
Abstract
This study investigated the potential of amniotic fluid stem cells (AFSCs) to synthesize mineralized extracellular matrix (ECM) within different porous scaffolds of collagen, poly-
Introduction
AFSCs osteogenic differentiation was previously described but the expression analysis of specific markers was carried out mainly by reverse transcription–polymerase chain reaction techniques. 4 This approach gives not enough information about proteins synthesized and secreted by differentiating AFSCs during the production of calcified extracellular matrix (ECM). Protein analysis can elucidate the timing of synthesis and secretion of specific bone proteins giving further information on the maturation degree of synthesized ECM.
Recently, a few articles described the use of AFSCs to colonize three-dimensional (3D) scaffolds for tissue engineering applications.3,4
Scaffolds play a pivotal role in tissue engineering by defining the 3D templates and synthetic ECM environments for tissue regeneration.5–7
Scaffolds can be made with different materials, mostly synthetic or natural polymers. The role of the material is to trigger and to control the crosstalk with the biological environment while imparting the scaffold the required mechanical and physical properties for the selected application.
Our previous studies have demonstrated that collagen scaffolds enhanced osteoblastic differentiation of dental pulp stem cells (DPSC). 8
In this study we first analyzed the osteogenic differentiation of AFSCs on two-dimensional (2D) surfaces, by western blot (WB) and immunofluorescence analysis, to give further evidence on their differentiation in osteoblast-like cells. The production of calcified ECM was also verified.
Afterward, we have compared the above-reported collagen scaffolds with scaffolds made by a synthetic polymer, poly-
Fibroin is one of the two proteins composing the silkworm filament. It has specific amminoacidic sequences in the backbone chain that behave like RGD sequences and are able to bind directly to cell receptors. 13
With different procedures, fibroin can be processed to fabricate films, foams, nonwoven micro- and nanonets, and solid or injectable gels. Various potential applications of fibroin for tissue engineering scaffolds have been proposed, thanks to unique properties that fibroin possesses in terms of biocompatibility and bioactivity.14–16
AFSCs seeded in these scaffolds were committed toward osteogenic lineage in vitro and then implanted in a rat subcutaneous model. The differentiation degree and the production of calcified matrix were than evaluated in comparison with DPSCs, considered as an example of cells easily committed to osteogenic lineage.8,17
Materials and Methods
Preparation of fibroin and PDLLA scaffolds
All scaffolds have been prepared starting from fibroin and PDLLA solutions containing salt crystals, by casting in a proper mold and subsequent salt leaching.
Fibroin water solution
Bombyx mori cocoons purchased from Cooperativa Sociolario were degummed by treating twice with 1.1 g/l Na2CO3 water solution at 98°C for 1 h, washing in deionized water, and air-drying. After degumming, silk was dissolved in 9.3 M (20% w/v) LiBr at 65°C for 2 h. To eliminate the salt, the solution was then dialyzed in a Slide-A-Lyzer Cassette (Pierce), (molecular weight cut-off [MWCO] 3500 Da) against distilled water for 3 days; the fibroin concentration was afterward adjusted to 8% w/v.
PDLLA solution
PDLLA (type RESOMER® 207, Mw=255 kDa) was purchased from Boehringer Ingelheim. The polymer was used without further purification. PDLLA was dissolved in a dichloromethane:dimethylformamide (70:30 v/v) at a 6.5%, w/v, concentration.
Preparation of scaffolds
Silk fibroin and PDLLA scaffolds were prepared by adding 8 and 15.2 g of granular NaCl (particle size≈315–425 μm) into 4 mL fibroin solution and 8 mL PDLLA-DCM/DMF solution, respectively, and then poured in cylindrical polystyrene plates (3.5 cm in diameter, 1.5 mm in height). The containers were covered, left at room temperature for 24 h, and then immersed in deionized water for 3 days with water change every 6 h to remove the porogen. 16
From the obtained materials, discs with a height of 1.5 mm and diameter of 5.2 mm were cut.
All samples were sterilized by Cobalt-60 gamma rays using facilities and control procedures validated in conformity with UNI EN ISO 9001, UNI EN ISO 13485, UNI EN ISO 11137 e GMP by Gammarad Italia S.p.A with a standard cycle of 25.00 kGy and a dosimeter batch Red Perspex 4034.
All sample characterization has been performed before and after the sterilization procedure.
Material characterization
Molecular weight measurements
Molecular weight (Mw) and number average molecular weight and polydispersity index (PI) of materials before and after gamma rays irradiation were determined by gel permeation chromatography (GPC). Silk proteins were dissolved in 9.3 M LiBr (Fluka Chemical) at a concentration of 10% w/V at 65°C for 5 h and filtered to eliminate impurities. LiBr was removed by dialysis against distilled water for 2 days at room temperature using a 3500 Da MWCO membrane (Slyde-A-Lyzer, Pierce). After dialysis the solution was filtered. Molecular weight analysis was performed with a high permeation liquid chromatography/GPC system equipped with a Shodex Protein KW-804 column, a Jasco1570 UV/Vis detector set at 274 nm, and a Spectra Physics P4000 isocratic pump. The calibration curve was obtained using protein standards (Pharmacia). Samples and standards were diluted with eluent (pH 7.5, composition: 4 M urea, 20 mM Tris HCl, and 0.15 M NaCl) at a concentration between 0.25% and 1.25% w/V and measurements were conducted with a flow rate of 1 mL/min. Chloroform was the solvent for PDLLA and the instrument was calibrated by using polystyrene standards.
Morphological analysis
Scanning electron microscopy (SEM; Supra 40–Zeiss:operating mode: high vacuum, secondary electron SE detector) has been used for morphological observations of scaffolds before and after sterilization. For SEM imaging, dried samples were sputter coated (SEM Coating Unit PS3, Assing S.p.A.) with a thin layer of gold in argon atmosphere (20 mA at 5×10−7 Pa for 30 s).
Thermal analysis
Thermal analysis has been used to determine the effect of sterilization on the materials thermodynamic parameters. A DSC 30 (Mettler-Toledo) has been used, with a nitrogen flux of 10 mL/min and two heating scans from 0°C to 200°C at 10°C/min with a rapid cooling in between (100°C/min) for PDLLA and a single heating scan from 0°C to 340°C at 10°C/min for fibroin.
Cell culture and seeding
Supernumerary amniocentesis samples were provided by the Laboratorio di Genetica. All samples were collected with informed consent of the patients according to Italian law and ethics committee guideline.
