
Abstract
Short-term thrombotic occlusion and compliance mismatch hamper clinical use of synthetic small-diameter tissue engineered vascular grafts. It is felt that preconditioning of the graft with intimal (endothelial) and medial (vascular smooth muscle) cells contributes to patency of the graft. Autologous, non-vessel-derived cells are preferred because of systemic vascular pathology and immunologic concerns. We tested in a porcine model whether cultured bone marrow–derived mononuclear cells, also referred to as mesenchymal stem cells (MSC), are a potential source of intimal or medial cells in vascular tissue engineering. We show that MSC cultured in endothelial medium do not gain an endothelial phenotype or functional characteristics, even after enrichment for CD31, culturing under flow, treatment with additional growth factors (vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF)-2), or co-culture with microvascular endothelial cells (EC). On the other hand, we show that MSC cultured in MSC medium, but not in smooth muscle cell medium, show phenotypical and functional characteristics of vascular smooth muscle cells. We conclude that bone marrow-derived MSCs can be used as a bona fide source of medial, but not EC in small-diameter vascular tissue engineering.
Introduction
To seed and precondition a small-diameter TEVG, a suitable—preferably autologous—cell source for endothelial and vascular smooth muscle cells (SMC) has to be defined. Unfortunately, autologous endothelial and vascular SMC derived from blood vessels are often not suitable because of systemic cardiovascular disease. Many alternative (stem or progenitor cell) sources have been proposed and tested, such as peripheral blood mononuclear cells3,4 and mesenchymal stem cells (MSC) derived from various sources such as muscle, 5 adipose tissue,6,7 and bone marrow,8–10 the last of which has also been implemented in a clinical trial. 11
Endothelial cells (EC) and vascular SMC are embryologically derived from the mesodermal germ layer, although they arise at different time points from different precursors. Endothelial cells arise relatively early and are primarily derived from hemangioblasts. 12 Single EC fuse and form a lumen (vasculogenesis), creating primitive tubes. The remainder of the endothelial population mainly arises from this early developed network through proliferation (angiogenesis), although additional “mesoangioblasts” and circulating cells are also thought to contribute. 13 Vascular SMC differentiate locally during development from mesenchymal cells of different origins, such as the neural crest or the epicardium. 14 During development, only the mesoangioblast has been proposed as a common precursor for endothelial and vascular SMC, but there is no evidence of its persistence in the adult. 14
Thus, a common mesenchymal precursor for endothelial and vascular SMC in adults seems unlikely, but blood- and bone marrow–derived MSCs have independently been described to differentiate toward either cell type.8,15,16 Suggesting blood as a source of endothelial (progenitor) cells17–19 or even of vascular smooth muscle (progenitor) cells 20 is attractive, because it can be harvested through a simple blood draw. Despite the current discussion on progenitor characteristics of these cells, 21 it is clear that circulating numbers are low, resulting in challenging harvest that is not robust and predictable.19,22 For this reason, a perhaps more feasible source might be the mesenchymal stem and progenitor pool residing in the bone marrow. To explore the ultimate capacities of these cells to differentiate into vascular cells that can be used in vascular tissue engineering, we isolated porcine bone marrow–derived mononuclear cells (MSCs) and stimulated them toward endothelial or SMC differentiation. We made use of a porcine model because this is the preferred preclinical model for testing of ultimate small-diameter TEVG.
We postulated that bone marrow–derived mononuclear cells (MSC) could be differentiated in vitro toward vascular SMC but not to endothelium. We put forward that the bone marrow can be used as a cellular source for gaining strength and stiffness in a TEVG, but that another source of cells should be identified to create a nonthrombogenic inner lining of small-caliber TEVG.
Materials and Methods
Isolation and cultivation of porcine aortic EC and SMC
The Maastricht University animal ethics committee approved this study. Procedures were in accordance with institutional guidelines. Dutch Landrace pigs (sus scrofa) weighing 40 to 50 kg were euthanized using pentobarbital. The thoracic aorta was removed, and EC and SMC were isolated.23,24 Primary porcine aortic EC (PAEC) were cultured in EC growth medium-2 of the microvascular type (5% fetal bovine serum (FBS); Lonza) and porcine aortic SMC (PSMC) in SMC growth medium-2 (5% FBS; Lonza). Cells were used between passages 3 and 10.
Isolation and cultivation bone marrow-derived mononuclear cells (MSC)
Femora and humeri were explanted, and capita were removed. Bone marrow was aspirated, diluted with phosphate buffered saline (PBS)/0.05% ethylenediaminetetraacetic acid (EDTA) and a single cell suspension was created using a 100 μM cell strainer (BD Bioscience). This was loaded on a density gradient cell separation medium (Histopaque 1.077, Sigma) and centrifuged to isolate mononuclear cells. 25 Cells were washed three times in PBS/0.05% EDTA and plated at a density of 2.5×105 cells/cm2 on fibronectin-coated culture flasks. Mononuclear cells were cultured in different culture media, namely EC growth medium, MSC growth medium (10% FBS), or SMC medium (all Lonza). Five days after seeding, non-adherent cells were removed, and between 10 and 12 days after seeding, cells were detached and subcultured. Outgrowing cells are referred to as MSC 25 and used for experiments between passages 3 and 7 (in EC and SMC medium) or between passages 3 and 10 (in MSC medium).
