
Abstract
The encapsulation of both cells and a surgical mesh in a polymerizing collagen hydrogel followed by mechanical compression, after polymerization, results in the rapid formation of a living dermal equivalent (LDE) with physical properties suitable for in vivo application. It was found in the current study that the LDE supported the attachment, growth, and differentiation of keratinocytes, allowing for the formation of living skin equivalents (LSEs) with a monolayer epidermis (LSE-M) and a stratified epidermis (LSE-S). The utility of the LDE for the fabrication of living wound dressings was further evaluated by testing the safety and efficacy of the LSE-M and LSE-S in a lapine model of an acute full-thickness skin defect. It was found that the LSE-S significantly stimulated blood vessel formation and accelerated epidermal wound closure compared with controls. The LSE-M showed similar trends but these were not significant. These findings indicate the clinical usefulness of the LDE in the treatment of acute and possibly chronic wounds, such as venous and diabetic ulcerations. The 1-h fabrication time of the LDE is a significant reduction compared with that of dermal components of current FDA-approved dressings, such as Dermagraft, Apligraf, and OrCel, which require days to weeks of in vitro culture. It is therefore proposed that the presented method could reduce the high cost associated with the production of living, tissue-engineered dressings.
Introduction
Cells embedded in a collagen hydrogel are able to contract the matrix and expulse the fluid content thereby improving the mechanical strength over a period of hours to days depending on cell type and cell number. 4 This lab has established a mechanical compression protocol 5 that can, without significantly affecting cell viability, improve the mechanical properties faster and to a greater extent than cells are able to. To illustrate, the break force of a collagen hydrogel compressed for 5 min is twice that of a gel contracted by 2.0×105 fibroblasts/mL over a period of 1 week. 6 In addition, these compressed collagen hydrogels persist significantly longer in vivo than uncompressed gels. 7
The compression protocol results in compressed collagen hydrogels with tensile strengths approaching those of some native tissues. 5 However, due to the small, postcompressive physical dimensions, the break strength is not adequate for practical applications (unpublished data). This issue was addressed by co-encapsulating a synthetic degradable polymer mesh during collagen hydrogel formation followed by mechanical compression, resulting in a living connective tissue (e.g., dermal) equivalent with adequate handling properties in a period of 1 h. 8 In the current study, the utility of this rapid fabrication technique for the creation of tissue-engineered, living wound dressings was tested by evaluating the safety and efficacy of living wound dressings produced from this technique in promoting wound closure in full-thickness skin defects in a lapine model compared with no treatment (NT) and acellular treatment.
Materials and Methods
Keratinocyte isolation
Keratinocytes were isolated from skin tissue taken from the ventral side of the ear lobe from adult male, outbred New-Zealand White (NZW) rabbits according to techniques previously described. 9 Briefly, the harvested tissue was divided into 5×5 mm2 pieces, which were incubated in 0.25% trypsin-EDTA (Invitrogen-Gibco) for 90 min at 37°C. The epidermis was separated from the underlying dermis with forceps and epidermis was incubated in 0.25% trypsin for an additional 30 min at room temperature. The resultant cell suspension was mixed with Dulbecco's modified Eagle's medium (DMEM; SIGMA) supplemented with 10% (v/v) fetal calf serum (FCS; First Link) (DMEM+10% FCS) to neutralize the trypsin, filtered through a 70 μm cell strainer, and centrifuged at 2000 RPM for 5 min. The cells were resuspended in keratinocyte growth medium (CnT-07; CellNTec), counted with the trypan blue dye exclusion assay, and plated at a density of 0.5–1.0×106 cells/25 cm2 for expansion.
Fibroblast explant culture
Dermal fibroblasts were explanted from dermal tissue obtained after the epidermis was separated from the skin using a previously described protocol. 10 The dermal tissue was cut into 2×2 mm2 squares, plated onto tissue culture plastic (approximately 15 pieces on a 6-well plate), and covered with a glass coverslip (22×26 mm2) weighed down by a 50 g stainless steel load and cultured in DMEM+10% FCS. Coverslips were removed after the outgrowth culture reached confluence and fibroblasts were trypsinized, resuspended, and plated at a density of 3–10×104 cells/25 cm2 for expansion.