AFSCs were isolated as previously described by De Coppi et al. 1 Back-up human amniocentesis cultures were harvested by trypsinization, and subjected to c-Kit immunoselection by MACS technology (Miltenyi Biotec). Human AFSCs (hAFSCs) were subcultured routinely at 1:6 dilution and not allowed to expand beyond the 70% of confluence.
Human DPSCs (hDPSCs) were isolated as previously described by Riccio et al. 8
hAFSCs and hDPSCs were grown in α minimum essential medium (αMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Invitrogen).
For the osteogenic differentiation studies on 2D petri dishes, cells were seeded at a density of 3000 cells/cm2 and cultured in osteogenic medium (culture medium supplemented with 100 nM DEX, 10 mM b-glycerophosphate, and 50 μg/mL ascorbic acid-2-phosphate; Sigma-Aldrich).
For the differentiation studies on 3D scaffolds, to improve their wettability, the gamma ray-sterilized PDLLA scaffolds were first soaked in ethanol for 30 min, and then exchanged with phosphate-buffered saline (PBS) for three times (30 min each one). All the scaffolds were then washed twice with αMEM containing 10% FBS (1 h for each rinse). Cells were seeded on each scaffold at density of 1500 cell/mm3 and transferred 24 h later into 6-well tissue culture plates containing 3 mL of medium per well. Cell/scaffold cultures were maintained in osteogenic medium for up to 4 weeks in an incubator at 37°C with 5% CO2.
Cell viability inside the scaffolds after 7 days of culture was tested by 6-carboxyfluorescein diacetate (6-CFDA) probe (Sigma-Aldrich) staining and analyzed by in vivo confocal microscopy.
Western blotting
Whole cell lysates were obtained from undifferentiated and differentiating AFSCs at various culture times. Cell lysates were obtained as previously described. 18 About 50 μg of protein (Bradford assay) for each sample was separated by 10% or 15% sodium dodecyl sulfate-poly acrylamide gel electrophoresis and then transferred to polyvinylidene di-fluoride membranes. Blots were incubated overnight with one of the following antibodies (Abs; diluted 1:1000 in Tris-buffered saline tween 20+2% bovine serum albumin [BSA], and 3% milk): rabbit anti-Runt-related transcription factor 2 (anti-Runx2) (AbCam); mouse anti-Osteopontin (anti-OPN; Santa Cruz); rabbit anti-Osterix (anti-Osx; GeneTex); mouse anti-Osteocalcin (anti-OCN; GeneTex). Membranes were then incubated with peroxidase-labeled anti-rabbit or anti-mouse secondary Abs diluted 1:5000, for 30 min at room temperature. All membranes were observed using Enhanced ChemioLuminescence (Amersham). Antiactin Ab was used as control of protein loading in timing experiments.
Densitometry was performed on three independent experiments by NIS software (Nikon). An equal area was selected inside each band and the mean of gray level (in a 0–256 scale) was calculated. Data were then normalized to values of background and of control actin band.
Subcutaneous implantation
hAFSC-scaffold and hDPSC-scaffold constructs were cultured in osteogenic medium for 1 week (medium changed twice) before implantation. For implantation, in the study male outbred rats with age ranging between 8 and 12 weeks (Charles River Laboratories) were used. One dorsal mid-sagittal incision was made on the dorsa and two subcutaneous pockets were created using blunt dissection. One scaffold with a volume of 600 mm3 (15 mm Ø×3.5 mm thickness) was implanted subcutaneously into each pocket. Each animal received two constructs. Three samples were implanted for each scaffold or scaffold/cell construct. After the scaffold implantation, the incisions were sewed with prolene 4–0 sutures (Ethicon). Animals were immunocompromised using Cyclosporine A at a dosage of 15 mg/Kg b. wt., administered 4 h before transplantation and then daily for 2 weeks. During the last 2 weeks the daily dosage was reduced gradually up to 6 mg/kg body weight.
Four weeks later the rats were sacrificed and the cell-scaffold constructs were rapidly fixed in 4% paraformaldehyde in PBS. The animal procedures were performed according to the guidelines approved by the Committee of Use and Care of Laboratory Animals of the University of Modena e Reggio Emilia.
Histology
Samples of 2D or 3D cultures were fixed in 4% paraformaldehyde in PBS at pH 7.4 for 15 or 60 min, respectively, and then processed for successive steps. Cells differentiated on Termanox coverslides were processed for immunofluorescence or common histological staining. Scaffold samples were embedded in paraffin and cut into 10-μm-thick histological sections. Routine hematoxylin/eosin (H&E) staining was performed on some samples to analyze morphological details. Fixed cells or histological sections were stained with von Kossa and Alizarin Red staining for light microscopic observation. Images of histological samples were obtained by a Zeiss Axiophot microscope equipped with a Nikon DS-5Mc CCD color camera.
The percentage of mineralized area in all specimens was measured using ImageJ software (NIH).
Immunofluorescence and confocal microscopy
Fixed monolayer cells and histological sections were processed as previously described by Riccio et al. 8 Rabbit anti-Runx2; mouse anti-OPN; rabbit anti-Osx; mouse anti-OCN; mouse anti-Human mitochondrial protein, Millipore; and rabbit anti-OCN, Millipore) diluted 1:50 for 1 h at RT were used as primary Abs. Secondary Abs were diluted 1:200 in PBS containing 3% BSA (sheep anti-mouse-fluorescein isothiocyanate, and donkey anti-rabbit-Cy3™, Jackson ImmunoResearch). After washing in PBS samples were stained with 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) in H2O for 1 min and then mounted with antifading medium (0.21 M DABCO and 90% glycerol in 0.02 M Tris, pH 8.0). Negative controls consisted of samples not incubated with the primary Ab. The multi-labeling immunofluorescence experiments were carried out avoiding cross-reactions between primary and secondary Abs.
Fluorescent samples were observed by a Nikon TE2000 microscope equipped with a CCD camera Hamamatsu ORCA 285. Images were captured and processed by NIS software (Nikon). Confocal imaging was performed on a Leica TCS SP2 AOBS confocal laser scanning microscope as previously described. 18 The confocal serial sections were processed with the Leica LCS or ImageJ software to obtain 3D projections, as previously described. 8 The image rendering was performed by Adobe Photoshop software.
Statistical analysis
For all experiments, values were reported as mean±standard deviation based on triplicate cell cultures. The experiments were performed twice to ensure reproducibility. To test the significance of observed differences between the study groups, an unpaired Student's t-test was applied. A p-value<0.05 was considered to be statistically significant.
