
Abstract
Hematopoietic stem and progenitor cells (HSPCs) have been cultured using a wide variety of cytokines to promote differentiation into megakaryocytic cells (Mks), the precursors to platelets. Greater Mk DNA content, or ploidy, has been correlated with increased platelet release. Gradients of pH, pO2, and signaling factors regulate megakaryopoiesis in the bone marrow niche. In this study, we demonstrate that a 3-phase culture process with increasing pH and pO2 and different cytokine cocktails greatly increases megakaryocyte production. CD34+ HSPCs were first cultured at 5% O2 and pH 7.2 with a cytokine cocktail previously shown to promote Mk progenitor production. At day 5, cells were shifted to 20% O2 and pH 7.4 and maintained in 1 of 17 cytokine cocktails identified using a 24 factorial design of experiments method to evaluate the effects of interleukin (IL)-3, IL-6, IL-9, and high- or low-dose stem cell factor (SCF), in conjunction with thrombopoietin (Tpo) and IL-11, on expansion of mature Mks from progenitors. The combination of Tpo, high-dose SCF, IL-3, IL-9, and IL-11 best promoted Mk expansion. IL-3 greatly increased total cell fold expansion, but this was partially offset by lower Mk purity. IL-9 promoted CD41 and CD42b expression. High-dose (100 ng/mL) SCF increased Mk production and ploidy. Different commercial media and IL-3 sources substantially impacted differentiation, and X-VIVO 10 serum-free media best supported mature Mk expansion. Shifting from pH 7.4 to pH 7.6 at day 7 increased Mk production by 30%. Treatment with nicotinamide at day 7 or day 8 more than doubled the fraction of high-ploidy (>4N) Mks. Ultimately, the 3-phase culture system gave rise to 44.5±8.1 Mks and 8.5±3.1 high-ploidy Mks per input HSPC. Further optimization was required to improve platelet production. Using Iscove's modified Dulbecco's medium (IMDM)+20% BSA, insulin and transferin (BIT) 9500 Serum Substitute greatly improved the frequency and quality of Mk proplatelet extensions without affecting Mk expansion, commitment, or polyploidization in the 3-phase process. Mks cultured in IMDM+20% BIT 9500 gave rise to platelets with functional activity similar to that of fresh platelets from normal donors, as evidenced by basal tubulin distribution and the expression of surface markers and spreading in response to platelet agonists.
Introduction
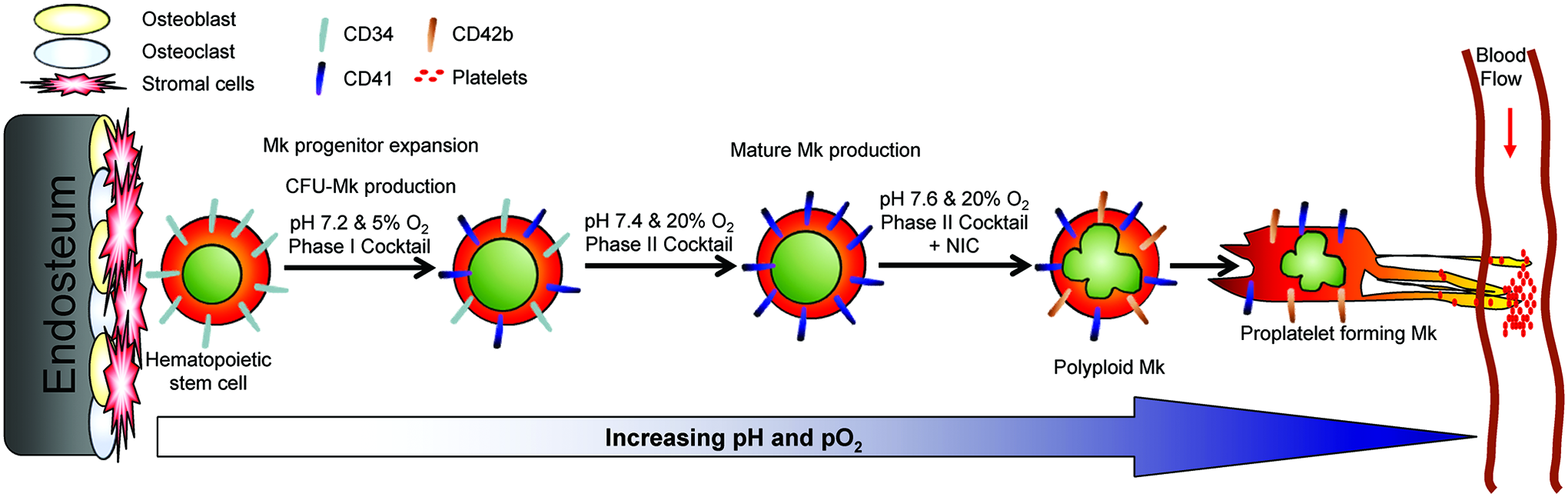
Schematic of Mk differentiation in vivo and in 3-phase culture. Hematopoietic stem and progenitor cells (HSPCs) reside close to the endosteal bone surface in the niche. During the course of differentiation, megakaryocytic cells (Mks) are exposed to continually increasing pH and pO2 until reaching the sinuses, where platelets are released from proplatelets aided by shear from the blood flow. Expression of CD34 decreases as Mks mature, while expression of CD41 and CD42b increases. Text above the arrows describes the niche-inspired culture system developed to optimize ex vivo Mk production from HSPCs. Color images available online at www.liebertpub.com/tea
CD34+ HSPCs that commit to the Mk lineage express surface CD41 and form Mk progenitors. Differentiating Mks undergo endomitosis, whereby cells replicate their DNA without completing cytokinesis to form polyploid cells with 4N, 8N, 16N, 32N, etc., multilobular nuclei. 10 Higher Mk DNA content is associated with greater platelet production.11,12 Finally, Mks extend proplatelets from which platelets are shed. Thrombocytopenia, a deficiency in blood platelets, is a consequence of several hematological malignancies and can be onset as a side effect of chemotherapy. 13 In vitro platelet production from HSPC-derived Mks could augment the supply and circumvent problems associated with bacterial and viral contamination, as well as immune rejection.14–16
Producing the 1–5×1011 platelets required for a single transfusion 17 presents a major challenge. CD34+ HSPCs from G-CSF-mobilized peripheral blood (mPB) or umbilical cord blood (CB) have been used to produce Mks by many investigators. Culture conditions that result in ≥50% Mks typically yield 5–10 Mks per input mPB CD34+ cell18–22 ; producing one transfusion dose under these conditions would require ∼250 million CD34+ cells. CB CD34+ cells typically yield 10-fold more Mks per CD34+ cell than mPB CD34+ cells.19,20,23 However, a CB harvest contains 2–5 million CD34+ cells,24,25 while a mPB harvest typically contains ∼200 million CD34+ cells.26–28 We recently achieved a yield of 14 Mks per mPB HSPC and obtained up to 45% high-ploidy (>4N) Mks. 29 This Mk productivity has the potential to allow production of one platelet transfusion from 125 million mPB CD34+ cells, but it is clear that the yield must be increased by another order of magnitude.
In order to maximize production of mature Mks from CD34+ HSPCs, we are optimizing cytokine combinations for different stages of Mk differentiation, as well as increasing pH and pO2 during culture to mimic gradients present in the BM niche where Mk differentiation takes place (Fig. 1). Our previous work focused on the production of Mk progenitors with the potential to yield high-ploidy (>4N) Mks. 29 Besides identifying an effective cytokine combination, we showed that shifting the Mk progenitors from 5% O2 and pH 7.2 to 20% O2 and pH 7.4 at day 5 greatly increased mature Mk production.
Several groups have produced small quantities of culture-derived human platelets, but have done so with co-culture,30–33 without regard to initial HSPC expansion,34,35 or with a focus on bioreactor design.36,37 The goal of this study was to first optimize expansion and differentiation of mature Mks from Mk progenitors produced in phase one, and then show that the resultant Mks could give rise to functional platelets. We used a 24 factorial design of experiments approach to evaluate the effects of interleukin (IL)-3, IL-6, IL-9, and high- and low-dose stem cell factor (SCF), in conjunction with thrombopoietin (Tpo) and IL-11 at 20% O2 and pH 7.4. The SCF level was investigated because it has been suggested that high-dose SCF inhibits Mk maturation.38,39 We also investigated shifting to supervascular pH 7.6 during terminal Mk differentiation, as well as the timing for adding nicotinamide (NIC), which greatly increases Mk ploidy.18,29,40
Materials and Methods
Unless noted, reagents were obtained from Sigma-Aldrich, antibodies from BD Biosciences, and cytokines from Peprotech. Cryopreserved CD34+ cells were purchased from the Fred Hutchinson Cancer Research Center with Northwestern University Institutional Review Board approval. Cells were obtained from healthy donors undergoing G-CSF mobilization following informed consent.
Cultures were initiated in T-flasks as described 29 with previously frozen mPB CD34-selected cells at 50,000 cells/mL with 100 ng/mL Tpo, 100 ng/mL SCF (Amgen), 2.5 ng/mL IL-3 (Peprotech or R&D Systems), 10 ng/mL IL-6, and 10 ng/mL IL-11 (Table 1, cocktail “c”) in media adjusted to pH 7.2 and maintained at 100,000–350,000 cells/mL via dilution feeding in a fully humidified chamber at 37°C, 5% CO2, and 5% O2 for 5 days.