Fabrication of the skin equivalents: the dermal component
The living dermal equivalent (LDE) studied in the current article was composed of a commercially available poly-[lactide-co-glycolide] (PLGA) (Vicryl) mesh coated with a cell-seeded collagen layer. The PLGA material provides strength to the LDE and degrades through simple hydrolysis. 11 It has been shown that the Vicryl mesh can persist in vivo for a period of 4–8 weeks.12,13
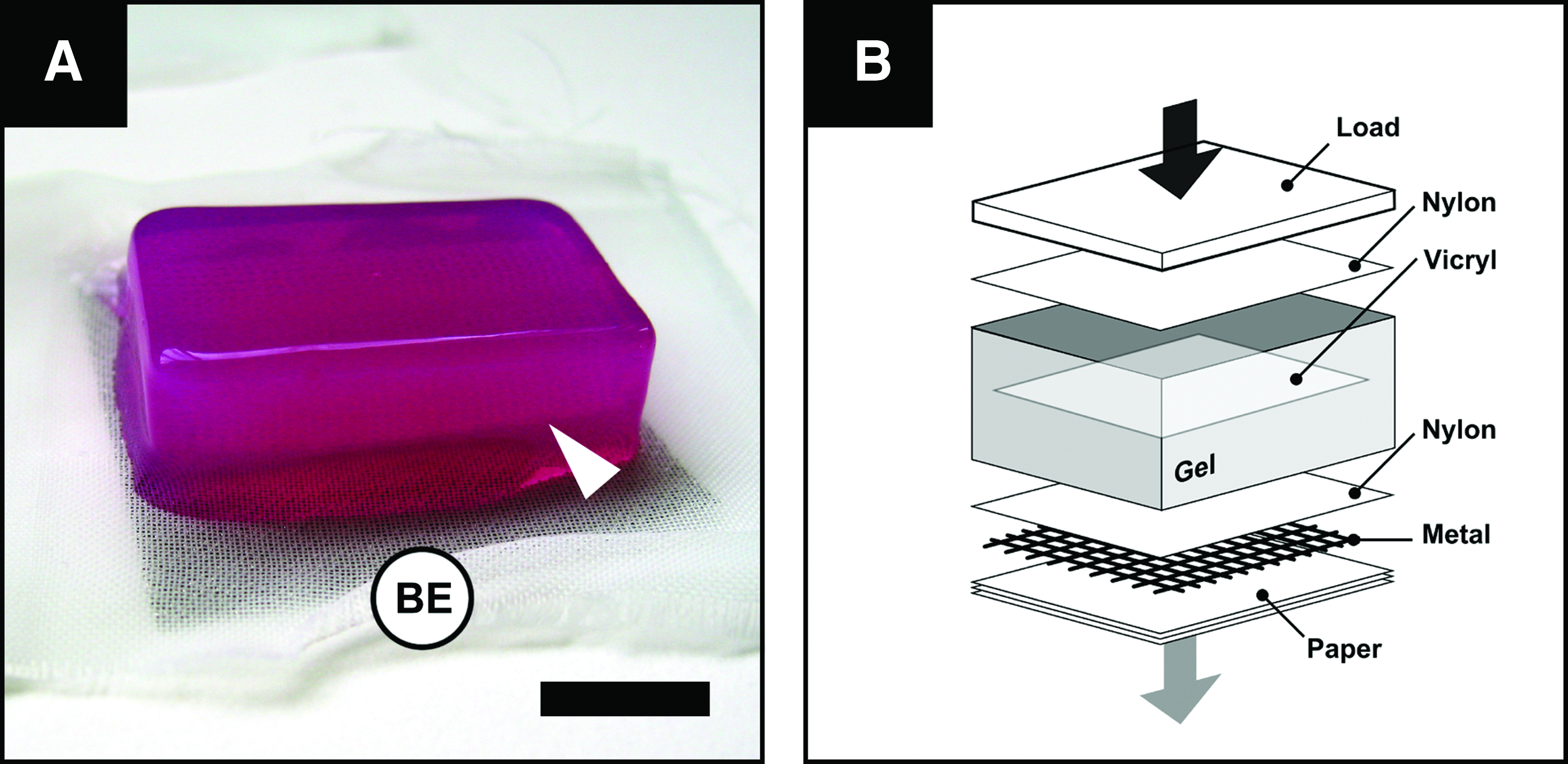
The LDE was fabricated within the space of an hour, as previously described. 8 Briefly, cold (4°C) 4 mL acid-soluble rat tail (type I) collagen (First Link UK Ltd.) was mixed with 0.5 mL of pH indicator (10×minimum Eagle's medium) and brought to neutral pH by titration with (1 M) NaOH. The collagen solution was mixed with a 0.5 mL suspension of 5×105 fibroblasts in DMEM and poured into a 33×22×20 mm3 stainless steel mould. A 30×20 mm2 cut rectangular piece of a commercially available surgical mesh (Vicryl; PLGA) was added onto the surface of the gel and a second volume of neutralized, cell-seeded collagen was added on top. The assembly (Fig. 1a) was allowed to set for 30 min at room temperature after which time it was transferred onto a standardized assembly of blotting elements, and compressed in an unconfined configuration with a gravitational load (stress equivalent 1.8 kN/m2) for a period of 5 min (Fig. 1b), resulting in an LDE that could directly be used for the fabrication of the skin equivalents (SEs).