Results
Mineral deposition on 2D surface
hAFSCs were isolated from amniotic fluid cell populations by MACS technology. Flow cytometry and immunofluorescence experiments were performed on selected cells by using a specific Ab to verify that sorted cells are mostly c-Kit+ (data not shown).
To obtain osteogenic differentiation, hAFSCs were plated on a plastic surface at the density of 3000 cell/cm2 and then cultured in osteogenic medium up to 30 days. Osteogenic differentiation was evaluated by morphological, biochemical, and immunocytochemical methods. Differentiating cultures and control cells were observed by phase-contrast microscopy to evaluate the morphological changes and the proliferation rate (Fig. 1). Seeded cells showed a fibroblast-like morphology, proliferated, and reached the confluence as monolayer at day 7 in both differentiating and control cultures. Differentiated cells were characterized by a decrease in proliferation activity and started to form nodular aggregates since day 14 (Fig. 1).
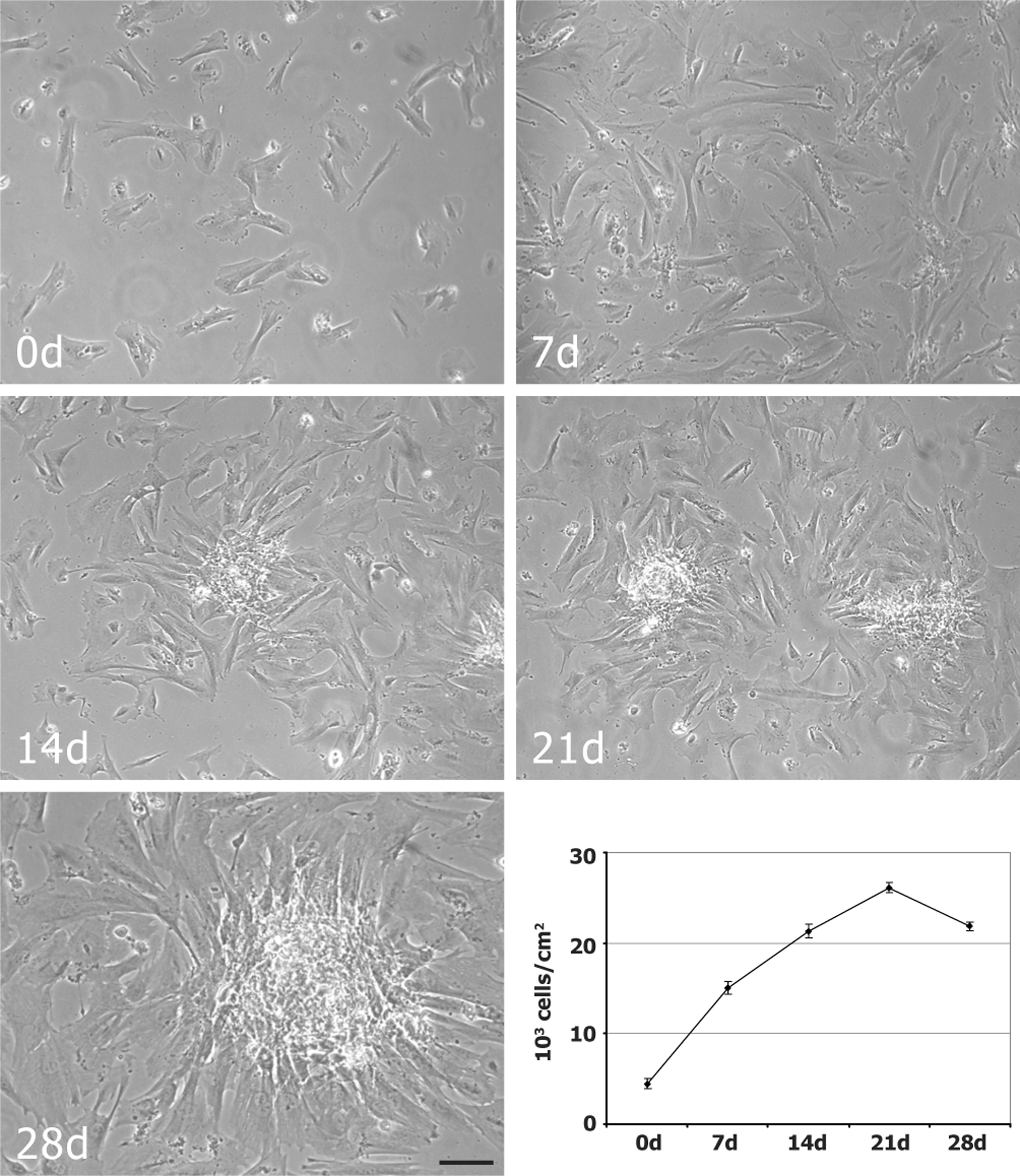
Growth dynamics of differentiating hAFSCs. Phase-contrast images of hAFSCs maintained in osteogenic medium for 0, 7, 14, 21, and 28 days. Scale bar: 50 μm. Diagram representing the proliferation rate of differentiating hAFSCs. Data were calculated by three independent experiments and presented as the mean±standard deviation (SD) of cell number for cm2. hAFSCs, human amniotic fluid stem cells.
To verify if hAFSCs were able to produce calcified matrix, cells were stained with Alizarin Red or von Kossa (Fig. 2). The presence of calcium deposits became evident after 14 days of culture when nodular aggregates appeared stained in red/orange or black.
