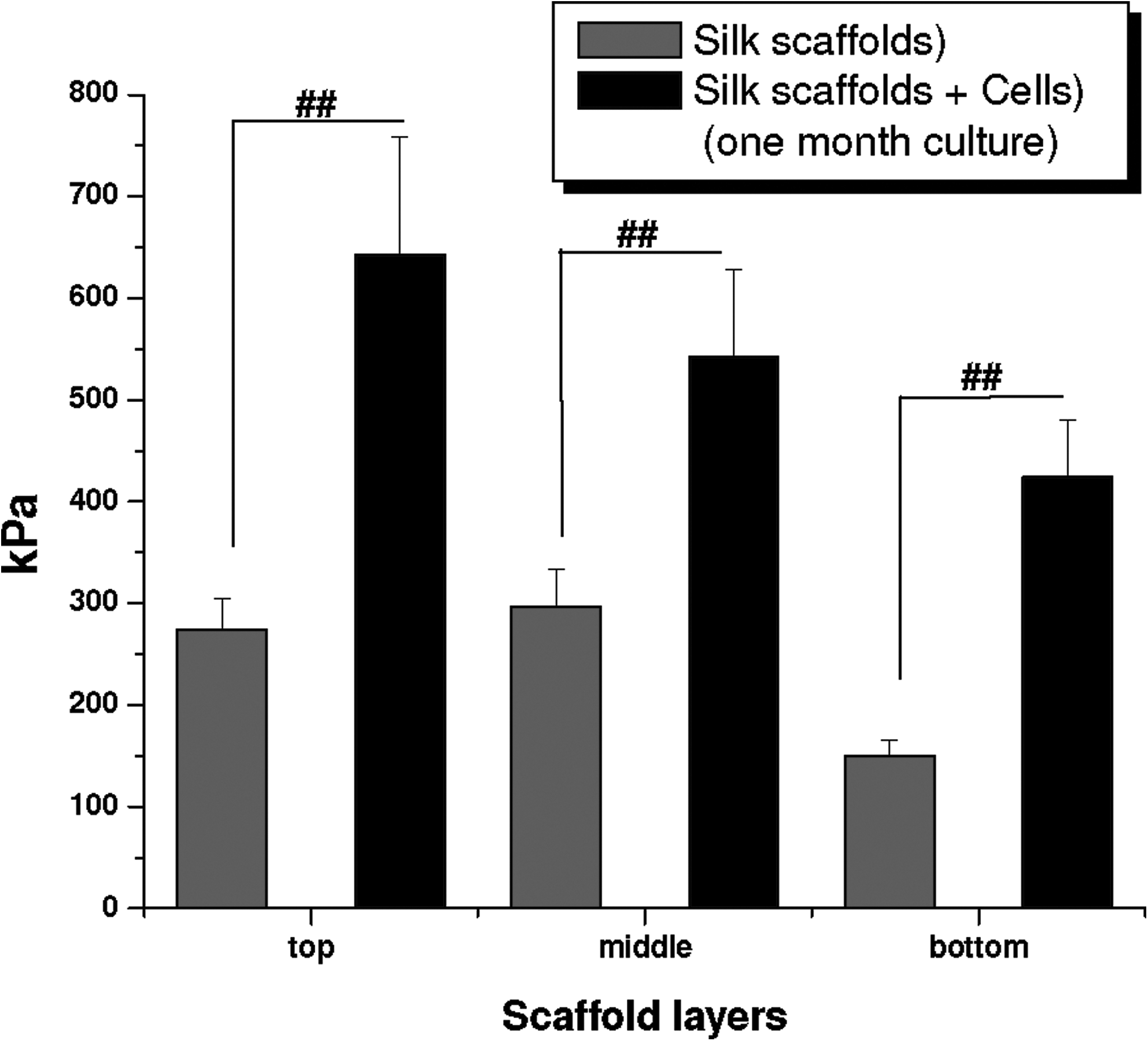
Comparative graph showing compressive modulus of individual silk meniscus scaffold layers with and without hMSCs cultured in differentiation media for 4 weeks. Data represent mean±standard deviation (n=4, ##p≤0.01).