Alamar Blue reduction assay
The effect of the LDE fabrication process on fibroblast viability was assessed directly and 7 days after compression with the Alamar Blue (AB) reduction assay. 14 Briefly, the LDE was washed and incubated in a 10% AB dye solution in phenol red–free DMEM (Gibco) for 4 h at 37°C and 5% CO2 after which time the amount of fluorescence was measured (Fluoroskan Ascent Labsystems International; 510 nm excitation, 590 nm emission). The fluorescence intensity was expressed as relative fluorescence units (RFU) and for each sample the obtained value was subtracted by the value representing the autofluorescence of the nonreduced 10% AB solution. AB reduction directly after compression was compared with that of (noncompressed) cells plated onto tissue culture plastic.
Fabrication of the SEs: the epidermal component
Two types of living skin equivalents (LSEs) were formed: (1) an LSE featuring a monolayer epithelium, and (2) an LSE featuring a stratified epithelium. Second-passage keratinocytes were seeded and cultured on the apical surface of the LDE (Fig. 2a), directly after fabrication, at an initial density of 5±0.4 (standard deviation [SD])×105 cells/cm2 forming an SE with a monolayer epithelium (LSE-M).
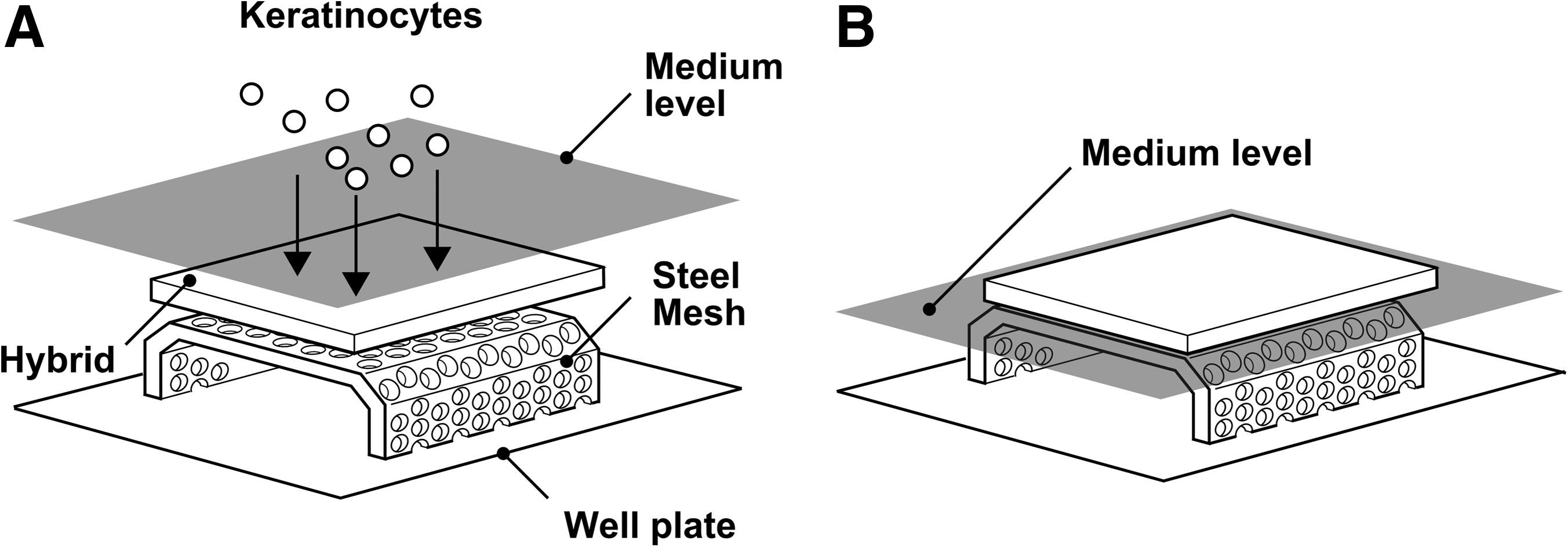
LSEs with a stratified epidermis (LSE-S) were created from LSE-M through an additional 2-week cultivation period. The LSE-M were cultured submerged in growth medium for 3 days to establish confluence after which the constructs were raised to the air–liquid interface (Fig. 2b) and cultured in differentiation medium (CnT-02-3D; CellNTec) for an additional 11 days.
Experimental design
To further evaluate the utility of the LDE for the creation of living wound dressings, the biological effect of the SEs, formed from the LDE seeded with allogeneic cells, was tested in an acute, full-thickness skin defect model. The LSE-M and LSE-S were compared to test whether the wound-healing benefit of a stratified epidermis over a monolayer epidermis warranted the extended in vitro culture period of the LSE. Controls were autologous full-thickness skin graft (FTSG) and open-wound defects (i.e. no treatment).