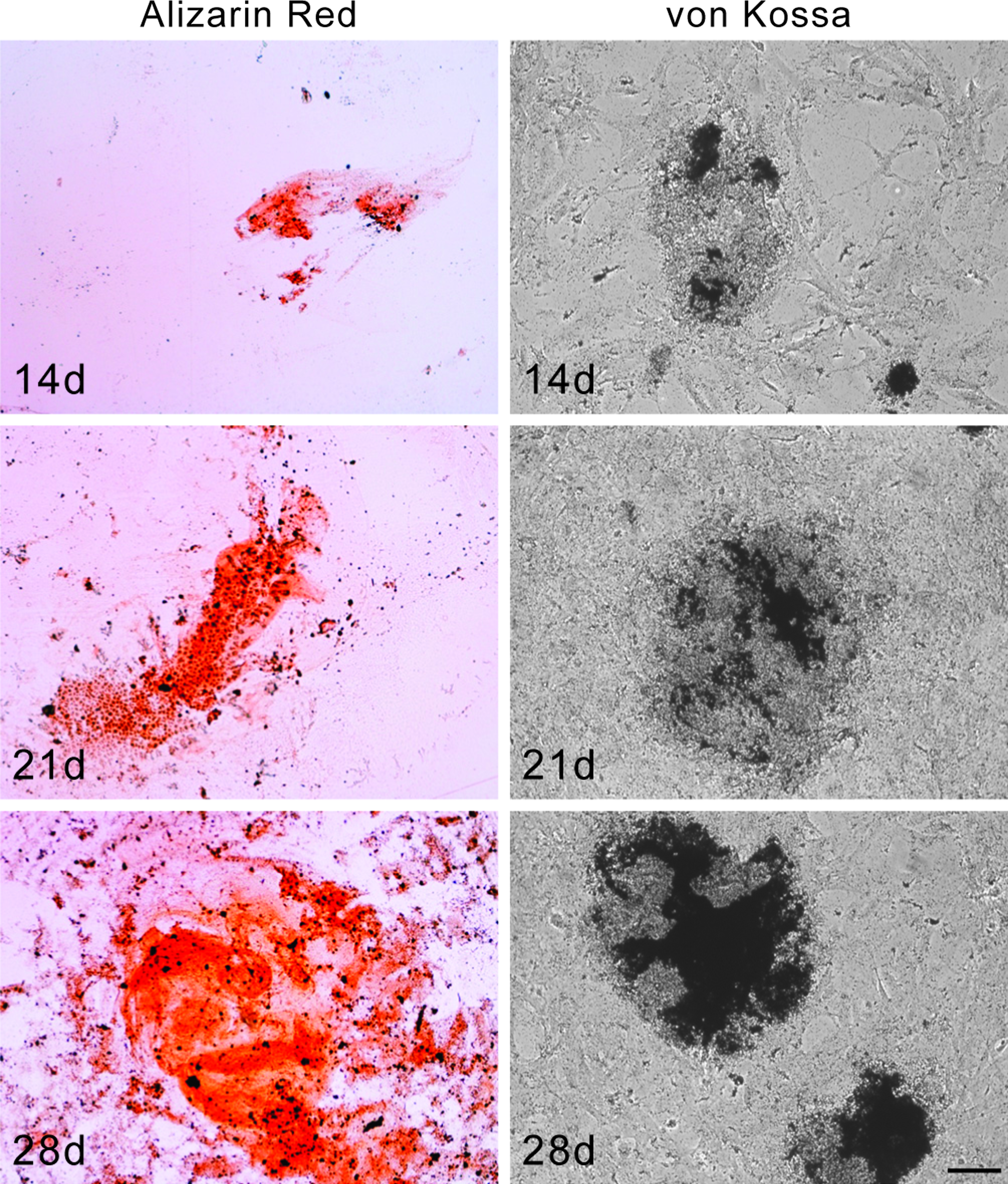
Histochemical analysis of hAFSCs grown in osteogenic medium. Images of 14, 21, and 28 days cultured hAFSCs stained with Alizarin red and von Kossa. Scale bar: 50 μm. Color images available online at www.liebertonline.com/tea
Expression of osteoblastic differentiation markers
To verify the commitment and differentiation of hAFSCs into osteoblast-like cells, the presence of specific markers, such as Runx2, Osx, OPN, and OCN, 8 was analyzed by immunofluorescence analysis and WB in whole cell lysates of hAFSCs.
Double immunofluorescence labeling was carried out to analyze simultaneously the localization of OPN and Runx2 in hAFSCs (Fig. 3). In the same manner it was observed the distribution of OCN and Osx (Fig. 3). During hAFSC osteogenic differentiation, OPN was localized in the cytoplasm, above all in the perinuclear region that normally contains the rough endoplasmic reticulum (RER) (green signal). Runx2 presented the nucleoplasmic distribution typical of transcription factors (red signal). At day 7 it was present approximately in all cell nuclei while at day 14, when a strong decrease was observed by WB, hAFSCs showed a lower labeling. At day 28 Runx2 signal increased again. OCN showed typical cytoplasmic localization with a pattern compatible with the distribution of RER (green signal). Similarly to Runx2, Osx showed a nucleoplasmic localization (red signal). The signals from two proteins strongly increased at days 14, having a peak at day 28.
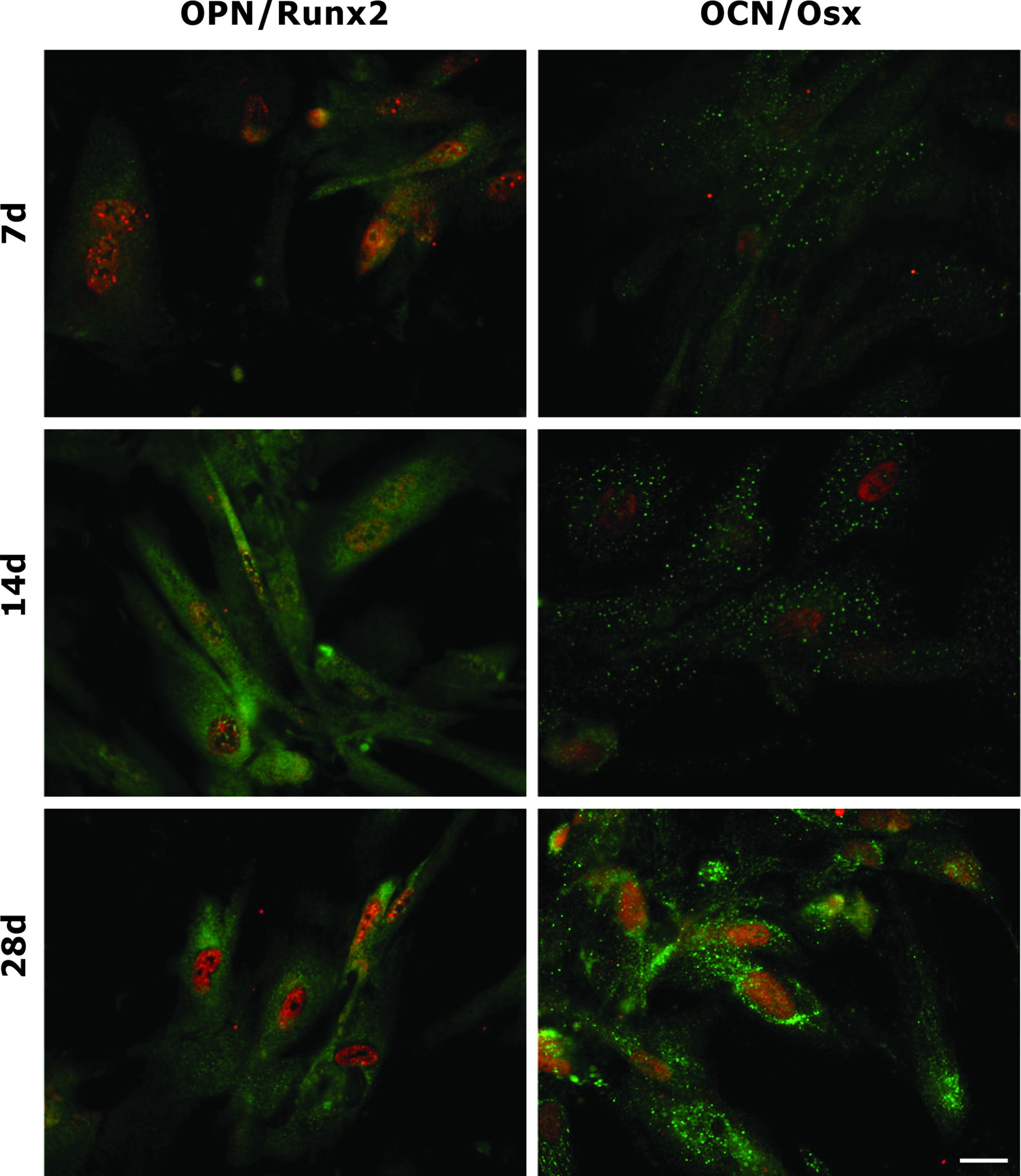
Immunofluorescence analysis of hAFSCs grown in osteogenic medium. Images showing superimposing between OPN and Runx2 signals of hAFSCs at 7, 14, and 28 days of culture. Double fluorescence is given from anti-OPN (green) and anti-Runx2 (red). Images showing superimposing between OCN and Osx signals of hAFSCs at 7, 14, and 28 days of culture. Double fluorescence is given from anti-OCN (green) and anti-Osx (red). Scale bar: 10 μm. OPN, Osteopontin; OCN, Osteocalcin; Runx2, Runt-related transcription factor 2. Color images available online at www.liebertonline.com/tea