CD34+ hematopoietic stem and progenitor cells were seeded in X-VIVO 20 with cocktail “c” at 5% O2 and pH 7.2. On day 5, cells were washed and resuspended in X-VIVO 20 at pH 7.4, supplemented with 1 of 17 cytokine cocktails using IL-3 from Peprotech, and maintained at 20% O2. Values represent concentrations in ng/mL. Symbols correspond to those in Figures 1, 2, and Supplementary Fig. S1.
Tpo, thrombopoietin; IL, interleukin; SCF, stem cell factor.
From day 5, cells were maintained at 20% O2 in media with various cytokines at 100,000–350,000 cells/mL. To shift to pH 7.4 or pH 7.6 at days 5 and/or 7, cells were centrifuged and resuspended in fresh media and cytokines.
Cytokine evaluation in 24 factorial design of experiments
Cells were maintained as noted above in X-VIVO 20 media (Lonza) with IL-3 from Peprotech for 13 days. At day 5, cells were centrifuged, resuspended in X-VIVO 20 adjusted to pH 7.4 and 20% O2, and cultured with 100 ng/mL Tpo, 10 ng/mL IL-11, and 1 of 16 cytokine cocktails comprising all combinations of SCF at 2.5 or 100 ng/mL and IL-3, IL-6, and IL-9 at 0 or 10 ng/mL (Table 1). A control flask containing cytokine cocktail “c” was also maintained in X-VIVO 20 at pH 7.4 and 20% O2. A flow chart of the culture process is shown in Supplementary Figure S1 (Supplementary Data are available online at www.liebertpub.com/tea).
Cell culture for media and IL-3 source evaluation
At day 0, cells were cultured in X-VIVO 10 (Lonza), X-VIVO 20, hematopoietic progenitor growth medium (HPGM; Lonza), StemSpan H3000 (StemCell Technologies), or StemLine II (Sigma-Aldrich) and supplemented with IL-3 from Peprotech or R&D Systems for 13 days. For days 0–5, cells were supplemented with cocktail “c” (Table 1) at pH 7.2 and 5% O2. At day 5, cells were centrifuged and resuspended in the basal media platforms used for the first 5 days, but adjusted to pH 7.4 and 20% O2 and supplemented with cytokine cocktail “3” (Table 1) with the same IL-3 source as for days 0–5. At day 7, cells were centrifuged and reseeded at 100,000 cells/mL in the same basal media platform adjusted to pH 7.6 and supplemented with the cytokine cocktail and IL-3 source used from days 5 to 7. For detection of colony-forming unit (CFU)-Mk, cells were seeded on day 0 in media supplemented with cocktail “c” (Table 1) at pH 7.2 and 5% O2. On day 5, cells were seeded into semi-solid MegaCult medium (StemCell Technologies) with 10 ng/mL IL-3 and 50 ng/mL Tpo and cultured at 5% O2 for 12 days. Colonies were then stained and characterized as described. 29
Cell culture for testing pH changes and NIC addition time
Cultures were initiated at pH 7.2 and 5% O2 using X-VIVO 10 and cytokine cocktail “c” with IL-3 from R&D Systems. From day 5, cultures were supplemented with cytokine cocktail “3” (Table 1) using IL-3 from R&D Systems. At days 5 and 7, cells were centrifuged and reseeded at 100,000 cells/mL in X-VIVO 10 adjusted to the indicated pH. When indicated, cultures were supplemented with 6.25 mM NIC.
pH adjustment and analysis of media
Media were adjusted to the desired pH level as previously described. 41 On each day of analysis, 3 mL of culture media was centrifuged at 200 g for 15 min to pellet cells. In initial experiments, the pH of the resultant supernatant was measured immediately (Method 1). In subsequent experiments, the supernatant was then centrifuged at 1000 g for 10 min to pellet any small particles. The resultant supernatant was stored overnight at 5% or 20% O2 before pH measurement (Method 2).
Cell culture for platelet production
Previously frozen mPB CD34+ HSPCs were seeded in either X-VIVO 10 or Iscove's modified Dulbecco's medium (IMDM)+20% BSA, insulin and transferin (BIT) (79% IMDM [Gibco], 20% BIT 9500 Serum Substitute [STEMCELL], 1% Glutamax [Gibco], 1 μg/mL low-density lipoproteins [Calbiochem]), pH 7.2, supplemented with cocktail “c” (Table 1) using IL-3 from R&D and SCF from Peprotech, and cultured at 5% O2. At day 5, cells were centrifuged and resuspended in X-VIVO 10 or IMDM+20% BIT, pH 7.4, supplemented with cocktail “3” (Table 1) and cultured at 20% O2 until day 7. To increase Mk frequency and concentration, Mks were enriched at day 7 using anti-CD61-conjugated magnetic beads (Miltenyi), resuspended in X-VIVO 10 or IMDM+20% BIT, pH 7.6, and supplemented with 100 ng/mL Tpo, 100 ng/mL SCF, and 6.25 mM NIC. Cells were seeded into tissue culture-treated dishes or fibrinogen-coated slides, and cultured at 20% O2.
Preparation of fresh and expired platelets
Fresh and expired platelets were collected as described in the Supplementary Methods.
Preparation of culture-derived platelets
One day before analysis, culture dishes containing Mks were placed on an orbital shaker set to 50 rpm in an incubator at 5% CO2 and 20% O2. The next day, the cell solution was transferred to a conical tube and the culture surface was washed with warm phosphate buffered saline (PBS). Nonenzymatic cell dissociation solution was added for 15 min at 37°C, 5% CO2, followed by another wash with warm PBS. The combined solutions were then pipetted, as described, 42 before centrifugation at 150 g for 10 min to pellet large cells. The supernatant was transferred to a fresh tube and centrifuged at 1000 g for 10 min. After discarding the supernatant, the platelet pellet was resuspended in HEPES/Tyrode's HT buffer. Before analysis, CaCl2 was added to a final concentration of 2 mM.
Flow cytometry
Cells were assayed for surface CD34, CD41, and CD42b, 29 and Mk ploidy 18 was determined as described. For platelet analysis, antibodies against CD41, CD42b, CD62P, and CD63, or corresponding isotype controls, were added to 0.65-mL polypropylene tubes. Twenty microliters of platelet suspension and phorbol-12-myristate-13-acetate (PMA; Calbiochem), Adenosine diphosphate (ADP) (Bio/Data), or thrombin were then added to final concentrations of 150 nM, 20 μM, and 3 U/mL, respectively. Samples were briefly vortexed to mix. After 15 min of incubation at room temperature, 1% paraformaldehyde in PBS was added to each tube for 15 min. Samples were placed on ice before acquisition on an LSRII flow cytometer (BD Biosciences). Culture-derived platelets were identified as CD41+ events that fell in the side scatter versus forward scatter gate corresponding to fresh platelets isolated from whole blood or recently expired platelets from apheresis units. Expression of activation markers was compared to an unactivated, stained sample.
Microscopy
Live, proplatelet-forming cells on tissue-culture-treated plates were observed with a DM IL LED inverted microscope (Leica) and imaged with a QICAM digital camera (QImaging). For immunofluorescence analysis of proplatelet formation on fibrinogen-coated slides, cells were fixed with 3.7% paraformaldehyde and permeabilized with 0.3% Triton X-100 before staining with 5 μg/mL mouse anti-β-tubulin primary antibody, 140 μg/mL of FITC-conjugated goat anti-mouse secondary antibody (Jackson Immunoresearch), and DAPI nuclear stain (Invitrogen). For analysis of platelet activation, glass chamber slides (Nunc) were coated with BSA or fibrinogen. Platelet suspension was added to each well in the presence or absence of 1 or 3 U/mL thrombin for 1 h at 37 °C, 5% CO2. Each condition was washed once with PBS containing Ca2+ and Mg2+ and stained for β-tubulin as described above. After removing the secondary antibody, cells were incubated with TRITC-phalloidin, washed, and stained with DAPI to confirm the absence of DNA+ particles. Proplatelet and platelet slides were imaged using a 63× or 100× oil objective, respectively, on an SP5 II Laser Scanning Confocal Microscope (Leica).
Statistical analysis
Results are expressed as mean±standard error. Statistical significance was determined using a paired two-tailed t-test for a particular day in culture. p-values for significant effects are noted. ReliaSoft DOE++ software was used for ANOVA to identify which individual cytokines or combination of cytokines yielded statistically significant (p<0.05) effects for each parameter in the factorial design.