Animals
The experiments were approved by a local ethical review committee and carried out under Home Office Project Licence PPL 80/2200 according to the Animals Scientific Procedures Act (1986). Fifteen female, outbred NZW rabbits were purchased from B&K Universal and housed in an approved animal facility. Rabbit chow and water were available ad libitum.
Preparation
On the day of surgery, the rabbits were anesthetized with hypnorm (0.3 mL/kg; 0.095 mg/kg fentanyl citrate, 3 mg/kg fluanisone, intramuscular) followed by diazepam (0.5 mg/kg, intravenous). Supplemental doses of hypnorm of 0.1 mL/kg were given every 30 min to maintain anesthesia. Rabbits were given oxygen (at 1 L/min) through a face mask with a rebreathing bag. Intraoperative fluid loss was compensated with a continuous infusion of 0.9% normal saline (at 10 mL/kg/h).
Surgical procedure
An outline of the areas to be excised was measured out and tattooed (Fig. 3a). Three 2×2 cm2 full-thickness skin defects (down to, but not including, the subcutaneous panniculus carnosus muscle; Fig. 3b) spaced 2 cm apart were created on both sides of the spine resulting in a total of six defects per rabbit. LSE or skin grafts were sutured into the wounds with eight interrupted stitches (3-0 Vicryl, W9507T; Ethicon) along the edges (four in the corners and four midway along the sides) and one central quilting suture (Fig. 3c). The implants were hydrated using 0.9% normal saline throughout the procedure.

Postoperative care
The operating time did not exceed 3 h. Postoperative analgesia was provided by subcutaneous injections of 2.2 mg/kg Rimadyl (carprofen; Pfiser Ltd.). The wounds were covered with semipermeable adhesive film (Op-Site; Smith and Nephew) that was secured around the four edges with surgical tape. Plastic cone collars were fitted onto the rabbits for the length of the study to prevent disruption of the wounds. The dressings were removed 2 days after surgery.
Tissue harvesting and handling
At 1, 3, and 5 weeks, the animals were euthanized with an overdose of sodium pentobarbitone (Lethabarb, 324 mg/mL; Arnolds). Wounds were retrieved with a margin of normal skin and immediately fixed in 10% (v/v) neutral-buffered formol saline (Genta Medical) overnight, embedded in paraffin, and sectioned at a thickness of 5 μm.
Histomorphometric analysis
To determine the percentage of epidermal closure and the extent of wound contraction, sections were stained with hematoxylin and eosin and captured with a digital camera (2.11 megapixel, 1600×1200 pixel; Olympus C-2020 Zoom) mounted on a light microscope (Olympus BH-2). The percent epithelialization was determined by the length of the new epithelial layer divided by the length of the wound. Because of the depth of the wound, no hair growth occurred in the newly laid down tissue. The wound length was therefore determined as the distance between hair follicles. Similarly, the percentage of wound contraction was determined by measuring the distance between the hair follicles and dividing it by the original wound length measured on the day of surgery.
Immunohistochemistry
To analyze the angiogenic and inflammatory response, sections were stained with mouse monoclonal antibodies against CD31 (an endothelial cell marker; ab9498; Abcam) and CD45 (a pan-leukocyte marker; MCA808G; Serotec). The staining was visualized with the VECTASTAIN Elite ABC Kit (PK-6102; Vector Labs) and 3,3-diaminobenzidine (DAB) peroxidase substrate solution (SK-4105; Vector Labs). Sections were counterstained with hematoxylin.
For both the CD31 and CD45 stain, up to 10 images were digitally captured from the newly formed connective tissue in each section through a 20× objective lens. The inflammatory response was quantified by counting the number of CD45+ cells per area and expressed as a percentage of the total number of hematoxylin-stained nuclei (i.e., cells) in that area. The angiogenic response was quantified in each field of view by counting the number of CD31+ blood vessel cross sections per area and measuring the blood vessel cross-sectional area as a percentage of the total in surface area.
Statistical analysis
The data were analyzed using the Statistical Package for the Social Sciences software program (SPSS, version 13.0). For all analyses, statistical significance was set at the 0.05 level.
A normal (or t) distribution (with a particular mean and variance) was assumed of the (in vitro) variables of interest, and parametric tests were used to test the difference between two (t-test for independent samples) or more means (analysis of variance [ANOVA]). When performing the t-test, the Levene's test was used to test the assumption of equal variances and if unequal variances were detected to correct for violations. If differences were detected by ANOVA, a Tukey-HSD (honestly significant difference) or a Bonferroni post hoc test was applied to identify those differences.