Immunofluorescence analysis was confirmed by WB experiments. A densitometric analysis was carried out on the WB bands to obtain a semi-quantitative analysis of the protein amount. Figure 4A, B shows that Osx is weakly present at day 7 and then increases in a time-dependent manner. On the contrary, Runx2 is detectable in all the experimental time points, but Runx2 amount decreases at days 14. The down-regulation of this protein is consistent with the role of Runx2 that acts as negative modulator of osteoblast proliferation. 19 OPN was weakly expressed until day 7, and then presents an expression peak at day 14 maintained until day 28. OCN was detectable during hAFSC osteogenic differentiation but increased at day 21 up to day 28.

Expression pattern of hAFSCs grown in osteogenic medium. Western blot analysis
Materials characterization
Comparison of the molecular weight indicates that gamma rays irradiation induced molecular mass degradation on both polymers, as shown in Table 1. Consistently with the literature, which is more abundant for PDLLA and scarce for fibroin,20,21 both polymers showed a sensible decrease of Mw, PDLLA approximately −40% (from 255 to 155 kDa) and fibroin approximately −70% (from 309 to 82 kDa), in both cases with a decrease of the PI. DSC analysis curves of PDLLA and fibroin before and after gamma rays sterilization revealed slight increase of the glass transition (from 58°C to 61°C) for PDLLA and a slight decrease of the degradation temperature (from 289°C to 287°C), for fibroin, with some other but not relevant changes. The results are consistent with the observed molecular weight decrease for fibroin and indicate that, due to the gamma rays irradiation, some crosslinking occurred in PDLLA.
PDLLA, poly-
SEM was used to evaluate the scaffold morphologies and to assess the effects of the sterilization process. In general, all scaffolds had a highly porous network with pore sizes in the range of approximately 100–300 μm, but PDLLA sponges showed a lamellar structure in contrast with the fibrous-like structure of fibroin sponges (Fig. 5A and C). Gamma rays treatment did not affect the general structure of the scaffolds, even if they caused the break of lamellae in the PDLLA sponges (see Fig. 5B arrow), and thickening of pore walls, shrinkage, and subsequent decrease of the pore sizes for fibroin (see Fig. 5D).
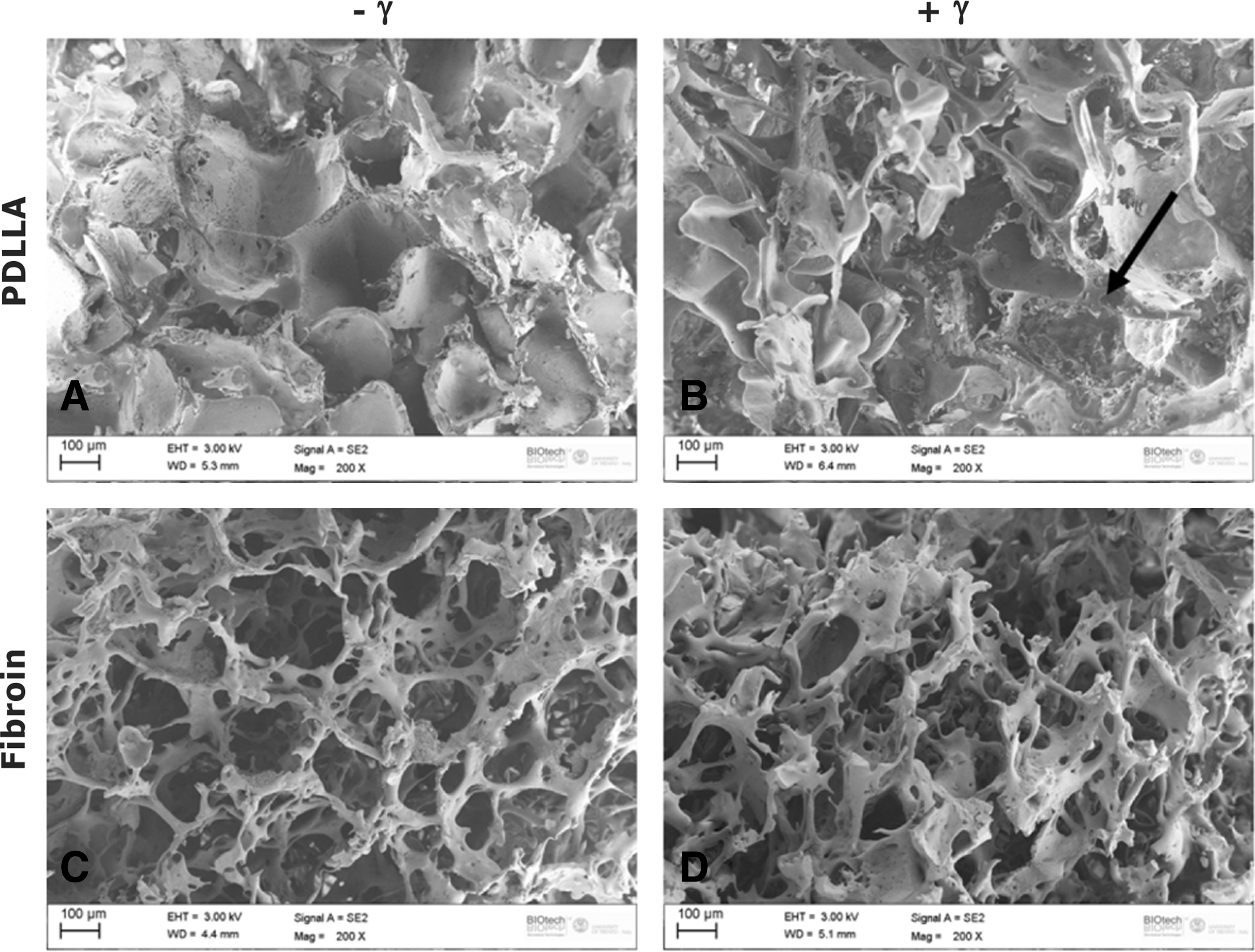
Scanning electron microscopy images of PDLLA and fibroin samples before