Reverse transcriptase quantitative polymerase chain reaction
Cells were detached and between 3.5×105 and 1×106 cells were spun and lysed using RLT-buffer (Qiagen). Reverse transcriptase quantitative polymerase chain reaction was performed as described. 26 All samples were normalized for input based on ß-actin and glyceraldehyde 3-phosphate dehydrogenase. A list of primer sets used can be found in Supplementary Table S1. (Supplementary Data are available online at www.liebertonline.com/tea).
Immunohistochemistry
Cells were grown on fibronectin-coated 4-well chamber slides (BD Bioscience) until confluence, fixed overnight at 4°C using 2% paraformaldehyde in phosphate buffered saline (PBS), and washed with 70% ethanol. Top chambers were removed, and endogenous peroxidase activity was blocked by incubating for 20 minutes in 0.3% hydrogen peroxide in PBS/0.05% Tween. Cells were blocked with 2% bovine serum albumin (BSA) in PBS/0.05% Tween, incubated overnight with the primary antibody in a humidified chamber (Supplementary Table S2), washed, and incubated with the corresponding biotin-labeled secondary antibody (Vector) for 1 hour. Then, cells were incubated for 45 minutes using the ABC-kit (Vector). Visualization was performed using 3,3′-diaminobenzidine tetrahydrochloride dehydrate, and hematoxylin was used for counterstaining. Slides were mounted with Entellan (Merck), and images were taken using a Zeiss Axioskop 2 plus microscope equipped with an AxioCam HRc (Zeiss).
Flow cytometry
Cells were detached using trypsin/EDTA (Lonza), washed, resuspended in PBS/1% BSA, and incubated with antibody (Supplementary Table S2) for 30 minutes at room temperature (RT) on a shaking plate. Then they were washed and fixed in 1% paraformaldehyde in PBS or, in case of a non-fluorescent labeled primary antibody, resuspended in PBS/1% BSA with fluorescent-labeled secondary antibody and again incubated for 30 minutes at RT on a shaking plate. After that, cells were washed and fixed. Fluorescence was determined using a flow cytometer (FACSCanto II, BD Bioscience), and data were analyzed using Kaluza-software (Beckman Coulter).
Western blotting
Using complete radio-immunoprecipitation assay lysis buffer (Santa-Cruz Technology), cells were lysed on ice for 15 minutes and scraped. The lysis mixture was sheared using a 21-gauge needle and incubated for 30 to 60 minutes on ice before being centrifuged at 10,000 g at 4°C for 10 minutes. Protein concentration of supernatants was determined using the bicinchoninic acid method (Pierce) and mixed with 4× sample loading buffer (BioRad), 20× reducing agent (BioRad), and distilled water to a final concentration of 35 μg protein in 40 μL. Samples were boiled for 5 minutes at 95°C, cooled to RT, centrifuged for 1 minute at 13,400 g, and loaded on a 4% to 12% Bis-Tris gel (BioRad). The gel was blotted on a polyvinylidene difluoride membrane (BioRad) overnight at 4°C at 30 V. Blotting membrane was blocked by incubating with 3% milk powder in 1× tris-buffered saline with Tween and incubated with primary antibodies (Supplementary Table S2), followed by a horseradish peroxidase or biotin-conjugated secondary antibody. When using a biotinylated antibody, an extra incubation step with the avidin–biotin complex (Pierce) was performed before detection of the protein bands using the SuperSignal West Femto or Pico substrate (Pierce). Visualization was performed using the ChemiDoc XRS system (BioRad).
AcLDL uptake
Cells were cultured until confluence, incubated for 4 hours at 37°C with 2.5 μg/mL acetylated low density lipoprotein (AcLDL) labeled with AlexaFluor594 (Molecular Probes), and washed with PBS. Photographs were taken using a Leica DMI3000B inverted fluorescence microscope equipped with a DFC350 FX camera (Leica).
Nitric oxide and prostacyclin production
Cells were seeded in a 6-well plate with a density of 6,000 cells/cm2 and grown for 3 days. Medium was removed, and in the case of the prostacyclin (PGI2)-production assay, 1.5 mL of fresh culture medium was added, and 300 μL was sampled at 0 and 1 hours. Medium was centrifuged for 2 minutes at 10,000 g to remove cell debris, and supernatant was analyzed using the 6-keto-Prostaglandin F1α Enzyme Immunoassay Kit (Assay Designs). In the case of determination of nitric oxide (NO) production, 1.5 mL of fresh culture medium with or without 10μM acetylcholine was added, and 250 μL was sampled at 0 and 1 hours. Medium was centrifuged using 10,000 Da molecular weight cut off (MWCO) polysulfone filters (Sartorius), and flow-through was analyzed using the Total Nitric Oxide Assay Kit (Assay Designs).