For the in vivo experiments, six repeats of one condition at one time point were tested in a single rabbit and, therefore, a two-way (treatment modality and time) multilevel repeated measures ANOVA (RM-ANOVA) was used to analyze the data. Significant multilevel main effects were followed up with a Bonferroni post hoc test. Significant interaction effects were followed up by testing the simple main effects (i.e., one-way ANOVAs for each factor, treatment modality, and time, conducted separately at each level of the other factor).
Results
In vitro characterization
Living dermal equivalent
The fabrication of the dermal component of the living wound dressing involved encapsulation of both cells and a surgical mesh in a collagen hydrogel followed by mechanical compression after a setting period. Evaluation with the AB reduction assay, directly after the fabrication process, showed good cell viability retention (Fig. 4) with a nonsignificant (8%) reduction in the number of viable cells, from 39.4±(SD) 6.3 RFU to 31.7±8.0 RFU (one-way ANOVA, Tukey's post hoc, p=0.630). Over the 7-day culture period, cells were found to have significantly increased threefold up to 105.2±22.6 RFU (one-way ANOVA, Tukey's, p<0.001).
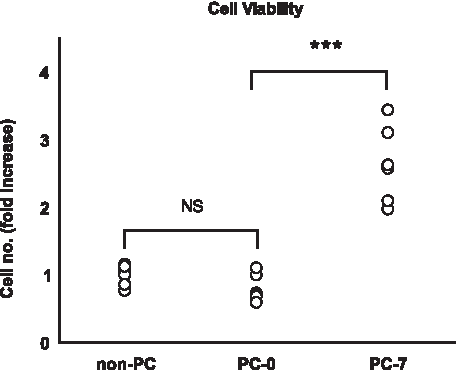
Scatter plot showing the change in viability and density of the cell inoculum (non-PC) seeded in the collagen component of the dermal equivalent, directly after compression (PC-0) and 7 days (PC-7) after compression (NS, not significant; Tukey's post hoc analysis; ***p<0.001; values are for six samples).
Living skin equivalent
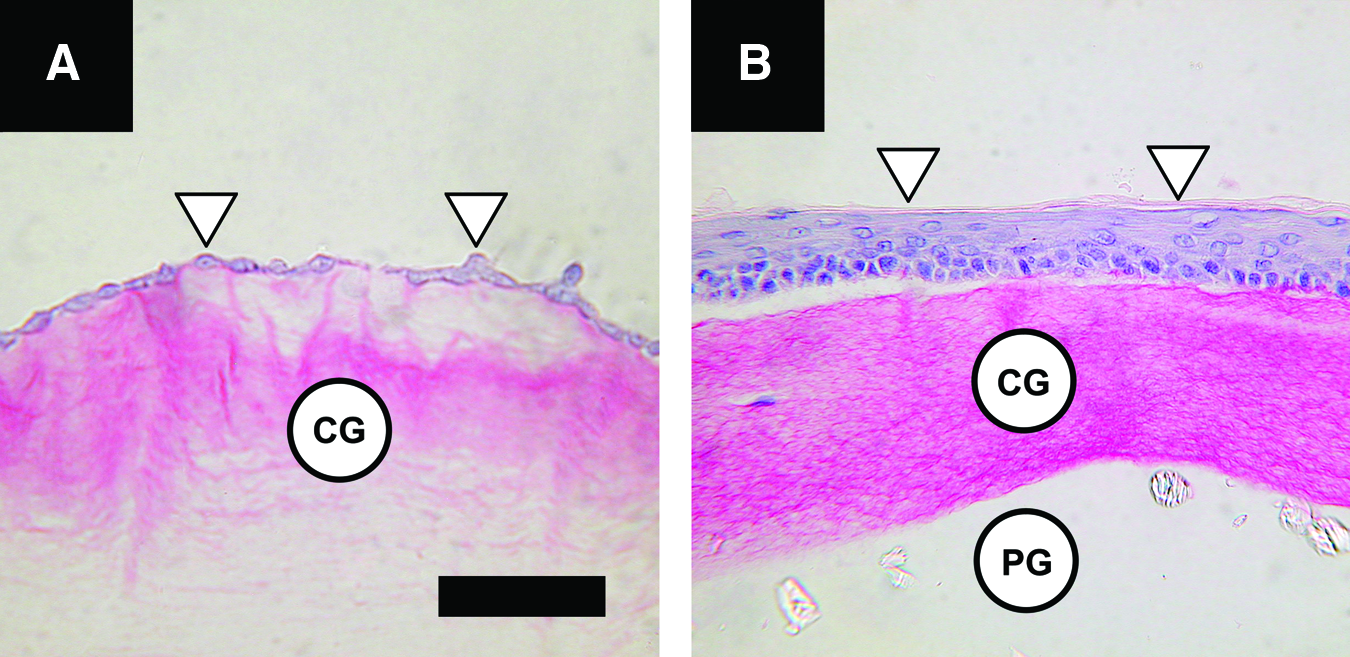
Finally, the living bilayered wound dressings were created by seeding keratinocytes onto the compressed dermal component at a density of 5±0.4 (SD)×105 cells/cm2, which, after a total fabrication time of 5 h, resulted in an LSE-M (Fig. 5a). These SEs were implanted directly (LSE-M) or after a 2-week cultivation period to differentiate the keratinocytes and form an LSE-S. The characteristic cuboïdal cell shape of keratinocytes in the basal layer and flattened cell morphology of keratinocytes in the suprabasal layers could be observed (Fig. 5b).