Fourier transform infrared spectroscopy–attenuated total reflectance measurements performed on fibroin scaffolds before and after gamma radiation did not show any changes in the pristine beta-sheet conformation of the protein conformation (data not shown).
Osteogenic differentiation on 3D surface for ectopic implant
To study the bone-forming ability of hAFSCs on collagen, fibroin, and PDLLA scaffolds, hAFSC-scaffolds were compared with hDPSC-scaffold constructs. Stem cell scaffold were induced in osteogenic medium for 7 days and were subcutaneously implanted into immune-suppressed rats. Figure 6 (A, B, K, L) shows cell distribution in the substrates 7 days after the cell seeding into the scaffold before implant. In both the scaffolds hAFSC (Fig. 6A, B) and hDPASC (Fig. 6K, L) are present and distributed within the micro-cavities delineated by the scaffold architecture. No difference in the cell density was observed between the scaffolds. Further, test performed by 6-CFDA probe demonstrated that before implant scaffolds are colonized by viable cells. Four weeks after subcutaneous implantation, collagen was completely reabsorbed (data not shown). Fibroin scaffold alone did not present mineralized matrix (data not shown), whereas a significant ectopic calcium deposition was found in hAFSC/fibroin construct, as noted by von Kossa and Alizarin staining (Fig. 6C, G). On the other hand, mineralization of PDLLA scaffolds was not evident by Alizarin staining (Fig. 6H), which was confirmed by von Kossa staining for the determination of calcium contents in the scaffolds (not shown).
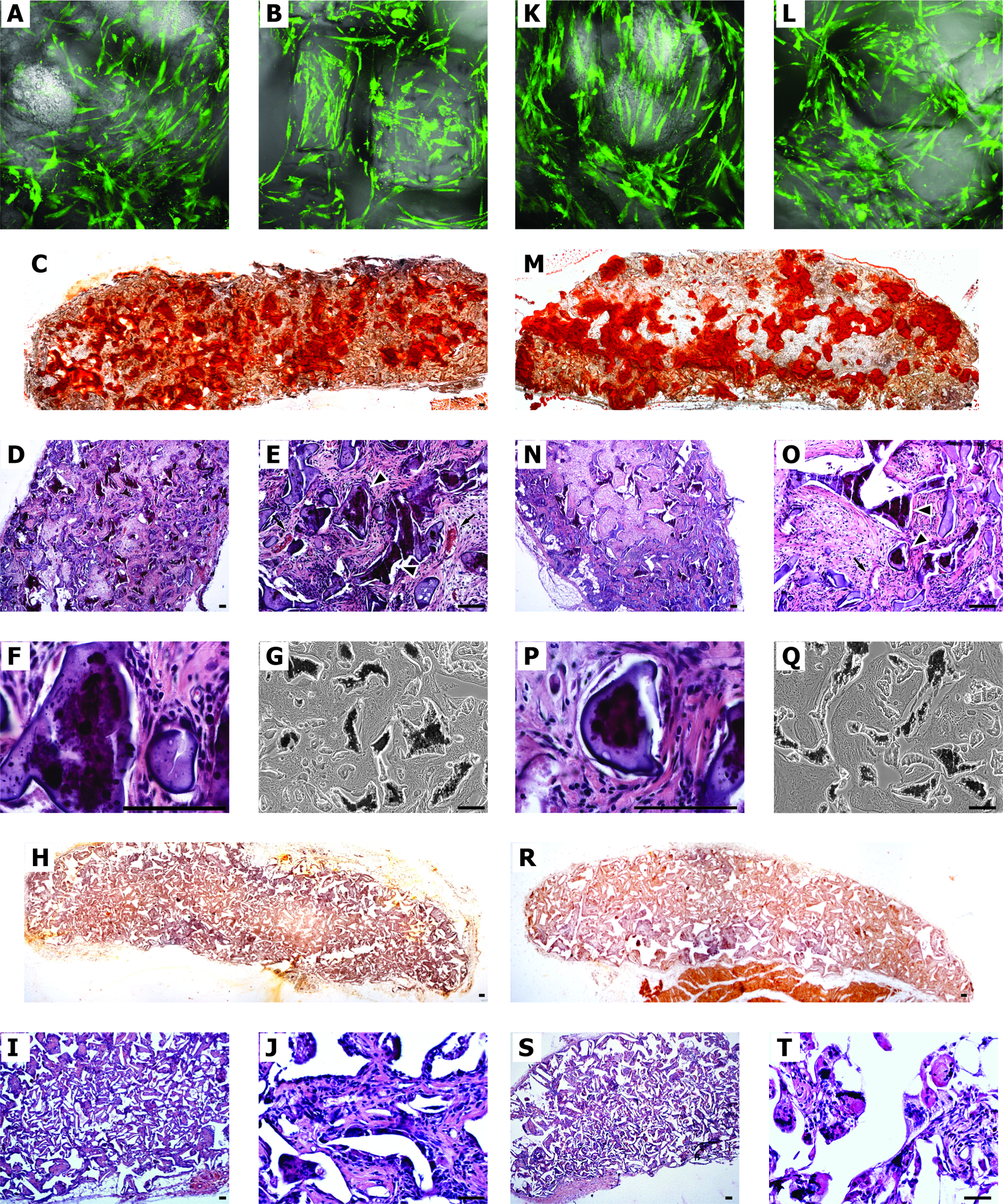
Microscopical analysis of hAFSCs cultured in three-dimensional (3D) scaffolds. Constructs of scaffold and stem cells [hAFSCs
Further, H&E staining shows bone-like tissue formation inside the scaffold area and a diffuse vascularization (Fig. 6D–F), indicating that fibroin is a suitable scaffold for bone regeneration by stem cells. The in vivo data showed that fibroin scaffolds provided hAFSCs with a more favorable microenvironment for their osteogenic differentiation and bone formation than PDLLA scaffolds did (Fig. 6I, J). Indeed, collagen scaffold could not be evaluated in ectopic implant, since the cell-scaffold construct was not evident after 4 weeks, suggesting a short-time reabsorption process.