Results
Developing cytokine cocktails for Mk expansion
We used a 24 full factorial design of experiments to investigate which combination of IL-3, IL-6, IL-9, and high- or low-dose SCF would most effectively generate mature Mks from Mk progenitors. Based on our previous work, 29 we cultured CD34+ mPB HSPCs with 100 ng/mL Tpo, 100 ng/mL SCF, 10 ng/mL IL-11, 10 ng/mL IL-6, and 2.5 ng/mL IL-3 (Table 1, cocktail “c”) at 5% O2 and pH 7.2 for 5 days in X-VIVO 20 before shifting the cells to 20% O2 and pH 7.4 and changing to 1 of 17 cytokine cocktails (Table 1), all of which contained saturating doses of Tpo (100 ng/mL) and IL-11 (10 ng/mL). IL-3 had the greatest positive effect on total cell expansion (Fig. 2A, B; p<10−14), but also the most detrimental effect on CD41 expression (Fig. 2C, D; p<10−14). The net effect of IL-3 on CD41+ cell production was strongly positive (Fig. 2E, F). About 2.5 ng/mL IL-3 had the same effect on CD41 expression as 10 ng/mL IL-3 (Fig. 2C; condition “c” vs. 2), but cell expansion was much lower (Fig. 2A). This suggests that Mk commitment is sensitive to even small doses of IL-3, and is consistent with results for enriched Mks cultured with 1 ng/mL IL-3 (not shown). IL-3 also caused CD34 expression to decrease more rapidly (Supplementary Fig. S2C, D; p<10−14), probably because cells with IL-3 divided more frequently. 100 ng/mL SCF significantly increased cell expansion compared to 2.5 ng/mL SCF (Fig. 2A, B; p<10−6), but, unlike IL-3, it did not decrease CD41 purity (Fig. 2C, D). As a result, SCF (p<10−4) and IL-3 (p<10−3) increased CD41+ cell production to a similar extent (Fig. 2E, F). IL-3 and SCF synergized to further increase the number of CD41+ cells (Fig. 2F; p<10−4). IL-6 did not significantly affect total cell expansion (Fig. 2B), Mk purity (Fig. 2D), or Mk yield (Fig. 2F). Cytokine effects on CD42b expression (Supplementary Fig. S2E, F) were similar to those for CD41 expression. IL-9 was the only cytokine to significantly increase expression of CD41 (Fig. 2D) and CD42b (Supplementary Fig. S2F). High-dose SCF (p<0.003), IL-6 (p<0.007), and especially IL-3 (p<10−14) significantly enhanced cell viability (Supplementary Fig. S2A, B).
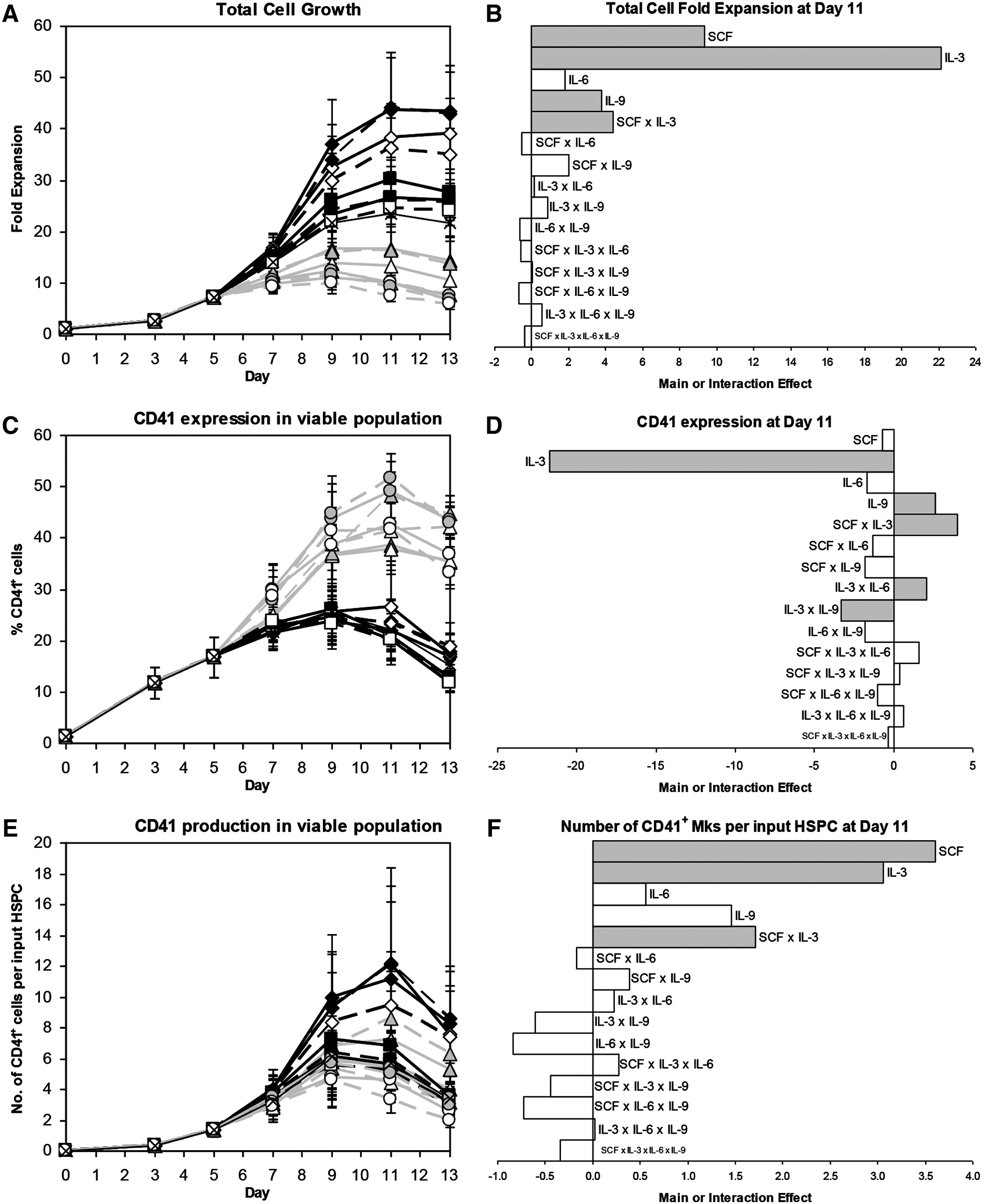
Mk production for 24 factorial design of experiments. Effects of 17 cocktails on
Cultures with 100 ng/mL SCF tended to have a higher percentage of high-ploidy (>4N) Mks at day 11 (Fig. 3A, B; p<0.07). In contrast, IL-3 (p<10−4), IL-6 (p<0.04), and IL-9 (p<0.02) all significantly decreased the day 11 ploidy (Fig. 3A, B). Since high-dose SCF increased cell expansion and Mk ploidy without affecting CD41 expression, SCF had the greatest positive effect on high-ploidy Mk production (Fig. 3D; p<10−4). The beneficial effect of IL-3 on CD41+ cell production (Fig. 2E, F) more than offset the lower percentage of high-ploidy (>4N) Mks such that IL-3 also increased high-ploidy Mk production (p<0.03). IL-3 synergized with SCF to further increase the yield (Fig. 3D; p<0.03). Cocktail 3 (Table 1) yielded the greatest number of Mks per input HSPC for four of five donors (not shown), and was selected as the cocktail for promoting Mk production from day 5 onward for the rest of the experiments in this study.
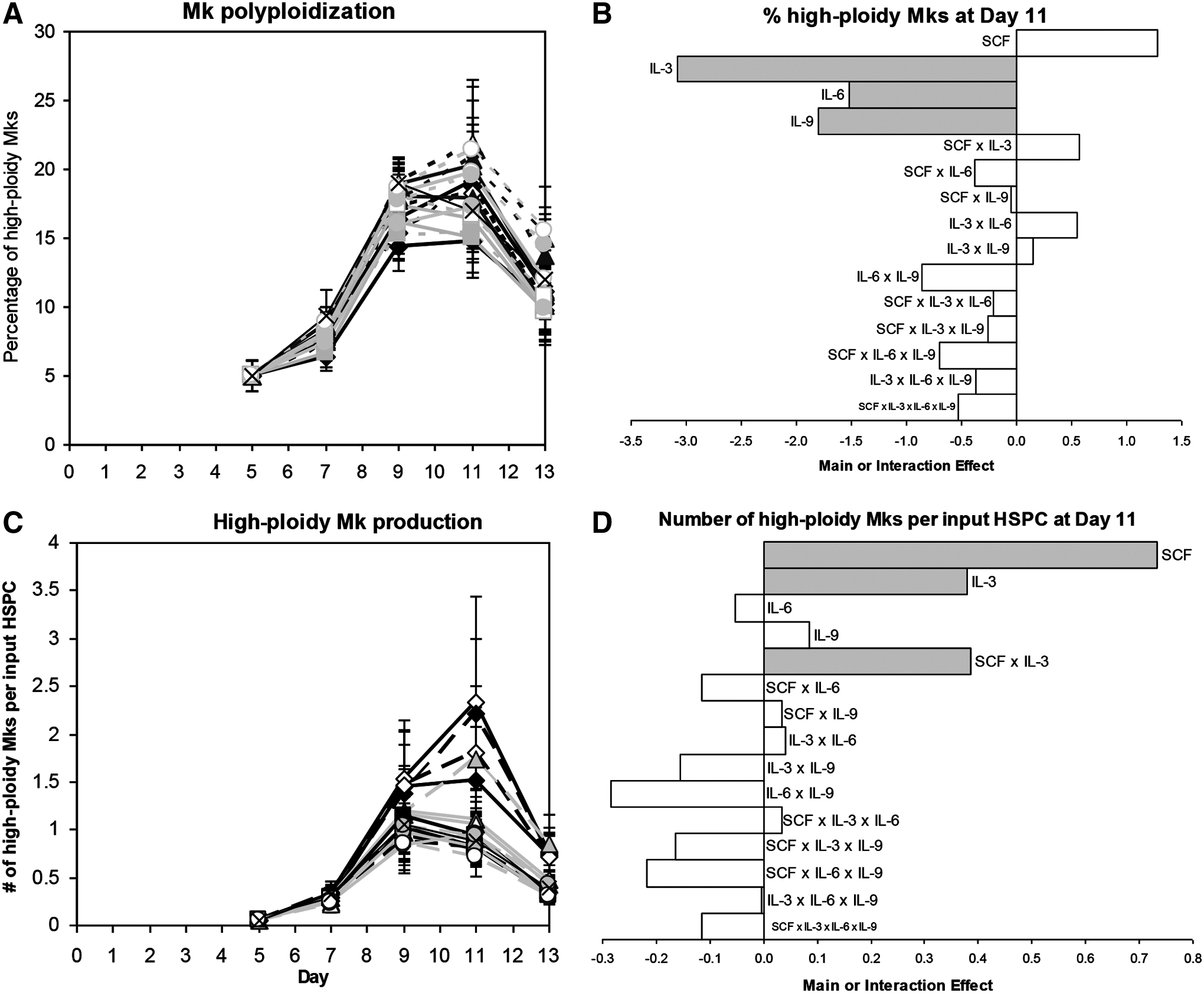
High-ploidy Mk production for 24 factorial design of experiments. Effects of 17 cocktails on
Optimizing commercial serum-free media and cytokine sources for ex vivo Mk maturation
We previously found that CD34+ cells cultured in X-VIVO 20 with cocktail “c” (Table 1) for the entire culture and shifted from 5% O2/pH 7.2 to 20% O2/pH 7.4 at day 5 yielded 12.6±4.5 CD41+ cells per input HSPC by day 13. 29 However, when cocktail “c” was used in the current study, only 3.4±0.7 CD41+ cells were produced per HSPC at day 13 (Fig. 2E). This decrease was due to lower total cell expansion and a substantial decrease in CD41 expression after day 9, and was consistent across five donor samples. We noticed that all conditions with IL-3 had very low terminal CD41 expression (Fig. 2C) and evaluated multiple lots of Peprotech IL-3 with no improvement. Cultures with Tpo alone (not shown) and all cytokine combinations without IL-3 (Fig. 2C) showed the expected increase in CD41 expression. This suggested that Peprotech IL-3 characteristics had changed and/or that a change in X-VIVO 20 medium components altered the IL-3 response.