Discussion
Meniscus allo/autograft represents a potential tissue engineering solution for meniscus defects to substitute for lost meniscal tissue. In this study native-like meniscus morphology was established in an in vitro 3D silk biomaterial model as originally proposed by Petersen and Tillmann (1998). We aimed at evaluating hMSC-scaffold interactions and changes related to hMSCs differentiation and ECM deposition toward achieving a biomechanically competent meniscus graft.24,25
In contrast to the available clinical option of allograft meniscus which are often associated with disadvantages like shape incongruency, disease transmission, and limited donor availability, custom designing for the lost/defective regions of the meniscal tissue using growth-factors, scaffolds, and their combinations, along with the possibility of using autologous hMSCs, makes current tissue-engineered meniscus approaches potentially useful.39,42 As previously shown in our earlier studies, the porous scaffold framework with pores of 350–400 and 500–600 μm provided initial mechanical support and guidance to the growing cells to deposit ECM and to generate a native-like fine meshwork 25 (Fig. 1). Further, the highly interconnected scaffold pores would provide conduits for hMSCs to migrate and nutrient and wastes to flow in and out (Figs. 1 and 2). This is an important design issue considering that earlier studies reported lack of cell migration into the inner core of the construct owing to smaller pores for migrating cells and increased cell death in inner core regions due to the hindrance of media transport.39,42 As per the Picogreen assay results, doubling of hMSCs within all scaffold layers, including the third bottom layer (with smaller laminar channels), confirms that cells survived and differentiated and also proliferated to double their initial seeding numbers, a sign of a congenial growth microenvironment within pores and lamina of the construct (Fig. 5).
Due to differences in scaffold pore size and orientation within layers, a difference in the ECM network was observed for growing and differentiating hMSCs, which tend to reorient and reorganize deposited ECM mimicking top and middle native meniscus architectures, confirmed by confocal and histology images (Fig. 2). Aligned and oriented ECM (mainly collagen and GAG) were observed in the present study by directed growth of hMSCs using the silk scaffolds as templates within bottom laminar channels (Figs. 1 and 2). High intrinsic tensile and compressive properties of the native meniscus are attributed to similar aligned and linearly organized collagen fibers lying at the bottom of the constructs.17,24,43 The deposited ECM further mimicked native tissue morphology and arrangement (mesh and laminar) as confirmed from confocal and histology sections (Figs. 2–4).
Mimicking meniscus fibrocartilaginous cellular phenotype is important to engineer clinically functional meniscus grafts. Successful recapitulation of native-like meniscus fibrocartilaginous structure was achieved within the scaffold layers using hMSCs differentiated toward a mature chondrocytic lineage using specific media and factors. From confocal images, biocompatibility of these silk scaffolds was further strengthened looking at confluent hMSCs occupying void scaffold pores accompanying good actin filament development and spreading, a sign of normal growth and development (Fig. 2). Similarly, enhanced cell proliferation within scaffolds layers as depicted by DNA assay results supported cell compatibility for future graft applications (p ≤0.01) (Fig. 5d). The late-passage hMSCs (P7) responded toward chondrogenic differentiation based on biochemical and gene expression studies toward a mature chondrocytic phenotype to attain features similar to native meniscus. Hence, the approach reported here further strengthens the scope of in vitro silk meniscus-like constructs to support late passage cells for growth and differentiation and suggests further investigation is warranted. This is an important observation as therefore only a small volume of a patients cells may be needed that could be passaged for seeding scaffold grafts in vitro for possible implantation.
From histology sections, intense staining to sGAG confirmed differentiation of seeded hMSCs to a mature chondrocyte phenotype with enhanced proteoglycan production within all individual silk layers, comparable to native tissue where proteoglycans consist of 2%–3% of the dry weight and form the cartilaginous zone of the meniscus (p≤0.01)3,44,45 (Fig. 3). GAGs play an important role in the maintenance of optimal visco-elastic behavior, compressive stiffness, and tissue hydration due to high water content (∼78%), further facilitating frictionless movement of the menisci over articular surfaces of the tibia and femur.46,47 Interestingly, the present study establishes the importance of cellular and cell–scaffold interactions on the cell shape of growing and differentiating hMSCs, as they appear more elongated within the larger pores but show a more compact morphology similar to mature chondrocytes when observed within third (bottom) laminar layers (Figs. 2–4). This is further supported by intense alcian blue and saffranin O staining of the bottom layer along with higher ECM content, where the bottom laminar layers produced higher sGAG compared to the two top layers with time in the presence of chondrogenic medium with TGF-β3 (p≤0.01) (Fig. 3b, c). This was possibly due to limited space available in the bottom layer where cells had a better chance to interact, resulting in morphological changes.