Evaluation of mineralized areas in fibroin scaffold revealed greater amounts of calcium produced in hAFSC samples compared to that in hDPSCs (Fig. 7). In fact, histology with Alizarin and von Kossa staining revealed that there was heavy mineral deposition by hAFSCs (Fig. 6C, G) throughout the fibroin scaffolds after 4 weeks of culture, whereas there was a lower mineralization by hDPSCs (Fig. 6M, Q).

Areas of mineralization on fibroin scaffolds. Evaluation of mineralized areas in fibroin/AFSC and fibroin/DPSC constructs after 4-week implants. For each scaffold the mean percentage of mineralized area was calculated on five transversal section cut at interval of 1 mm in the mid-region of fibroin, fibroin/AFSC, and fibroin/DPSC constructs stained by Alizarin Red. Data showed in the graph represent the mean value (+SD) of the three different experiments.
To check if cells inside the scaffold were still from human origin and not from the host, immunofluorescence experiments with anti-Human mitochondria Ab were carried out. This Ab recognizes only mitochondrial protein from human origin. Data demonstrate that most of cells are clearly labeled by anti-Human mitochondria Ab (Fig. 8), indicating that the human stem cells remained in the fibroin and PDLLA subcutaneous implant up to the end of the experiment.
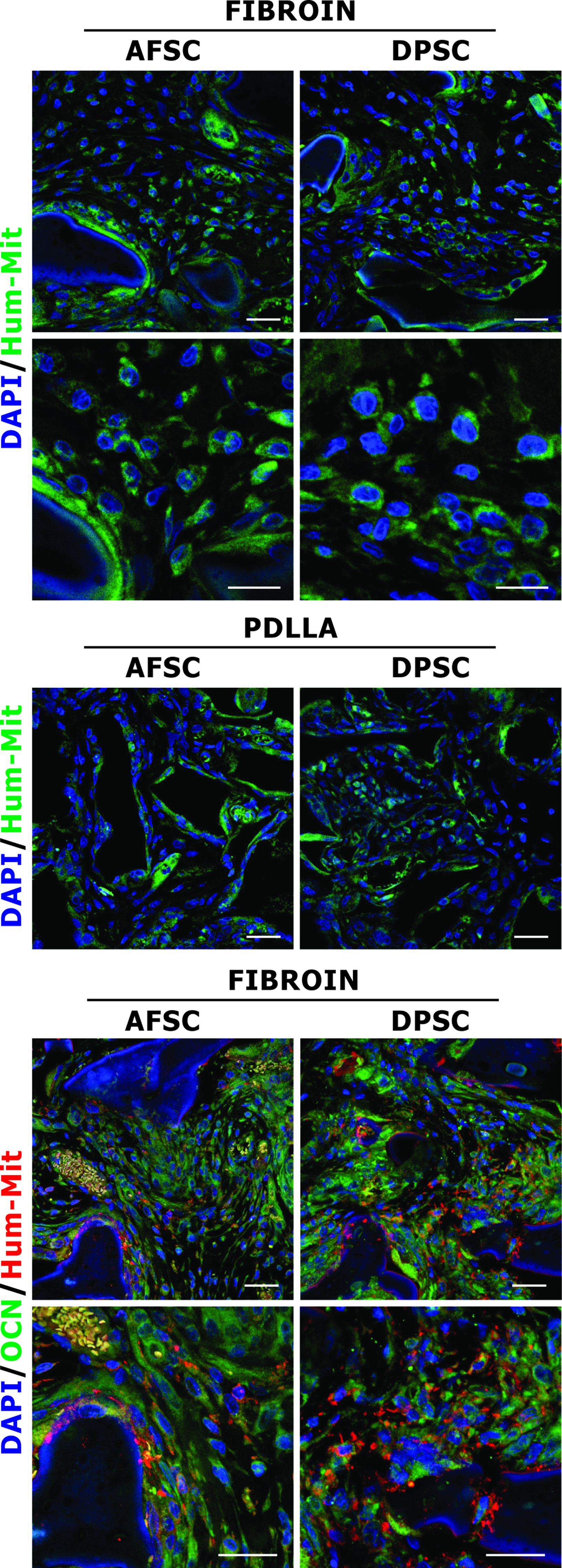
Immunofluorescence analysis of hAFSCs cultured in 3D scaffolds. Confocal images of double fluorescence signals from DAPI (blue) and anti-Human mitochondria protein (green) antibody are given for fibroin and PDLLA scaffolds, colonized with hAFSCs and hDPSCs, 4 weeks after subcutaneous implantation. Triple fluorescence images showing superimposing between DAPI (blue), OCN (green), and anti-Human mitochondria protein (red) signals in fibroin scaffold, colonized with hAFSCs and hDPSCs. Scale bar: 30 μm. DAPI, 4′,6-diamidino-2-phenylindole. Color images available online at www.liebertonline.com/tea
Contemporary expression of OCN and Human mitochondria was examined on fibroin scaffolds to verify if mineral deposition was effectively due to hAFSCs and hDPSCs (Fig. 8). Cells positive to OCN and Human mitochondria Abs are strictly adherent to fibroin fibres modifying to mineralized matrix, as also observed by H&E staining (Fig. 6B–D, L–N). Consistent with osteoblast activity and mineralization, expression of OCN was much higher in cells located around the matrix deposition area.