Platelet adhesion in vitro
Cells were grown until confluence in a 24-well plate (static platelet adhesion experiments) or in μ-slides I0.4 Luer (Ibidi; platelet adhesion under flow experiments). Twenty-four hours before the start of the experiment, cells were stimulated with 80 ng/mL of tumor necrosis factor-alpha (TNF-α) when required. From healthy volunteers, 60 mL of peripheral blood was drawn with citrate and 40 mL with 80mM trisodium citrate, 52mM citric acid, and 183mM D-glucose (ACD) as anticoagulant. Blood with citrate was centrifuged for 10 minutes at 2,150 g at RT and supernatant was transferred to a new tube and centrifuged again (10 minutes, 2,150 g, RT), yielding platelet-poor plasma (PPP). Blood with ACD was centrifuged for 15 minutes at 240 g at RT, yielding platelet-rich plasma (PRP). Platelet count was determined using a Coulter Counter (Beckman Coulter)., One mL of ACD was added per 15 mL of PRP. This was centrifuged for 2 minutes at 3,000 g at RT, and the pellet was resuspended in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid with the same volume as starting volume. Apyrase (Sigma) was added at a concentration of 0.1 U/mL, and this was again diluted with 1 mL of ACD per 15 mL of PRP. Platelets were labeled with 6 μg/mL of carboxyfluorescein succinimidyl ester (Invitrogen) for 10 minutes at RT and centrifuged for 2 minutes at 3,000 g. For the static platelet adhesion experiments, the pellet was resuspended in PPP in a final concentration of 1×108 platelets/mL. Half of the platelets were activated with 10μM 2-(methylthio)adenosine 5’-diphosphate trisodium salt hydrate (MES-ADP, Sigma). Five hundred μL of platelet suspension was added per well, with or without MES-ADP. Plates were incubated for 1 hour at 37°C, washed twice with PBS, and three photographs were taken per well using a Leica DMI3000B inverted fluorescence microscope equipped with a DFC350 FX camera (Leica) using the 10× objective and fluorescein isothiocyanate filter. In the case of platelet adhesion under flow experiments, platelet count was determined again and the pellet was resuspended in PPP in a concentration of 1.82×108 platelets/mL. To isolate red blood cells (RBC), the remaining blood from the citrate and ACD tubes was centrifuged for 5 minutes at 850 g without brake. The supernatant and the upper layer of RBC were removed, and an equal volume of PBS was added to the lower layer. This was centrifuged for 10 minutes at 1,920 g. The supernatant was discarded, and the RBC were added to the PRP, resulting in a final concentration of 1.0×108 platelets/mL, corresponding to a hematocrit of 45%. Platelets were activated with 10μM MES-ADP. The platelet mixture was then flowed over the cells using a syringe pump (NE 1600, ProSense, Oosterhout, The Netherlands) with a flow rate of 2.28 mL/min, after which the slides were washed with a plasma substitute (Gelofusine, Braun, Melsungen, Germany) for the same amount of time. Ten photographs were taken per slide. Surface coverage with platelets was determined per photograph using ImageJ 1.41o (National Institutes of Health).
Flow experiments
Cells were seeded at a density of 30,000 cells/cm2 on μ-slides I0.4 Luer (Ibidi) and allowed to attach for 2 hours, after which they were cultured for 72 hours under a shear stress of 2 Pa (20 dyn/cm2) using the Ibidi pump system (Ibidi) or under static conditions (static control). Cells were lysed on the slide using RLT buffer (Qiagen), and RNA was isolated using the RNeasy micro kit (Qiagen).
Co-culture experiments
MSC in EC medium, human coronary microvascular EC (HCMVEC, Lonza), or both (ratio MSC:HCMVEC=1:2) were seeded with a total density of 8,000 cells/cm2 on fibronectin-coated 4-well chamber slides (BD Bioscience) and grown for 72 hours. Processing for immunohistochemistry was performed as described above.
CD31-selection
Freshly isolated bone marrow-derived MSC were immunolabeled for CD31 (as described above). Cells were sorted 27 in MSC-CD31+ and MSC-CD31– cells using a FACSAriaI (BD Bioscience) and cultured in EC medium on fibronectin-coated plastics and treated as described above. Cells from passage 7 were used for messenger RNA (mRNA) analysis.
Growth factor stimulation
MSC in EC medium were cultured in parallel with MSC in EC medium supplemented with five times excess (50 instead of 10 ng/mL) VEGF and FGF-2 (Lonza) for 14 days, 16 both on fibronectin-coated plastics. Medium was changed every 2 to 3 days. Processing for mRNA analysis was performed as described above.
Migration experiments
Cells were seeded with 8.3×104 cells/cm2 on a fibronectin-coated ThinCert BD FluoroBlok (24 wells) and cultured overnight. Cells were labeled with 1 mg/mL of calcein (Invitrogen) for 1 hour at 37°C and stimulated with or without 10 ng/mL of platelet-derived growth factor (PDGF)-BB in the lower chamber in OptiMEM (Invitrogen). All conditions were executed in quadruple, and fluorescence of migrated cells was measured using a Victor plate reader (PerkinElmer) at different time points.