In vivo characterization
To test for safety and efficacy of the SEs, at 1, 3, and 5 weeks postinjury, the wounds were evaluated for inflammation, angiogenesis, and epidermalization.
Inflammation
The inflammatory response was quantified by counting the number of CD45+ cells per microscopic field of vision (Fig. 6a). No differences were detected between subsets and the data were pooled in an LSE group and an acellular group. The RM-ANOVA revealed a significant main effect of treatment, such that over the 5-week study period the average inflammatory response was 10% higher in the LSE group than in the acellular group [14.1±(SD) 8% vs. 12.8%±5%; F (1, 6)=8.0, p=0.03]. There was a significant main effect of time [RM-ANOVA, time, F (2, 6)=75.8, p<0.001], such that inflammation decreased over time for both groups. Additionally, a significant interaction effect was found between treatment and time [RM-ANOVA, cells×treatment, F (2, 6)=14.2, p=0.005; Fig. 6b], such that the inflammation was higher only in the first week by 20% in the LSE group than the acellular group [23%±4% vs. 19%±4%; one-way ANOVA, F (1, 2)=24.1, p=0.03].

Wound contraction
The data between LSE and acellular SEs did not differ and were pooled in an SE group and compared with the FTSG and NT groups. RM-ANOVA revealed a significant main effect of treatment on postoperative wound size [RM-ANOVA, treatment, F (2, 6)=9.3, p=0.014], such that over the 5-week study period the FTSG group had on average a twofold greater size than in the NT group (95.6%±13.8% vs. 50.8%±37.1%; Tukey's post hoc analysis; p=0.012; Fig. 6c). Wounds in the SE group had a marginally significant larger size (72.0%±30.0%) than those in the NT group (Tukey's post hoc analysis; p=0.071). A significant main effect was found for time [RM-ANOVA, time, F (2, 6)=18, p=0.003], such that wound size decreased over time in all groups. Additionally, no significant interaction effects were found between treatment and time, F (4, 6)=2.4, p=0.164.
Angiogenesis
The angiogenic response was determined from the number of vessel lumen and the total vessel luminal area in tissue sections stained for CD31 (Fig. 7a). The acellular data were pooled into one group and compared with the LSE-M and LSE-S groups.

For the number of lumen, a significant main effect of treatment was found, such that over the 5-week period on average a fourfold greater number of lumen was found in the LSE-S group than in the acellular group [94.4±105.7 vs. 24.8±8.2 lumen/area; RM-ANOVA, treatment, F (2, 3)=220, p=0.001]. There was a significant main effect of time [RM-ANOVA, time, F (2, 3)=384, p=0.001], such that the number of lumen decreased over time for all groups. Additionally, a significant interaction was found between treatment and time [RM-ANOVA, treatment×time, F (4, 3)=208, p<0.001; Fig. 7b]. Simple effect analysis showed significance for the LSE-S group [one-way ANOVA, F (2, 1)=438.4, p=0.034], with pairwise comparisons revealing that in the first week, the number of lumen was eightfold higher than the acellular group (236.6±38.3 vs. 24.8±8.2 lumen/area; LSD post hoc analysis, p=0.022).
For the luminal area, a significant main effect of treatment was found, such that the luminal area over the 5-week period on average was twice greater in the LSE-S group than the acellular group [5.5%±4.7% vs. 2.7%±1.2%; RM-ANOVA, treatment, F (2, 3)=24, p=0.014]. A significant effect of time was found, such that for all groups the total luminal area decreased as time progressed [RM-ANOVA, time, F (2, 3)=84.3, p=0.002]. Additionally a significant interaction effect was found between treatment and time [RM-ANOVA, treatment×time, F (4, 3)=19.8, p=0.017; Fig. 7c]. Simple effect analysis showed significance for the LSE-S group [F (2, 1)=756.0, p=0.026], with pairwise comparisons indicating that the total luminal area was threefold greater than the acellular group (11.8%±2.2% vs. 3.8%±0.9%; Bonferroni post hoc analysis, p=0.05).