Different proprietary media have diverse effects on hematopoietic cell differentiation,43–45 so we evaluated five commercial serum-free media platforms commonly used for Mk culture. Because cultures with IL-3 showed the greatest decrease in CD41 expression, we also evaluated IL-3 supplied by R&D Systems. Cells were cultured for 5 days at 5% O2/pH 7.2 using cocktail “c” (Table 1) and then shifted to 20% O2/pH 7.4 with cocktail 3. Based on preliminary experiments with X-VIVO 20 and Peprotech IL-3 showing that Mk production was increased by shifting from pH 7.4 to 7.6 at day 7 (not shown), pH was maintained at 7.4 for days 5–7 and 7.6 for days 7–13. Peprotech IL-3 tended to support a greater number of large-colony CFU-Mks than R&D Systems IL-3, while StemLine II and especially StemSpan H3000 tended to yield the lowest CFU-Mk content (Supplementary Fig. S3).
IL-3 from R&D Systems promoted greater cell expansion in all five media, and X-VIVO 20, by far, was the medium most sensitive to the IL-3 source (Supplementary Fig. S4A). The initial rate of CD34 expression loss was similar for all combinations of media and IL-3 (Supplementary Fig. S4B). On day 7, cultures with R&D Systems IL-3 had significantly lower CD34 expression (p<10−4). From days 7 to 11, cultures with R&D Systems IL-3 had significantly lower CD41 expression than the same media with Peprotech IL-3 (Supplementary Fig. S4C; p<0.02). However, the number of CD41+ cells at day 13 was higher for R&D Systems IL-3 for all media (Supplementary Fig. S4D; p<0.02). StemLine II yielded the lowest CD41 purity (p<10−6; days 7 to 13), and the number of CD41+ cells at day 13 for either IL-3 source was lowest for StemLine II (p<0.01). StemSpan H3000 consistently yielded the most rapid rise in CD41 expression (Supplementary Fig. S4C; p<0.05 at day 5). Unfortunately, using StemSpan H3000 for 5 days and then shifting to X-VIVO 10 (with R&D Systems IL-3) did not increase Mk yields (not shown). X-VIVO 10, X-VIVO 20, and HPGM were much more effective at increasing Mk ploidy than StemLine II or StemSpan H3000 (p<10−5 at day 11), although the percentage of high-ploidy (>4N) Mks decreased more rapidly after day 11 (Supplementary Fig. S4E). The greatest number of high-ploidy Mks (8.1±2.4 per input HSPC) was produced at day 11 using X-VIVO 20 with R&D Systems IL-3, although similar levels were obtained using X-VIVO 10 with R&D Systems IL-3 or HPGM with either IL-3 source (Supplementary Fig. S4F). We selected X-VIVO 10 with R&D Systems IL-3 for the rest of our experiments because cell expansion in X-VIVO 20 was very sensitive to the IL-3 source (Supplementary Fig. S4A) and X-VIVO 10 tended to support greater Mk purity than HPGM (Supplementary Fig. S4C).
Promoting high-ploidy Mk expansion by increasing pH and adding NIC
Shifting from pH 7.4 to 7.6 at day 7 slightly, but consistently, increased total cell expansion (Fig. 4A; p<0.005 at days 11–13) without affecting CD41 (Supplementary Fig. S5A) or CD42b (Supplementary Fig. S5C) expression or Mk ploidy (Supplementary Fig. S5E). Therefore, the numbers of CD41+ cells (Fig. 4C; p<0.07 for days 11–13) and high-ploidy (>4N) Mks (Fig. 4E; p<0.05 for days 11–13) produced per input HSPC were greater in cultures shifted to pH 7.6 at day 7. Shifting directly from pH 7.2 to 7.6 at day 5 caused a rapid and consistent decrease in cell viability and accumulation of debris (not shown), indicating that more gradual pH changes are necessary for optimal Mk production. Measurements of the culture pH showed substantial drift in the cultures over time (Supplementary Fig. S6), which was likely a result of pH changes due to cell expansion and metabolism. While it is not logistically feasible to regulate pH in small-scale cultures, our previous work 29 in combination with this data demonstrates the advantages of shifting Mks to progressively higher pH levels. Improved pH control, which is possible in a large-scale platform such as a cell culture bioreactor, may further enhance the benefits for Mk production and maturation.
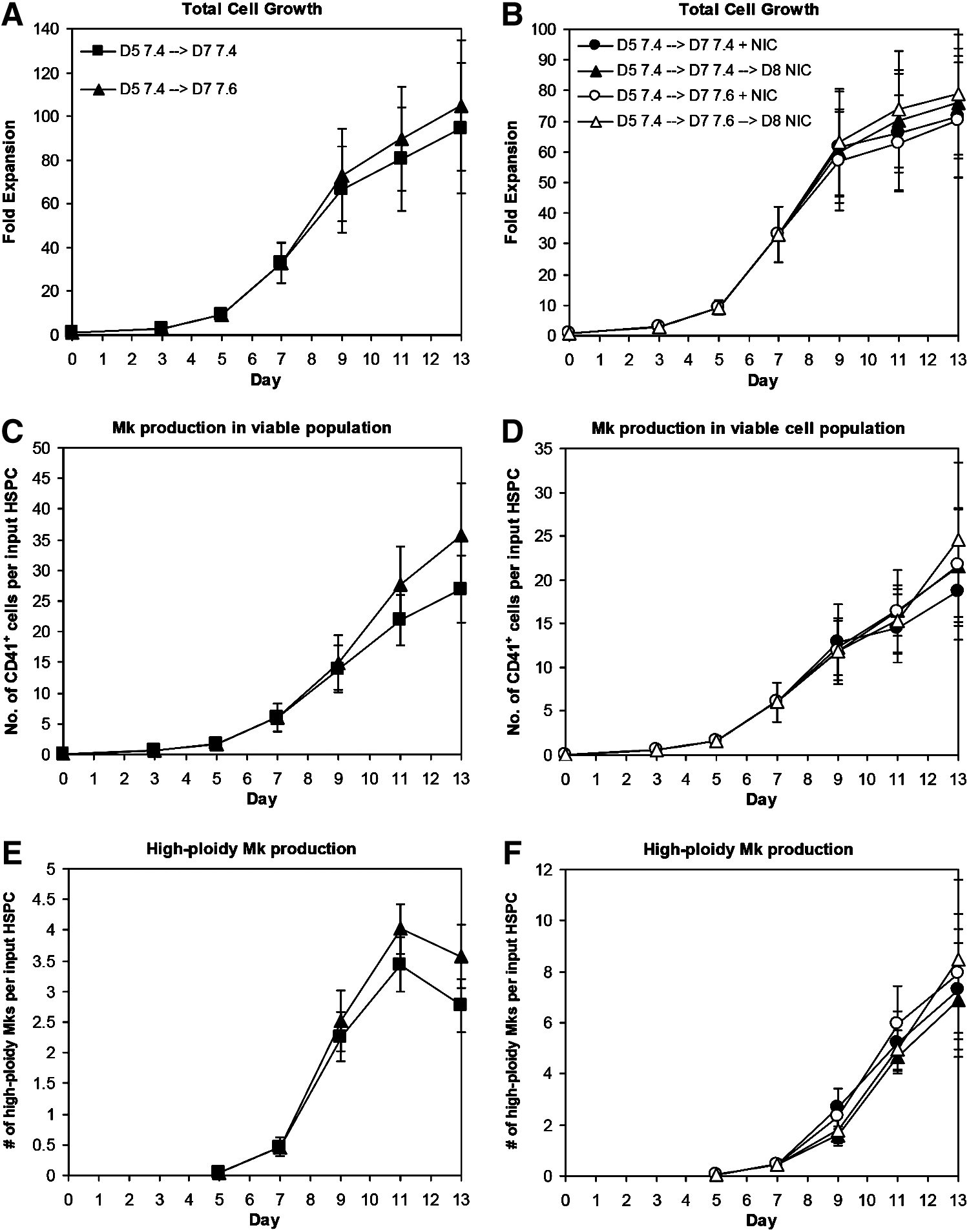
Increasing pH and adding nicotinamide (NIC) increase high-ploidy Mk production. CD34+ HSPCs were seeded in X-VIVO 10 with cocktail “c” (Table 1) using interleukin (IL)-3 from R&D Systems and maintained at 5% O2 and pH 7.2. On day 5, cells were washed, resuspended at pH 7.4/20% O2, and supplemented with cocktail 3.