Discussion
In recent years, stem and progenitor cells have emerged as a promising regenerative medicine tool. Their characteristics of self-renewal and pluripotency suggest that stem cells may be useful to repair injured tissue and reconstruct damaged organs. 22
Amniotic fluid recently raised the interest of scientists as an alternative source of stem cells. From both surface markers and the potential ability, the AFS cell lines are different both from pluripotential ES cells and from multipotential adult stem cells, and may represent a new class of stem cells whose properties of plasticity exist somewhere between embryonic and adult stem cell types.1,2
These AFSs are easily obtained, largely avoid the ethical concerns associated with ESC use, and propagate in culture without difficulty, maintaining their pluripotential capacity. 23 In this study, we have confirmed that SC can be isolated and maintained in culture from human AF using magnetic microspheres reacting with c-Kit, a common marker for multipotential stem cells.
We then analyzed the osteogenic differentiation features of hAFSCs in vitro cultured on 2D surface and in vivo in 3D scaffolds. According to data previously described, osteogenic differentiation of hAFSCs was obtained by treatment with DEX to stimulate cell differentiation, ascorbic acid to stimulate ECM synthesis, and β-glycerophosphate to promote ECM mineralization.1,3 hAFSCs respond to this osteogenic condition growing as monolayer and forming typical mineral deposits and nodules described from other authors. 24 After day 21, the number of hAFSCs shows a slightly reduction probably due to the increase of cell death that normally occurs during late cell differentiation. Alizarin Red staining demonstrates that calcified ECM deposition occurred.
To verify if morphological and histological observations really indicate a hAFSC osteogenic differentiation, we have investigated in hAFSCs the expression of bone related proteins. Different transcription factors and proteins typical of bone ECM are expressed during osteoblast differentiation. Only a small number of these can be used as stringent markers because they are strongly expressed in bone tissue or present typical expression peaks during osteoblast differentiation.
Starting from day 14, hAFSCs express higher amount of Osx and OPN maintained until day 28. The increase of Osx occurring after Runx2 peaks is according to data that describe Osx positively governed by Runx2 during osteoblast differentiation.25,26
Previous data indicate that OCN is the most specific marker of late osteoblast differentiation because it is synthesized only by mature osteoblast at the end of matrix maturation process.27,28 After 21 days of culture, the regular increase of intracellular OCN levels indicates that hAFSCs undergo a late differentiation phase characterized by an ECM maturation process.
It is encouraging that hAFSCs could be induced by the classic osteogenic medium to differentiate along the osteogenic lineage with a stronger and faster mineralization by hAFSCs than that by hDPSCs. In our experimental conditions, it seems that 2 weeks of in vitro culture in conditioned medium are sufficient for hAFSCs to start the osteogenic differentiation process, whereas a longer (3 weeks) treatment is needed for hDPSCs. 8
All these considerations were useful for the second part of the study, since the identification of an appropriate time of predifferentiation in vitro could be pivotal for obtaining an optimal result in vivo. In the prospective of a future clinical application, it is important to identify the conditions to obtain a population already committed to osteogenic differentiation but still able to proliferate under regulation of the microenvironment of the implanting site.
Previous studies indicated that osteogenic differentiation is improved by culture in 3D scaffolds that mime the physiological condition.29–31 Biomaterials play central roles as designable biophysical and biochemical milieus that direct cellular behavior and function in regenerative medicine and tissue engineering.6,7 Calcified tissue can be produced in vitro using bone marrow MSC cultured on collagen sponge or similar bio-scaffolds, already utilized in surgery to support and improve bone regeneration.32,33 Here we compared collagen sponge with fibroin and PDLLA scaffolds seeded with hADFSCs or hDPSCs.
As specifically described with bone marrow-derived SC34,35 or by De Coppi et al. 1 with AF c-Kit+ SC, a good approach may be to initiate differentiation in vitro for a lineage-specific pathway, allowing these cells to complete their development and acquire some specialized functions in vivo.
In fact, we cultured in vitro for 1 week stem cells/scaffold constructs, before subcutaneous implant in rat. Scaffolds contain a similar number and distribution of stem cells at the day of the implant, therefore the in vivo osteogenic differentiation process for hAFSCs and hDPSCs could be promoted by the scaffold/cell micro environment.
We demonstrate that mineralization of hAFSCs was enhanced substantially on the fibroin scaffolds compared to collagen and PDLLA scaffolds.
Collagen scaffold as an ectopic implant is completely absorbed, and then this experimental approach does not give information about the employ of this scaffold for engineered osseous grafts. On the other hand, silk fibroin can be categorized as nondegradable or slowly degradable material, depending on the used processing methods and on the materials conformation. In fact, silk can be gradually digested by proteolytic enzymes. Therefore, this scaffold is more favorable for the osteogenic differentiation in ectopic implant leaving enough time for mineral deposition by hAFSCs. Even if there are only few reports of silk fibroin degradation, particularly by gamma radiation, 21 interestingly, the fibroin Mw decreasing due to gamma rays radiation seems to reinforce the effects regarding its antitumor activity and immune response. 36 Here we showed that gamma rays treatment did not affect the general structure of the scaffolds, even if they caused the break of lamellae in the PDLLA sponges.
In conclusion, the present study confirmed that the biomimetic microenvironment provided by the fibroin scaffolds is an important element besides the morphogenetic factors for osteogenic differentiation of hAFSCs in vivo.
Footnotes
Acknowledgments
We thank Dr. Gianfranco Croci (Laboratorio di Genetica, Ospedale Santa Maria Nuova, Viale Risorgimento, 80–42100 Reggio Emilia, Italy) for providing sample of amniotic fluid cells. This work was supported by grants from “Progetto Strategico per lo sviluppo nella sede di Reggio Emilia della Facoltà di Medicina e Chirurgia” Prot: 2010 0007725, Arcispedale S. Maria Nuova di Reggio Emilia and MIUR FIRB Accordi di Programma 2010 Prot: RBAP10Z7FS. For this study was utilized the confocal microscope Leica TCS SP2 of the C.I.G.S. (Centro Interdipartimentale Grandi Strumenti) of University of Modena and Reggio Emilia, financed from Fondazione Cassa di Risparmio di Modena.
Disclosure Statement
All the authors state that no competing financial interests exist.