Collagen-production assay
Cells in passage 10 (to allow induction of a synthetic phenotype 28 ) were seeded with 25,000 cells/cm2 in a 12-well plate and grown to confluence within 1 week. After that, cells were stimulated with or without 10 ng/mL of TGF-β1 (R&D Systems) in SMC medium with low (0.5%) serum and no FGF2 for 3 days. Cells were isolated at 0 or 72 hours by washing twice in PBS, incubating overnight with 0.5M acetic acid at 4°C, and scraping to make a cellular extract. After isolation, samples were centrifuged 10 minutes at 3,000 g. Supernatant was analyzed using a collagen assay (QuickZyme BioSciences, Leiden, The Netherlands).
Statistics
The unpaired t-test was used for statistical analysis, except for the static platelet adhesion assay, for which two-way analysis of variance with Bonferroni post hoc testing was performed. Differences were considered significant at p<0.05. Analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA).
Results
Lack of endothelial phenotype of MSC cultured in EC medium
To induce an endothelial phenotype in bone marrow–derived MSC, cells were seeded and subcultured in EC medium containing growth factors involved in endothelial differentiation (VEGF, FGF-2). 29 With respect to morphology, mRNA, and protein expression, MSC cultured in EC medium (MSC-EC) were compared with PAEC. MSC-EC presented with a cobblestone appearance and grew in a monolayer, even at high confluence (Fig. 1A), although mRNA expression of the endothelial marker CD31 was low (a 0.005 fraction of the expression in PAEC). Expression of CD34, VEGF receptor-2 (VEGFR-2), von Willebrand factor (vWF), NOTCH4, and Delta-like 4 (DLL4), five other endothelial markers, was below detection level (Fig. 1B). In contrast, expression of alpha smooth muscle actin (αSMA), an early SMC marker, was 30 times as high in MSC-EC, and the pro-thrombotic tissue factor (TF) was almost 50 times as high. CD39, a transmembrane NTPDase expressed in EC and in immune cells, 30 was 15 times as high expressed in MSC-EC as in PAEC (Fig. 1B).
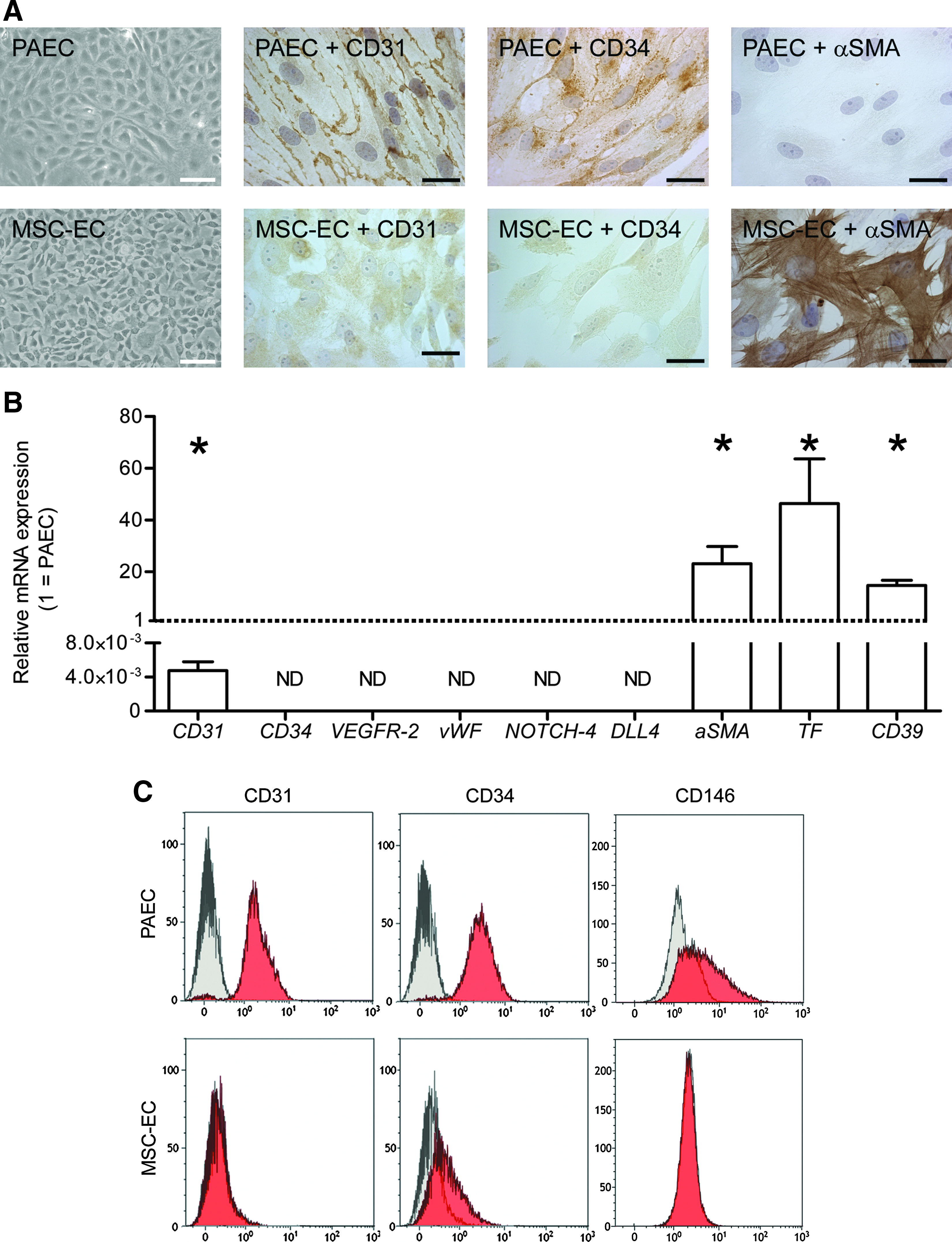
Phenotypical analyses of porcine bone marrow–derived mesenchymal stem cells cultured in endothelial medium (MSC-EC).