We evaluated the timing of NIC addition on Mk ploidy in cultures with X-VIVO 10 and R&D Systems IL-3. Although NIC increases Mk ploidy, it also inhibits total cell expansion. 18 Using cocktail “c,” we previously showed that NIC was similarly effective at increasing Mk ploidy at days 5 and 7, with a lesser effect when added at day 9. 29 Depending on the donor, extensive Mk apoptosis begins to occur between days 11 and 13. NIC addition at day 9 did not substantially increase Mk ploidy (not shown). We added NIC at days 7 or 8 to cultures either maintained at pH 7.4 after day 5 or switched from pH 7.4 to 7.6 at day 7 to decouple the effects of NIC from the pH shift. As expected, viability (not shown) and total cell expansion at day 13 tended to be greater for cultures treated with NIC at day 8 (Fig. 4B), while the percentage of high-ploidy (>4N) cells tended to be lower (Supplementary Fig. S5F). There was no consistent effect of pH on these responses. Trends in expression of CD41 (Supplementary Fig. S5B) and CD42b (Supplementary Fig. S5D) were similar for all conditions, with Mk surface marker expression increasing steadily from day 5. By day 11, 75%–85% of CD41+ cells treated with NIC at days 7 or 8 were also CD42b+. Cells treated with NIC at day 7 or 8 at pH 7.6 tended to have higher day 13 yields of total Mks (Fig. 4D) and high-ploidy Mks (Fig. 4F) per input HSPC. This is consistent with greater Mk (Fig. 4C) and high-ploidy Mk (Fig. 4E) production in cultures at pH 7.6 without NIC. Although total cell fold-expansion in the cocktail culture was inhibited by addition of NIC at day 7, the production of 8N, 16N, 32N, and 64N Mks by day 11 was greater (Fig. 5).
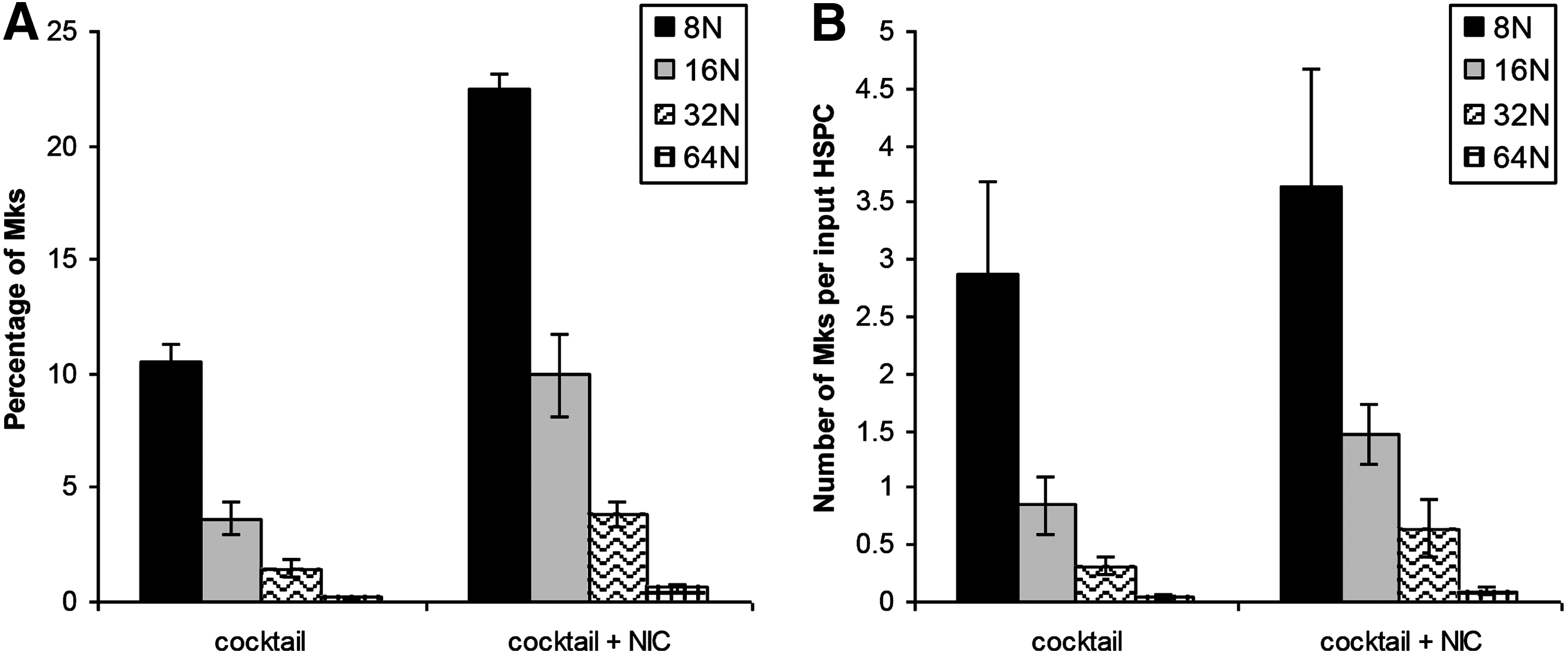
Distribution of high-ploidy Mks. CD34+ HSPCs were seeded in X-VIVO 10 with cocktail “c” (Table 1) using IL-3 from R&D Systems and maintained at 5% O2 and pH 7.2. On day 5, cells were washed, resuspended at pH 7.4/20% O2, and supplemented with cocktail 3 (Table 1). On day 7, cells were washed and resuspended in media adjusted to pH 7.6 and supplemented with cocktail 3 with or without 6.25 mM NIC.
Despite the generation of high-ploidy, CD42b+ Mks, only about half of the platelet-like particles (PLPs) we collected were CD41+ (Supplementary Fig. S7A) and of those effectively none were CD42b+ (not shown). Although we observed occasional Mks with extensive proplatelet formation (Supplementary Fig. S7B), the majority of proplatelet-forming cells in X-VIVO 10 cultures were abnormal, as evidenced by spread cell bodies and few, short extensions. We observed the same phenotype in X-VIVO 10 cultures treated with Tpo and SCF with NIC addition at day 5, without pH changes, suggesting that the media, not our multistage process, was responsible (Supplementary Fig. S7C). Therefore, the poor platelet production we observed was likely the result of irregular proplatelet formation.
In an effort to increase proplatelet formation, we employed a second serum-free medium, IMDM+20% BIT. This medium had previously promoted proplatelet formation in CB-derived Mks in our lab. To validate that we could achieve similar cell expansion, as well as Mk production and maturation, with this new media, we compared CD34+ mPB HSPCs cultured in IMDM+20% BIT or X-VIVO 10 media, using the optimized, 3-phase culture developed with X-VIVO 10 media. Cells were cultured in cocktail “c” (Table 1) at 5% O2 and pH 7.2 until day 5 and then in cocktail “3” (Table 1) at 20% O2 and pH 7.4 until day 7. To increase Mk frequency and concentration, we selected for CD61+ cells at day 7. Cells were then resuspended in media adjusted to pH 7.6 and supplemented with 100 ng/mL Tpo, 100 ng/mL SCF, and 6.25 mM NIC. IMDM+20% BIT gave similar or better fold-expansion and commitment to the Mk lineage (Supplementary Fig. S8A, B) and similarly supported Mk maturation, as demonstrated by CD42b expression and polyploidization (Supplementary Fig. S9A, B). Cultures with IMDM+20% BIT also had a similar pH profile compared to those with X-VIVO 10 (Supplementary Fig. S6).
Proplatelet formation
The first experiment with IMDM+20% BIT resulted in Mks with extensive proplatelet formation, as characterized by long, thin extensions with platelet-sized beads along their length (Supplementary Fig. S10A). Although nearly all of the PLPs were CD41+, the few PLPs harvested showed minimal activation with agonist treatment (Supplementary Fig S10B). To improve the number and functionality of culture-derived platelets, we optimized the culture process for platelet generation. In order to minimize handling and the potential for platelet preactivation, we seeded selected Mks in separate 60-mm, tissue-culture-treated dishes at day 7. Because shear force has been shown to increase platelet shedding from proplatelets, 46 we transferred one dish per condition to an orbital shaker 1 day before platelet harvest. To recover any PLPs that were attached to the culture surface, we incubated with cell dissociation solution containing EDTA after removing the supernatant. To dissociate any platelet-cell clumps and further promote shedding of platelets from proplatelets before centrifugation, we pipetted the combined supernatant and cell dissociation solution washes. 42
In subsequent experiments with cells from two different donors, we again observed that Mks produced in IMDM+20% BIT yielded extensive proplatelet formation (Figs. 6 and 7), while those produced in X-VIVO 10 tended to have poor proplatelet formation (Fig. 6). Due to the low frequency of proplatelets with X-VIVO 10, we proceeded to harvest and analyze only PLPs from cultures with IMDM+20% BIT.