Increased total collagen further supported hMSCs growing within congenial microenvironments in pores and lamina of all individual silk layers to allow optimum growth and ECM deposition similar to native-like meniscus (p≤0.01). Knee meniscal fibrocartilaginous tissue contains mainly water (72%), collagens (22%), and glycosaminoglycans (0.8%).48,49 Of the total collagen content, Type I collagen accounts for over 90%, with the remaining 10% of meniscal collagens consisting of Type II, III, and V collagen.45,50 Immunostaining showed similar ratios of collagen type I and II within the silk scaffolds, where collagen I was found more abundant compared to collagen type II, a feature similar to the native meniscus (Fig. 4).44,45,50 Further, the presence of numerous collagen type I bundles observed at day 28, particularly in the bottom third layer (Fig. 4c, f), is expected to impart improved tensile properties. Oriented in a circumferential direction (similar to native meniscus) could prevent radial extrusion of the meniscus and help to maintain structural integrity during load bearing.17,24,43,46,47
Using real-time studies, insight on gene expression within all silk scaffold layers was gained to support hMSC differentiation. Transcript levels of cartilage-related ECM gene markers such as Col-I-α1, AGC, and SOX 9 were induced or significantly upregulated in hMSC-silk scaffolds cultured in chondrogenic medium supplemented with TGF-β3 (Fig. 7). Upregulation of collagen type I gene further supports the biochemical assays, as the major constituent of native meniscus.45,50 Similarly, SOX9 a key transcription factor for chondrogenic differentiation and cartilage formation was upregulated and is believed to precede the upregulation of cartilage-specific genes during in vitro chondrogenesis.51,52 In silk meniscus constructs, the expression of SOX9 coincided well with higher AGC expression, suggesting its induction (Fig. 7b, c).
Mechanical integrity is an essential prerequisite for a load-bearing structure like the knee meniscus to withstand in vivo stresses. In our previous study, we reported compressive modulus values approximately one-third (top and middle) and one-sixth (third-bottom layer) for nonseeded silk meniscus scaffolds compared to reported native human meniscus.25,53 Compressive values observed in the present study for control scaffolds (nonseeded) were as per our earlier findings (Fig. 8). Lower values for the bottom layer were attributed to the thinner walls within smaller pores compared to thicker pore walls formed in the larger scaffold pores during salt leaching. 25 Importantly, upon seeding hMSCs and culturing for a month on silk meniscus scaffolds enhanced compressive modulus was found, with values comparable to those reported for the native meniscus (Fig. 8). Any subsequent change in mechanics due to formalin fixation of cell laden scaffolds was negated by treating the control scaffolds in a similar way. Further, based on prior publications for this type of fixation, it has been reported that the impact is insignificant when fixed for a shorter period.54,55 Compared to human native medial meniscus with 718 and 605 kPa, respectively, for axial and radial compressive moduli at physiological strain rate, cell-ECM-laden silk constructs showed comparable values of 643, 542, and 424 kPa for top-bottom layers, respectively. 53 A two-fold increase in compressive modulus was observed due to ECM deposition upon differentiation of the seeded MSCs. Similar observations were reported for other meniscal studies where cell-ECM deposition contributed toward enhanced biomechanical values compared to controls.39,42 Similarly, on comparing anterior (1048 kPa) and posterior (329 kPa) modulus values for native human meniscus, the cell-laden silk constructs showed higher compressive modulus to reported posterior values, whereas for the anterior, they reached nearly a half for top two layers (643 and 542 kPa, respectively) and one-third (424 kPa) for the bottom layer. 53 We expect the final compressive values to be further enhance and match the native meniscus after prolonged cell culture with associated higher ECM deposition on the silk layers.
The present study is a step forward toward achieving biomechanically competent functional meniscus equivalents using stem cells. However, vascular and avascular zones within scaffolds using stem cells remained unaddressed in this report and it is unclear if this is a design feature that must be implemented in vitro, or something that in vivo will be accounted for by native tissue remodeling, although at slow rates. Finally, evaluating these new constructs for longer in vitro and finally in vivo studies is needed to fully evaluate utility in moving forward toward clinically relevant options.
Conclusions
A stem cell-based multilamellar/multiporous silk meniscus-like tissue has been demonstrated. Along with native-like compressive properties, mature chondrocytic phenotype mimicking native tissue structure was achieved. Further, cultured hMSCs showed higher cellularization with improved ECM deposition, including higher collagen and proteoglycans levels along with higher gene expression levels, supporting chondrocytic differentiation. Due to the utility of silk protein biomaterials, along with the positive tissue outcomes and biochemical features found in the present systems, potential utility toward meniscus repairs should be pursued.
Footnotes
Acknowledgments
We thank the NIH P41 EB002520 (Tissue Engineering Resource Center) for support of this research. Assistance of Sung Jun Kim, East-West Neo Medical Center, Kyung Hee University, Korea, in histology is greatly acknowledged.
Disclosure Statement
No competing financial interests exist.