These apparent differences were confirmed at the protein level (Fig. 1A,C). According to immunohistochemistry, CD31 and CD34 were expressed on the cell membrane of PAEC, whereas only a faint cytoplasmic staining above background could be observed in MSC-EC, suggesting that CD31 might be translated in low quantities but not transported to the proper cellular compartment (the cell membrane). In contrast, αSMA staining could not be seen in PAEC, whereas the majority of MSC-EC showed strong αSMA staining in stress fibers. Lack of or low protein levels of CD31, CD34, and another endothelial marker, CD146, in MSC-EC were also confirmed using flow cytometry. PAEC and MSC-EC also failed to express the leukocyte marker CD45 (data not shown), confirming the absence of hematopoietic differentiation.
Lack of functional endothelial properties of MSC-EC
Because MSC-EC grew in a monolayer, we still considered the possibility that these cells could be used as a nonthrombogenic inner cell layer of a small-diameter TEVG. To test whether these cells had EC properties, we investigated the uptake of AcLDL and the production of PGI2 and NO. Platelet adhesion was also determined under static and flow conditions. Although MSC-EC were able to take up AcLDL (Fig. 2A), their production of PGI2 was significantly lower than that of PAEC (Fig. 2B). NO production could not be detected at all, with or without stimulation with 10μM acetylcholine, whereas a 10-times increase in NO-production was observed in PAEC upon stimulation (t=1 hour, Griess assay; data not shown). Moreover, MSC-EC showed platelet adhesion comparable with PSMC rather than PAEC under stimulated and unstimulated conditions and under static conditions and flow (Fig. 2C, D), suggesting that MSC-EC lack essential endothelial functions.
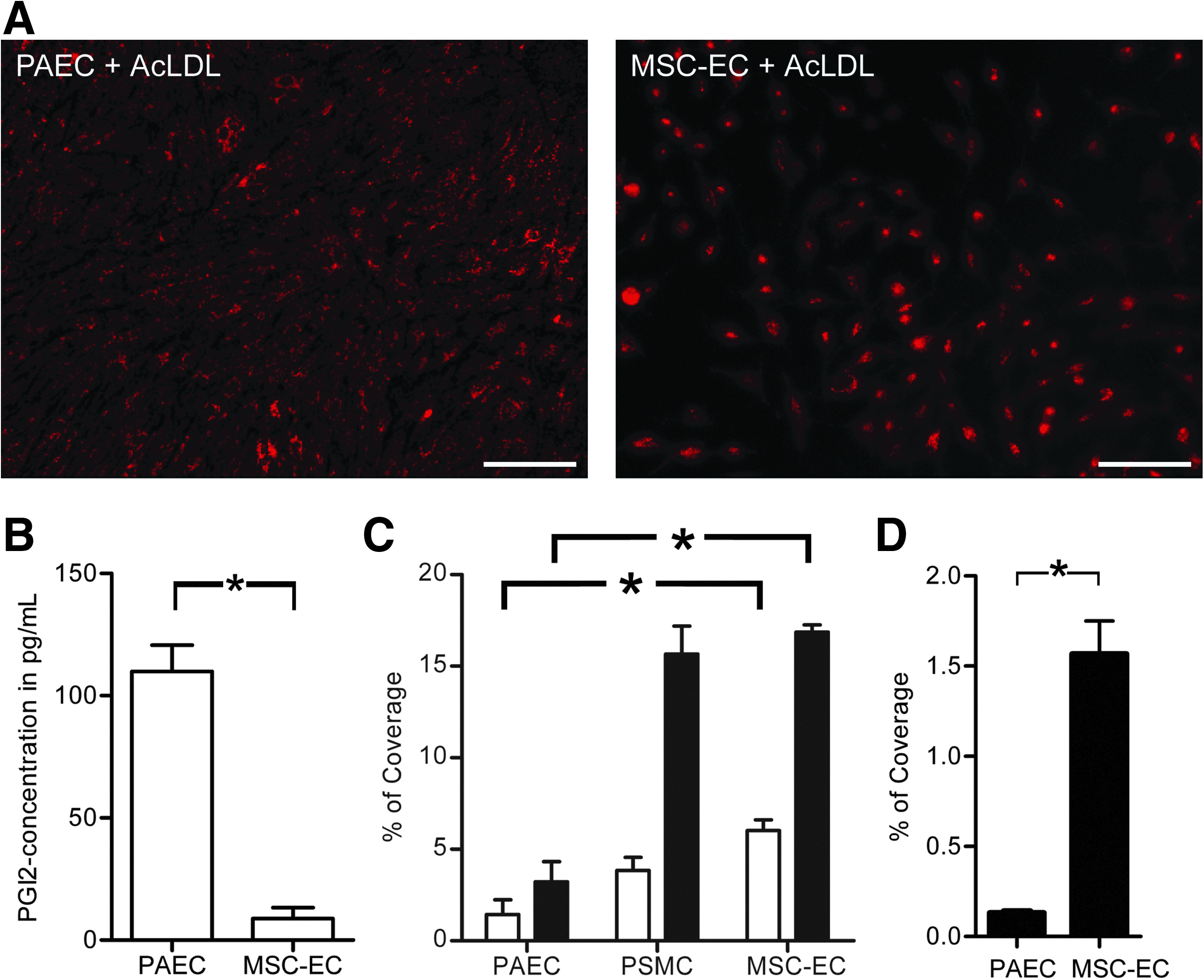
Functional analyses of MSC-EC.