Mk proplatelet formation in X-VIVO 10 and Iscove's modified Dulbecco's medium (IMDM)+20% BSA, insulin and transferin (BIT). Proplatelet-forming Mks were observed on day 11 of culture in tissue-culture-treated dishes via phase contrast microscopy. Scale bar=20 μm.
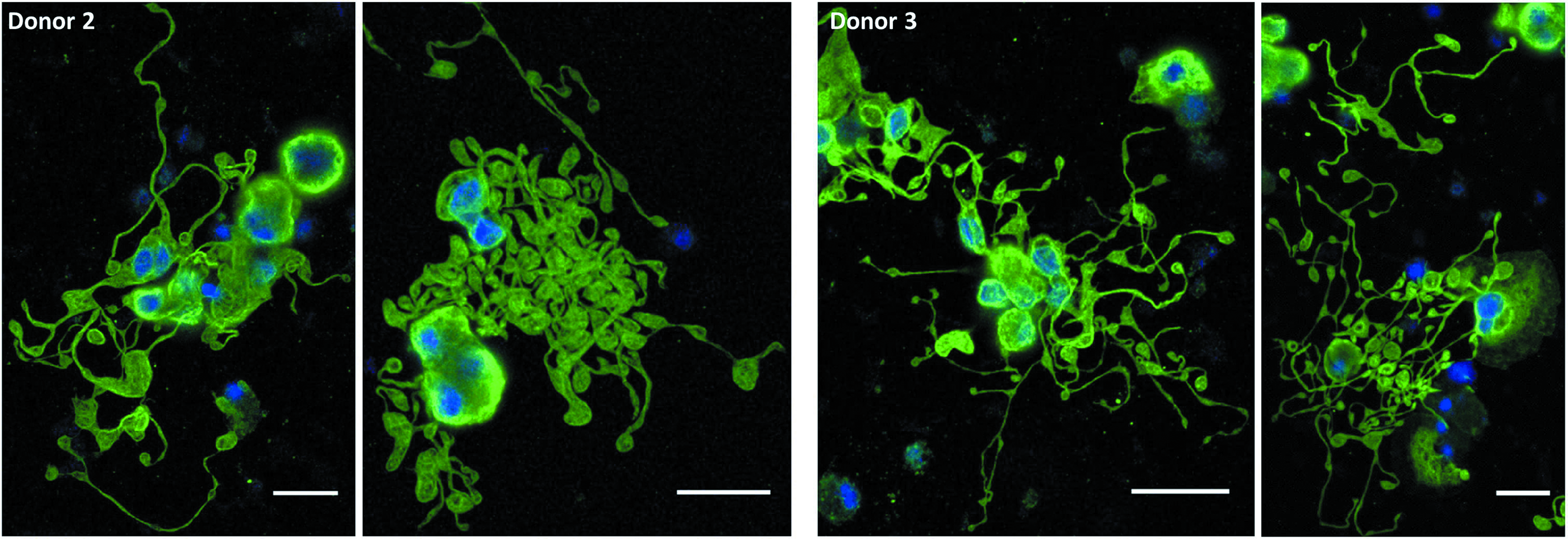
Mk proplatelet formation in IMDM+20% BIT on a fibrinogen-coated surface. On day 7, selected Mks were resuspended in IMDM+20% BIT supplemented with 100 ng/mL thrombopoietin and 100 ng/mL SCF and seeded on fibrinogen-coated slides. On day 11, cells were fixed and stained for β-tubulin (green) and DNA (blue) before imaging. Scale bar=20 μm. Color images available online at www.liebertpub.com/tea
Assessment of culture-derived platelet morphology, surface markers, and function
Platelets are CD41+CD42b+ anucleate cells that have a discoid shape in their resting state. Upon activation at the site of an injury, platelets translocate alpha granules to their surface, resulting in plasma membrane expression of CD62P (P-selectin) and CD63. CD62P mediates the inflammatory response through platelet binding to endothelial cells, monocytes, lymphocytes, and neutrophils, 47 while CD63 is a marker of lysosomal release 48 and is thought to facilitate platelet spreading. 49 Activated platelets also enlarge and extend filopodia and lamellipodia. To demonstrate the soundness of the optimized culture conditions for Mk production, we evaluated the ability of the resulting mature Mks to produce functional platelets. We first isolated culture-derived platelets via successive centrifugation and then identified them on a flow cytometer using the scatter properties of fresh and recently expired platelets. We identified the population that fell outside the platelet gate as proplatelets and preplatelets 50 (Fig. 8). About 2/3 of the culture-derived, platelet-sized particles were double-positive for CD41 and CD42b and showed similar antibody staining intensity to that observed with fresh and recently expired platelets (Fig. 8). This suggests that there was only a small amount of platelet-sized debris in the culture and that Mk-produced matrix metalloproteinase activity was not sufficient to cleave the CD42b receptor, as another group has reported. 51 All of the culture-derived platelets were capable of activation, as evidenced by increased expression of CD62P (Fig. 9) and CD63 (Fig. 10) in CD41+ platelet-sized particles upon treatment with the agonists PMA and thrombin. The shift in expression was similar to that observed with fresh and expired platelets. We found that incubating fresh platelets for 15 min with 3 U/mL thrombin increased nonspecific binding more than anti-CD62P binding, so we switched to a 5-min incubation (Fig. 9, inset). Although not present in vivo, PMA activates the protein kinase C pathway, which is known to be important in platelet activation. 52 PMA has previously been shown to increase surface expression of CD62P and CD63 in platelets from peripheral blood. 53 ADP, which is known to be a weaker agonist than thrombin, 54 increased activation-dependent marker expression in fresh and recently expired platelets, but not in culture-derived platelets.