Epidermal wound closure
Epidermal wound closure was quantified as the length of newly formed epidermis expressed as a percentage of the length of the wound (Fig. 8). The acellular data were pooled into one group and compared with the LSE-M and LSE-S groups. A two-way RM-ANOVA (treatment type, three levels; time, three levels) revealed a significant main effect of treatment on epidermal wound closure [F (2, 3)=39.3, p=0.007], such that closure was on average over the 5-week period, 16% higher in the LSE-S group (81±[SD] 28%) than in the acellular group (70%±43%) (Tukey; p=0.009). A significant main effect was found for time, such that for all groups wound closure increased with time [RM-ANOVA, time, F (2, 3)=2246.3, p<0.001]. In addition, a significant interaction effect was found between treatment and time [RM-ANOVA, treatment×time, F (4, 3)=39.3, p=0.006; Fig. 9]; in the first week, epidermal regeneration was fourfold higher in the LSE-S group compared with the acellular group.
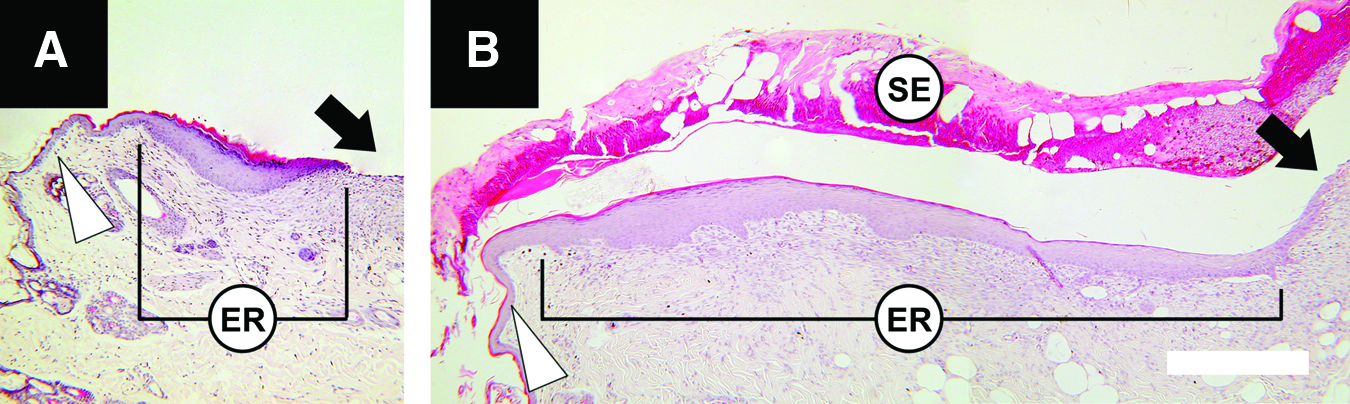
Micrographs of wound edges, 1 week after injury, showing the difference in epidermal regeneration (ER) in wounds receiving
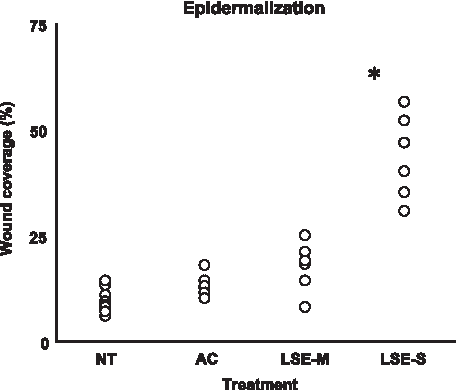
Scatter plots for the percentage epidermal wound closure, 1 week postinjury, in NT wounds, wounds treated with AC, and wounds treated with LSE-M and LSE-S.
Discussion
Our findings show that the tissue-engineered, living wound dressing, consisting of an ultrarapid fabricated LDE and a stratified epidermis, accelerated wound healing in acute full-thickness skin defects. The LDE was fabricated using a previously developed technique 8 involving collagen hydrogel encapsulation of allogeneic fibroblasts and a surgical mesh (Fig. 1a) followed by mechanical compression (Fig. 1b) forming, in the space of a single hour, a living dermal tissue with appropriate handling properties and a precise number of cells.
The LDE supported keratinocyte attachment and differentiation for the creation of an LSE with an undifferentiated (monolayer; Fig. 5a) and a differentiated (stratified; Fig. 5b) epidermis. Only the LSE-S had a significant stimulating effect on wound healing. This involved both an increased angiogenic response (Fig. 7) and accelerated epidermal wound closure (Fig. 9) compared with no-treatment, acellular dressings and the LSE-M.
Currently, the only FDA-approved tissue-engineered wound dressings featuring an LDE include Dermagraft,15,16 Apligraf,17,18 and OrCel.19,20 These products are available off-the-shelf for which they rely on the use of allogeneic cells. In the current study their use is associated with an increased inflammatory response (Fig. 6b), an absence of LSE integration into the wound bed, and therefore no effect of the LSE on scar cosmesis (Fig. 6c). These findings are consistent with the clinical experience with commercially available biological dressings which due to their temporary nature require multiple applications for a sustained therapeutic effect.