CD31-enrichment cannot improve endothelial yield
Culturing MSC in EC medium was not sufficient to induce endothelial phenotype or function. To increase purity, we enriched the bone marrow–derived MSC fraction for CD31hi cells. Subsequently, unselected MSC and CD31hi MSC were cultured in parallel in EC medium. Morphologically, these two types of MSC-EC were largely comparable with PAEC (Fig. 3A), and as a result of selection, CD31-expression was high in CD31hi MSC-EC, although no further endothelial phenotype could be confirmed on the basis of CD34 and VEGFR-2 expression (Fig. 3B), suggesting that CD31 enrichment did not result in higher yields of endothelial (progenitor) cells.

CD31hi selection of MSC, flow experiment with MSC-EC and co-culture of MSC-EC with endothelial cells (EC). (
MSC do not gain endothelial phenotype even after stimulation of differentiation
It has been published that culturing MSC under flow can induce an endothelial phenotype. 15 Thus, we cultured MSC for 72 hours under flow (2 Pa) and observed no effect of flow on mRNA expression of endothelial-specific genes (CD31, CD34, VEGFR-2; Fig. 3C and data not shown). Differences with PAEC even became more prominent than under the static conditions (data not shown), indicating that culturing under flow does not induce expression of endothelial markers in MSC-EC.
Reportedly, high levels of pro-angiogenic growth factors such as VEGF and FGF-2 can induce endothelial differentiation of human MSC. 16 To test this, porcine MSC-EC were cultured for 14 days in EC medium containing 50 ng/mL of VEGF and FGF-2. Of all endothelial markers tested (CD31, CD34, VEGFR-2, vWF, NOTCH4 and DLL4), only mRNA expression of CD31 exceeded detection level but showed a a 0.0003 fraction of the expression in PAEC (Fig. 3D and data not shown). αSMA and TF levels were five and 53 times as high as in PAEC, respectively (Fig. 3D). Also, when mRNA expression levels of MSC-EC cultured with high levels of growth factors was compared with MSC-EC cultured in regular EC-medium, no induction of expression of endothelial markers could be observed (data not shown).
Finally, we tried to induce endothelial phenotype in porcine MSC-EC by co-culturing them with HCMVEC. 31 Immunostaining with an antibody raised against porcine CD31, not cross-reacting with human CD31, should reveal endothelial phenotype of porcine MSC-EC, but no such cells could be detected (Fig. 3D).
SMC phenotype of MSC cultured in SMC or MSC medium
To induce vascular SMC phenotype in MSC, bone marrow-derived MSC were cultured in MSC medium (MSC-M) 32 or SMC medium (MSC-S) and compared with primary PSMC (cultured in SMC medium). Morphologically, MSC-M were more akin to PSMC than MSC-S (Fig. 4A). PSMC and MSC-M were elongated with multiple protrusions and were able to grow in multiple layers, whereas MSC-S were shorter, had no protrusions, and stayed in a monolayer, even at high confluence. mRNA expression of several SMC-markers such as αSMA, CALPONIN, and DESMIN (Fig. 4B, C) confirmed similarity between MSC-M and PSMC, although αSMA and CALPONIN levels were still somewhat lower (Fig. 4B). MSC-S failed to show SMC induction.

Phenotypes of porcine bone marrow–derived MSC cultured in MSC medium (MSC-M) or smooth muscle cell medium (MSC-S). (
Protein levels of αSMA, CALPONIN, and smooth muscle myosin heavy chain isotope II (SMMHC-II) were comparable between PSMC and MSC-M using Western blotting (Fig. 4D,E). Immunohistochemistry confirmed comparable protein expression patterns between PSMC and MSC-M, whereas MSC-S lacked expression of SMC markers (Fig. 5). Not only quantitative differences in protein expression could be confirmed, but also the appearance of stress fibers was observed in PSMC and MSC-M and only minimally in MSC-S (Fig. 5; most obvious in SMMHC-II staining).