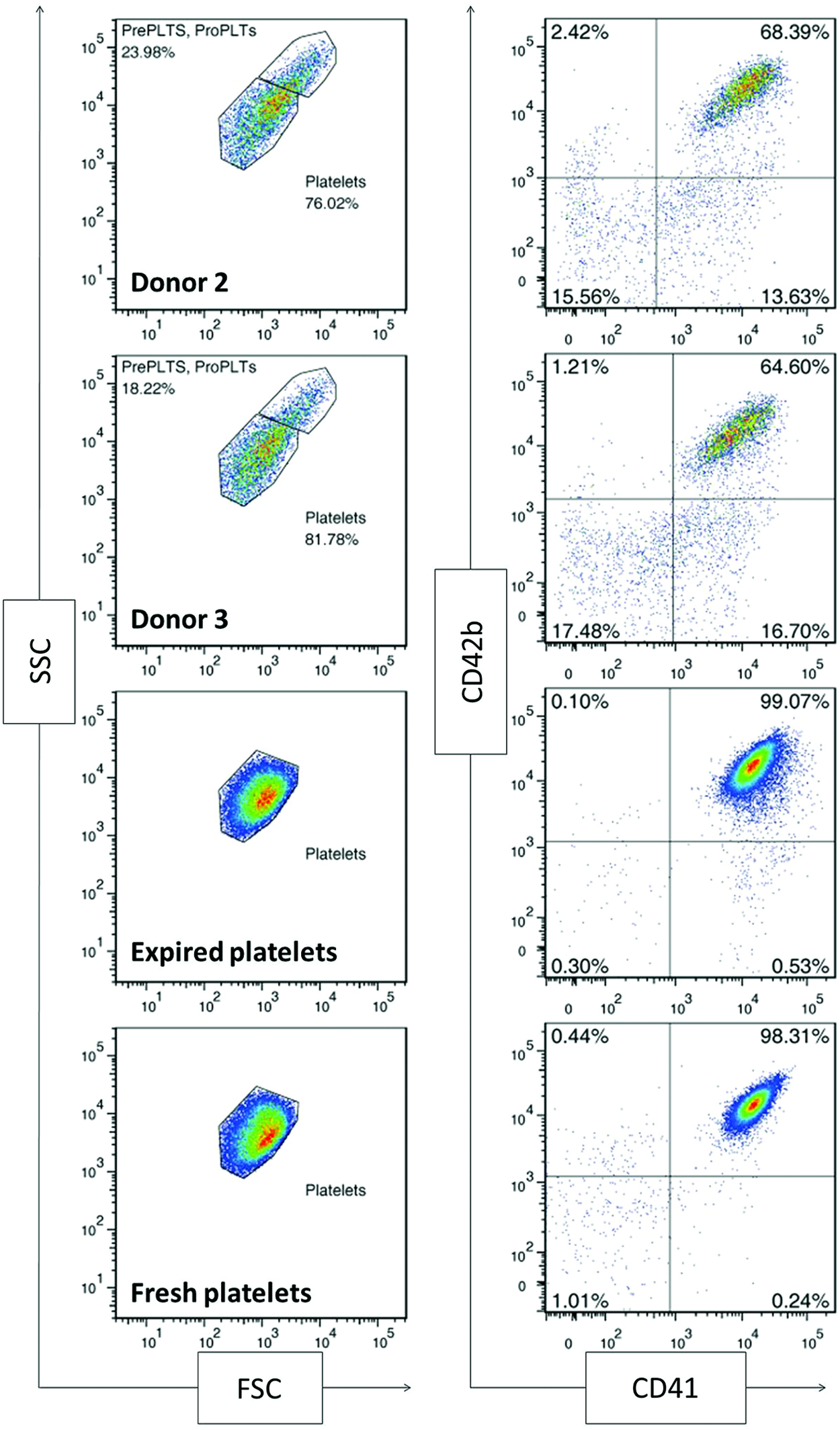
Scatter properties and surface marker expression of unactivated, culture-derived platelets. All samples were analyzed using the same forward scatter (FSC) and side scatter (SSC) voltage settings on the flow cytometer. Platelet-sized particles were identified with an SSC versus FSC gate drawn on fresh and recently expired platelets. CD41 and CD42b expression was analyzed for particles in this characteristic size range. An isotype control was used to determine positive events for each sample (not shown) Pre PLTs, preplatelets; Pro PLTs, proplatelets. Color images available online at www.liebertpub.com/tea
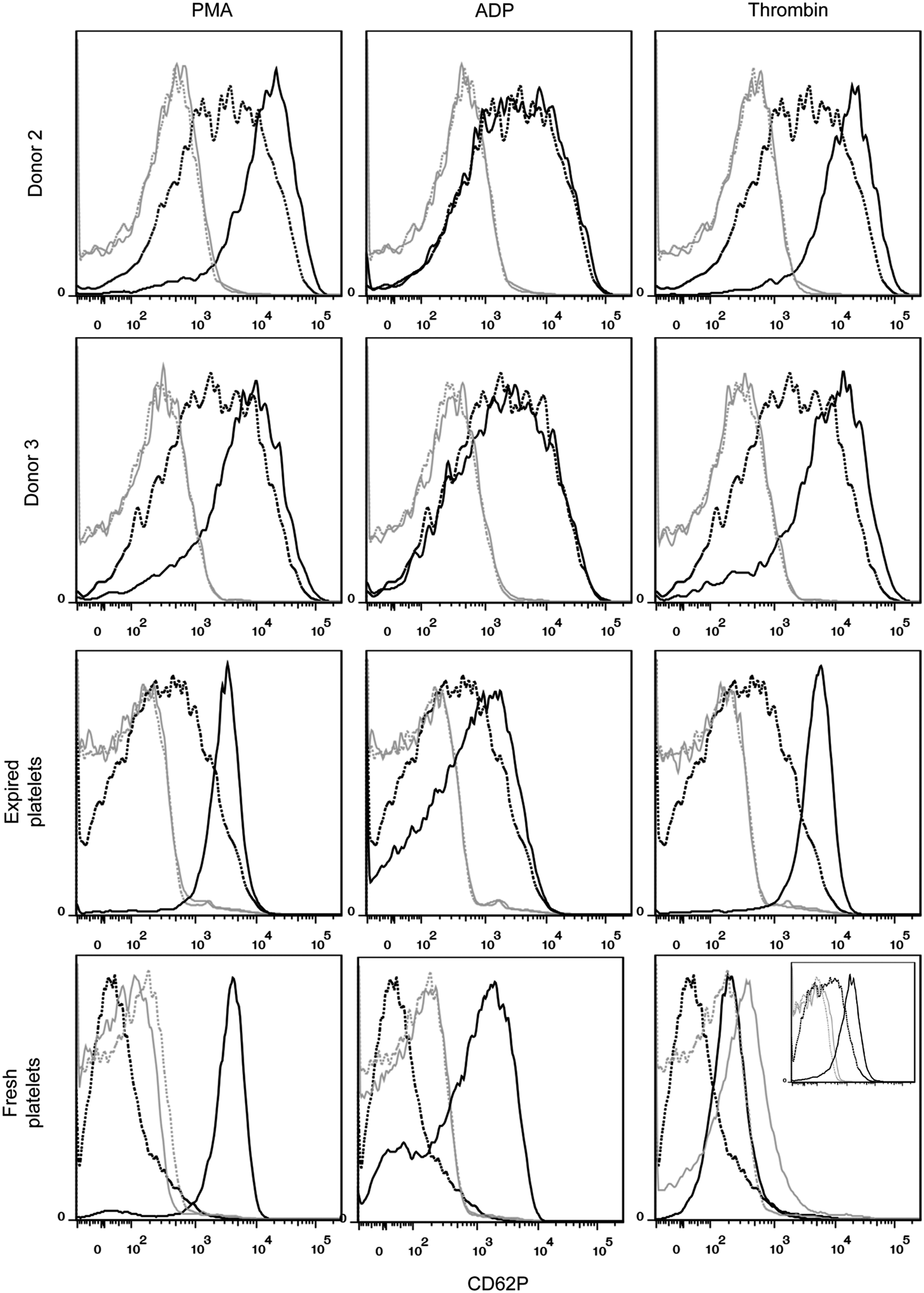
CD62P expression in unactivated and activated culture-derived platelets. Platelets were identified as CD41+ events that fell in the SSC versus FSC gate drawn on fresh and recently expired platelets. Marker expression was compared for unactivated (dotted black line) and activated (solid black line) platelets for 15-min incubation with one of three agonists: 150 nM phorbol-12-myristate-13-acetate (PMA), 20 μM adenosine diphosphate (ADP), and 3 U/mL thrombin. Isotype controls are shown for unactivated (dotted gray line) and activated (solid gray line) platelets. Inset for fresh platelets with thrombin shows the results of a 5-min incubation.
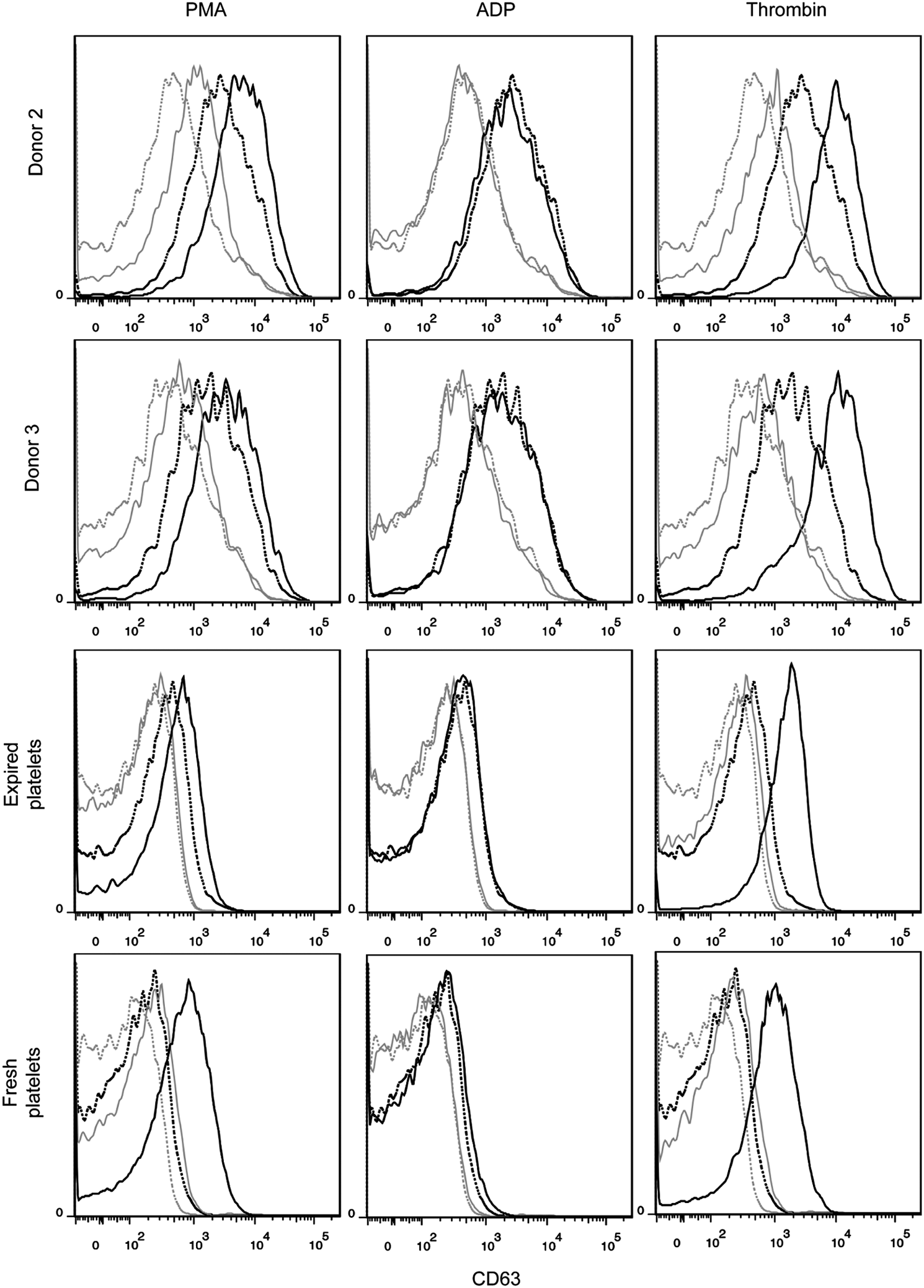
CD63 expression in unactivated and activated culture-derived platelets. Platelets were identified as CD41+ events that fell in the SSC versus FSC gate corresponding to fresh and recently expired platelets. Marker expression was compared for unactivated (dotted line) and activated (solid line) platelets for three agonists: 150 nM PMA, 20 μM ADP, and 3 U/mL thrombin. Isotype controls are shown for unactivated (dotted gray line) and activated (solid gray line) platelets.
Microscopy analysis of the culture-derived platelet fraction on inert BSA-coated surfaces showed 2–4 μm particles with the characteristic outer band of β-tubulin and minimal F-actin, which matched that observed with both fresh and recently expired platelets (Fig. 11). Virtually all of the culture-derived particles stained negative for DNA, indicating that the PLPs are anucleate. We also saw evidence of preplatelets 50 and proplatelets in the platelet supernatant, suggesting that the conversion to platelets in culture was not complete. Incubation on fibrinogen increased the presence of F-actin-containing filopodia, and the addition of thrombin resulted in spread platelets up to 20 μm in diameter with clear stress fibers and lamellipodia (Fig. 11).