Tissue-engineered, living wound dressings have proven to be more effective in healing chronic wounds, such as venous leg and diabetic foot ulcers (Dermagraft and Apligraf)15,17 and acute split-thickness skin defects (OrCel), 20 than conventional nonsurgical treatment modalities, but the cost associated with their lengthy manufacture limits their clinical application to hard-to-heal wounds. To illustrate, Dermagraft (USD $38/cm2), 19 an LDE, is fabricated by seeding and maintaining fibroblasts in vitro on a Vicryl surgical mesh (the same used in this study) for a period of 2–3 weeks to achieve a sufficient number of cells for a therapeutic effect.21,22 The dermal component of OrCel (USD 28/cm2), 23 an LSE, is formed in 2 days and is established, like Dermagraft, by seeding allogeneic fibroblasts onto a preformed scaffold, in this case a collagen sponge. 20 The dermal component of Apligraf (USD 32/cm2), 19 an LSE, is also fabricated through cell encapsulation.24–26 However, a 6-day culture period is required for the embedded cells to strengthen the dermal matrix sufficiently to allow for clinical handling.
The technique presented in this article allows for an over 90% reduction in LDE fabrication time compared with OrCel, Dermagraft, and Apligraf and results in a 30% reduction in the formation of an LSE-S (Table 1). It is proposed that the presented technique could result in a lowering of the purchase cost of tissue-engineered dressings, allowing their application in cases in which they are currently considered more expensive than conventional treatments.27,28
LDE, living dermal equivalent; LSE-M, living skin equivalents with a monolayer epidermis; LSE-S, living skin equivalents with a stratified epidermis.
Dermagraft and Apligraf rely on cell activity for the formation of a stable dermal equivalent. The cell-independent technique presented in this article, however, allows for instantaneous handling with good cell viability retention (Fig. 4). Other cell-independent means to improve the handling properties of collagen hydrogels have been described and involve exposure to aldehydes (e.g., glutaraldehyde 29 or aldehyde sugars), enzymatic treatment (e.g., lysyl oxidase 30 and transglutaminase 31 ), nonenzymatic glycation, 32 and photo-polymerization using UVA light either alone 33 or with riboflavin. 34 Although these methods are effective in strengthening collagen hydrogels, these approaches are either too cytotoxic (e.g., glutaraldehyde, 29 UVA, 33 and riboflavin 34 ), too time-consuming (e.g., ribose 32 ), or too costly and impractical for bulk processing (e.g., lysyl oxidase 30 and transglutaminase 31 ).
From the experimental setup it is not possible to determine whether the improved response of the LSE-S was mainly due to the larger number of fibroblasts and keratinocytes or due to the presence of differentiated keratinocytes. Previous in vivo experiments have shown that the number of transplanted fibroblasts and keratinocytes is directly related to the size of the wound healing stimulus35–38 and that, for keratinocytes, this effect is mainly mediated by proliferating (i.e., undifferentiated) rather than differentiated cells. 39 Previous work from our lab has shown that the increased fibroblast densities obtained in compressed collagen hydrogels induce gene and protein expression of vascular endothelial growth factor that stimulates blood vessel formation both in vitro 40 and in vivo.41,42 The evidence suggests that the presence of a differentiated or even an undifferentiated epidermis in a tissue-engineered dressing is not essential for a wound healing effect. Indeed, although comparative studies are yet to be performed, Dermagraft (an LDE) shows similar wound closure rates as Apligraf (an LSE).15,43 Further in vivo analysis will therefore be performed to test the hypothesis that for a significant wound healing effect the need for an epidermis and an extended in vitro culture period can be obviated by inoculating the LDE, presented here, with an increased number of fibroblasts.
Conclusion
In the current study, a novel LSE was shown to accelerate wound healing, indicating its potential utility as a therapeutic wound dressing. This LSE consisted of a stable LDE and a stratified epidermis. The LDE is formed within 1 h which is over 90% quicker than currently available dermal tissue engineering strategies. The addition of a stratified epidermis, however, reduces this time advantage to 30%. It is proposed that the LDE fabrication technique can be modified to yield a wound dressing that does not feature an epidermis but maintains its effectiveness. It is therefore speculated that this method could help reduce the cost associated with the use of tissue-engineered wound dressings.
Footnotes
Acknowledgments
It is with pleasure that we acknowledge the invaluable technical assistance of Mrs. C. Gray, Ms. L. Fletcher, Ms. B. Kazmi, and Ms. C. Frederiksson. We gratefully acknowledge funding from the EU Framework VI program (3G-Scaff reference: 013603-3G SCAFF), EPSRC, BBSRC, and RNOH Special Trustees.
Disclosure Statement
No competing financial interests exist.