Protein expression patterns as determined using immunohistochemistry of PSMC, MSC-M, or MSC-S. PSMC and MSC-M show expression of αSMA, CALDESMON, CALPONIN, DESMIN, and SMMHC-II in stress fiber orientation, whereas MSC-S show low or no levels of expression. Fibers can be observed in MSC-S only in the αSMA staining. Expression of collagen 1 is observed in all cell types, but is distinctly lower in MSC-S. Scale bar=200 μm.
Functional SMC properties of MSC cultured in MSC medium
To study functional SMC properties of MSC-M, we studied migration toward platelet-derived growth factor-BB (PDGF-BB). Migration of MSC-M was comparable with that of PSMC (Fig. 6A). Second, the ability of MSC-M to produce collagen, at the mRNA level and the protein level was assessed. MSC-M mRNA expression of COLLAGEN1A1 was similar to that of PSMC, and expression of COLLAGEN1A2 was significantly greater than that of PSMC (Fig. 6B). Protein levels of collagen as determined using the QuickZyme assay were comparable between cell types (Fig. 6C).
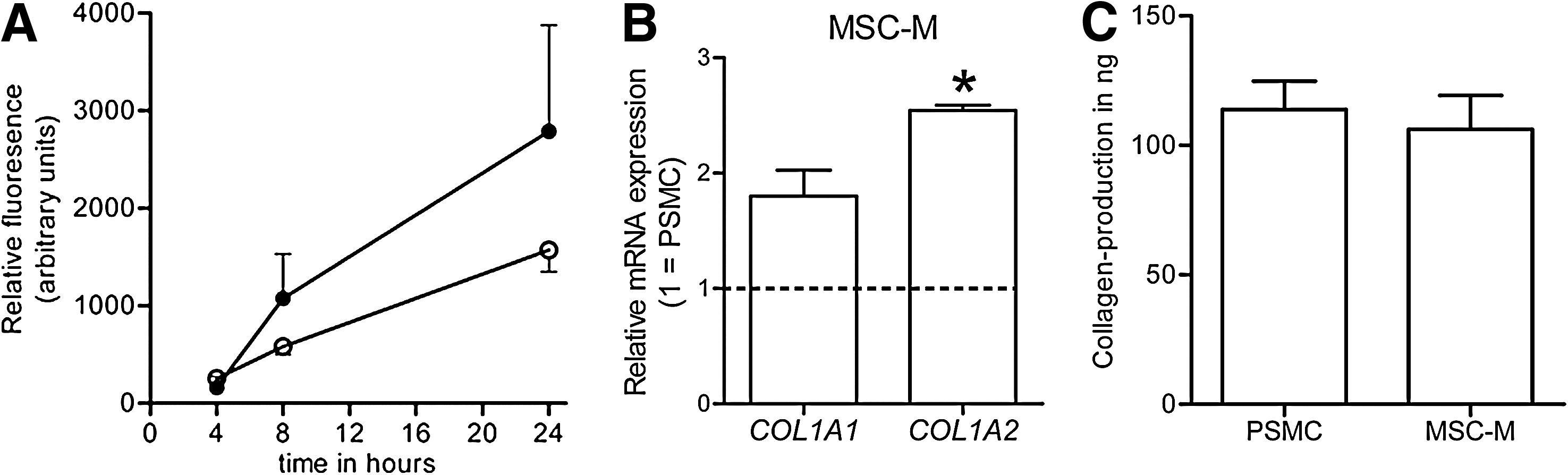
Functional analyses of porcine bone marrow–derived MSC cultured in MSC-M. (
Discussion
Usage of bone marrow–derived MSCs in (vascular) tissue engineering has been suggested,8–10 and several clinical trials have been successfully performed,11,33,34 although no detailed description of their ultimate vascular performance has been reported. We show here that bone marrow–derived MSCs do not differentiate into EC; nevertheless, they are able to gain favorable vascular SMC characteristics in vitro.
In vitro, proliferative bone marrow–derived MSCs 35 are able to differentiate into at least three different cell types (adipocytes, chondrocytes, and osteoblasts) 36 and are therefore most commonly referred to as MSC. These cells can also differentiate into skeletal muscle cells, 37 cardiomyocytes, 38 and hepatocytes, 39 although it is unclear whether these, mainly in vitro, findings have relevance during (patho)physiological processes in vivo.
Regarding their vascular potencies, MSC have independently been described to differentiate into endothelial and vascular SMC in different species in vitro.15,16,32 In vivo, it is thought that these cells are recruited from the bone marrow upon vascular damage and travel to the injured artery to contribute structurally to vascular repair. 40 Many different factors and pathways have been shown to play a role in recruitment and differentiation of these MSC. 41 Re-creating those pathways in vitro to produce differentiated vascular cells for vascular tissue engineering will therefore be challenging. Moreover, the observation that bone marrow–derived cells contribute structurally to the endothelium after vascular injury has recently been questioned and suggested to be possible microscopic artifacts.42,43 This corresponds with our observation that bone marrow–derived MSC are unable to differentiate into EC.