Microscopy analysis of culture-derived platelets. Platelets were seeded on slides coated with BSA or fibrinogen, in the presence or absence of thrombin for 1 h at 37°C. Platelets were then fixed and stained for β-tubulin (green), F-actin (red), and DNA (blue). The characteristic platelet band of β-tubulin (arrow), proplatelets (a), preplatelets (b), filopodia (c), actin stress fibers (d), and lamellipodia (e) are highlighted. Scale bar=5 μm. Color images available online at www.liebertpub.com/tea
Platelet preactivation
The culture-derived platelets showed signs of preactivation, as evidenced by flow cytometry analysis. In contrast to fresh unactivated platelets that showed very low, uniform CD62P expression that is similar to the staining observed for the isotype control, unactivated culture-derived platelets and (to a lesser extent) recently expired platelets exhibited a wide CD62p peak (Fig. 9) that was clearly higher than the background staining. Basal CD63 expression was also greater for culture-derived platelets (Fig. 10). Finally, basal spreading on BSA and fibrinogen, as well as after treatment with thrombin, was more extensive for culture-derived platelets (Fig. 11). We confirmed that even with longer incubation times, fresh and expired platelets treated with thrombin did not spread to reach the size of thrombin-treated, culture-derived platelets. Although greater basal surface marker expression and spreading may be partially attributed to the larger size (Supplementary Fig. S11) of culture-derived platelets, some preactivation was clearly present. In addition to the shear effects, the preactivation may result from platelets being produced in culture several days before they were harvested and analyzed. In this regard, platelet preactivation is likely to occur much more rapidly at 37°C than during normal storage at 22°C. 55
Discussion
We and others have shown the benefits of separating in vitro megakaryopoiesis into multiple phases and developing distinct culture conditions for each phase.29,56–58 However, this is the first study to comprehensively explore the effects of environmental factors such as pH and pO2, as well as cytokines, and also demonstrate that Mks derived from mPB CD34+ HSPCs can form proplatelets and functional platelets. Our previous work demonstrated the importance of pH in Mk maturation and consistently showed that culturing primary Mks at higher pH in the range from 7.2 to 7.6 increased production of high-ploidy (>4N) Mks.9,29 pH also modulates the differentiation of granulocytes,41,59,60 erythrocytes,60,61 and BM stromal cells. 62 Hematopoietic stem cells reside close to the bone surface and migrate toward the sinuses during maturation (Fig. 1). Consistent with the BM architecture, granulocytic cells, which mature away from the sinuses, show maximal differentiation at pH 7.1–7.2,41,59 while Mks, which extend proplatelets between sinus endothelial cells to release platelets into the circulation, and erythrocytes, which must be mature when they reach the sinus, exhibit much lower differentiation rates at pH 7.1–7.2.9,60,61 Increasing pH from 7.4 to 7.6 at day 7 tended to increase production of total and high-ploidy Mks beyond the benefit previously attributed to shifting from pH 7.2/5% O2 to pH 7.4/20% O2 at day 5. 29
We used a full 24 factorial design of experiments to evaluate the main and interaction effects of four cytokines (SCF, IL-3, IL-6, and IL-9) in conjunction with Tpo and IL-11 on producing mature Mks from progenitors. We previously used a 24 factorial design to evaluate the effects of IL-3, IL-6, IL-11, and Flt3 ligand (together with Tpo and SCF) on Mk progenitor production from mPB HSPCs. High-dose SCF is commonly used in HSPC culture to enhance progenitor expansion and increase viability. However, in contrast with previous studies using CB CD34+ cells which showed that limiting amounts (0.6 to 1.25 ng/mL) of SCF promoted Mk maturation, while higher doses inhibited maturation,38,39 we found that high-dose SCF greatly increased production of CD41+ cells and high-ploidy (>4N) Mks. The different results may be due to our use of mPB versus CB CD34+ HSPCs. NIC greatly increases Mk polyploidization, but also inhibits cell expansion.18,29,40 Thus, one must optimize the timing of NIC addition to produce the greatest number of high-ploidy Mks. In one-phase mPB CD34+ cell cultures carried out at 20% O2 and pH 7.4 with Tpo as the only cytokine, Mks mature quickly such that NIC addition at day 5 is optimal.18,40 However, expression of CD41 and CD42b on Mks was delayed in the multi-phase culture system, so that NIC addition was optimal between days 7 and 8. Delaying NIC addition until day 9 did not allow enough time for extensive Mk polyploidization before apoptosis.
Our results for five commercial media differed by as much as 2-fold for total cell expansion and 2.8-fold for Mk purity. We also found that proplatelet formation was highly sensitive to the serum-free medium used. While many media components are proprietary, the media typically contain serum-derived proteins of human or animal origin. HSPCs are sensitive to serum and xenogenic materials, particularly with respect to Mk differentiation.63–65 In addition, the concentrations of insulin, transferrin, and β-mercaptoethanol in serum-free media can significantly affect HSPC expansion and differentiation.66–68 Lot-to-lot differences in media components may also be problematic. IL-3 from Peprotech did not have any N-terminal methionylation, whereas different lots of IL-3 from R&D Systems varied in the extent of N-terminal methionylation. Otherwise, both were produced in E. coli and were reported to have a 133-amino-acid sequence (Ala–Phe). Previous work comparing N-terminal-methionylated and nonmethionylated wild-type IL-1β showed significant differences in the isoelectric point and receptor binding activity between the two versions of this cytokine. 69 It is possible that the activity and/or cellular uptake of IL-3 were also strongly affected by N-terminal methionylation and led to significant differences in Mk expansion.
We previously demonstrated production of 13.1±2.1 Mks and 2.2±0.8 high-ploidy (>4N) Mks per input HSPC. 29 By optimizing the cytokines employed after day 5, changing the commercial basal medium platform and the IL-3 supplier, and increasing pH from 7.4 to 7.6 at day 7, the Mk yield was increased to 44.5±8.1 Mks per input HSPC (Fig. 4C and Supplementary Fig. S4D; n=6). This represents an almost 4000-fold average expansion of Mks from the initial population at day 0. After supplementing the culture with 6.25 mM NIC at day 8, the number of high-ploidy Mks produced per input HSPC by day 13 reached 8.5±3.1 (Fig. 4F; n=3) with one donor yielding 13.8 high-ploidy Mks per input HSPC (not shown). This would decrease the theoretical number of mPB CD34+ cells required to produce one platelet transfusion to 45 million. Although further improvement is clearly required, this represents a substantial advance. It is important to note that the majority of the cytokine dosages applied in these studies were at saturating levels commonly used in the literature. A dose-dependence study 70 and use of mass manufactured pharmaceutical grade cytokines would decrease the costs associated with expansion of Mks from HSPCs. Given the substantial effect that IL-3 has on fold expansion and Mk purity, a dose-response curve for IL-3 may help clarify the trade-off between those parameters. A Tpo mimetic peptide agonist (Nplate; Amgen) and recombinant human IL-11 (Neumega; Pfizer) are both commercially available as pharmaceutical-grade cytokines for human consumption. Given that Tpo and IL-11 are applied at a high concentration in all three stages of the Mk culture process, use of pharmaceutical-grade cytokines, rather than commercially available recombinant cytokines, may be more cost effective and move the culture process in a direction more conducive to producing large quantities of Mks and platelets. These cytokines would also be more consistent in quality due to stringent standards in testing and quality assurance.
Proplatelet and platelet formation by cultured Mks is promising and suggests that, with further modification of the shearing and harvest protocols, it should be possible to achieve large-scale platelet production. We generated proplatelets on tissue-culture-treated plastic, as well as glass slides coated with fibrinogen, indicating that a protein coating is not necessary for this process. We collected culture-derived platelets that were larger on average than fresh and expired platelets (Supplementary Fig. S11), perhaps indicating incomplete conversion from proplatelet and preplatelet precursors. 50 Introduction of shear forces, like those present in microcapillaries in vivo, 71 may promote this conversion. The platelets produced were partially preactivated, which is consistent with other studies assessing the properties of culture-derived platelets.72,73 However, exposure to PMA and thrombin increased activation-dependent surface marker expression on all CD41+ platelet-sized particles, indicating that they retained the potential for activation. We observed hyporesponsiveness of culture-derived platelets to ADP, which may be due to partial preactivation. We hypothesize that resting the platelets after centrifugation may restore this response. 74 We compared culture-derived platelets to both recently expired and fresh platelets because expired platelets are likely to more closely resemble the platelets that are transfused into patients. Due to extensive testing for pathogens, a unit of platelets may not be cleared for transfusion use until 3 days after collection. In addition, patients are often given the oldest, pre-expiration platelet unit available to prolong the supply. Expired platelets also showed signs of preactivation, suggesting that low levels of preactivation may not substantially decrease the post-transfusion benefit. Notably, other investigators have transfused culture-derived platelets, with similar in vitro functional activity to that described in this study, into mice and observed culture-derived platelets participating in growing blood clots in response to injury.30,33,51,75
Footnotes
Acknowledgments
We are grateful to Amgen for the SCF donation. We thank Mark Duncan for assistance with confocal microscopy and Northwestern Memorial Hospital Blood Bank for providing expired apheresis platelets. Supported by NSF Grant CBET-0853603. Alaina Schlinker was supported in part by NIH/NCI training grant T32CA09560.
Disclosure Statement
The authors declare no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.