Developmentally, the main source of EC is VEGFR-2–expressing hemangioblasts. 13 Upon stimulation with VEGF, hemangioblasts differentiate into EC.44,45 The presence of hemangioblasts in adults is a matter of debate. It can be expected that, if they exist, they would still be dependent on VEGF for endothelial differentiation, 46 which findings in embryonic stem cells in vitro underscores. 47 Flow cytometric analysis on freshly isolated bone marrow–derived mononuclear cells showed that a subset (∼10%) of these cells expresses VEGFR-2 (data not shown). VEGF-A is supplemented in the endothelial growth medium used, suggesting that VEGF signaling can take place but apparently does not lead to endothelial differentiation, indicating that no functional hemangioblasts are present in the bone marrow–derived MSC fraction. When MSC-EC were cultured for a long period of time (up to 14 passages), the differences with PAEC became even more prominent, and when compared with freshly isolated bone marrow–derived MSCs, no increase in expression of endothelial markers could be seen (data not shown). Even addition of 5 times greater levels of the pro-angiogenic growth factors VEGF and FGF-2 for 14 days did not induce endothelial differentiation. Additionally, we tested whether another endothelial culture medium, human endothelial serum-free medium (Gibco), would result in outgrowth or differentiation of MSC-derived EC, but this medium did not result in any outgrowth of cells (data not shown). Although (partial) SMC differentiation of these cells is seen in human and animal models and in datasets from different research groups, endothelial differentiation seems to be extraordinary and is therefore not easy to translate to a clinical setting. These and other recent findings42,43 question the plasticity of bone marrow–derived MSC.
To determine whether the bone marrow–derived MSC population might be suitable as a source of primary (already differentiated) EC, we enriched the mononuclear cell fraction for CD31hi cells using fluorescence-activated cell sorting. 27 A small fraction (∼1%) of cells could be isolated in this way. Nevertheless, these cells showed good proliferation, but only CD31 expression was prominent, whereas there was no expression of other endothelial markers (CD34 and VEGFR-2). Even after culturing these cells for 7 passages in endothelial medium, gain of endothelial phenotype was not observed, suggesting that these cells are not endothelial progenitor cells or (hem)angioblasts. We consider the CD31hi-MSC-EC to be partially differentiated hematopoietic stem cells that could represent a proliferative precursor of monocytes, granulocytes, or B-cells. 48 To further investigate this assumption, it might be worthwhile to explore the expression of different mRNA splice variants of CD31, because it it has been published that these differ between endothelial and hematopoietic cells. 48 Unfortunately, this is not possible for porcine cells, because the sequences of the different CD31 splice variants in pigs are unidentified.
It has been published previously that culturing of MSC under flow could induce endothelial differentiation,15,46 but different outcomes have been published for the effect of low (1 Pa) versus high (2 Pa; as used in our study) and short (24 hours) versus long (48–72 hours) time exposure. Dong et al. 15 showed endothelial differentiation to be prominent after shear stress exposure, whereas Bai et al. 46 found that endothelial characteristics (alignment with flow, VEGFR-2 expression) decrease with greater flow rate and longer exposure time in a VEGF-dependent manner. We chose to expose our cells to 2 Pa for 72 hours in VEGF-containing culture medium, because the ultimate usage for these cells in a small vascular tissue engineered graft will expose them to chronic high-flow conditions. When doing this, we could not reproduce endothelial differentiation in our experimental setting. When EC are cultured under flow, cells align in the direction of flow, 49 VEGFR-2 is activated independently of its ligand, 50 and its expression is increased. 51 We observed no alignment of MSC under flow (data not shown). Moreover, VEGFR-2 expression patterns remained absent, and expression of CD31 was not induced. We conclude that—at least when using porcine MSC—flow-induced endothelial differentiation does not take place. Likewise, co-culturing of MSC with microvascular EC, which according to previous studies could induce EC differentiation of MSC, 31 did not lead to the development of porcine MSC-derived EC in our hands. It is unlikely that this is due to species differences, because high homology (96–99%) of proteins important in endothelial differentiation such as VEGF, VEGFR-2, FGF receptor 1 (FGFR1) and FGFR2 are present. It might be that co-culturing of MSC with microvascular EC for a longer time (>72 hours) is necessary, although Lozito et al. already showed a strong increase in CD31 expression after 48 hours. 31
Functionally, MSC cultured in endothelial medium grow in a monolayer, show cobblestone morphology comparable with that of PAEC, and are able to take up acetylated low-density lipoprotein (Ac-LDL). When porcine skin fibroblasts are cultured in the same EC medium (EGM-2MV, Lonza), uptake of Ac-LDL can also be observed (data not shown), suggesting that this is an artifact induced by the culture medium rather than bona fide endothelial differentiation. Although it is unlikely that porcine MSC-EC can substitute fully for EC, we considered the possibility that they adopt nonthrombogenic properties. We therefore tested the thrombogenic status of the MSC-EC. Morphologically, MSC-EC showed a quiescent monolayer of cells and presented with relatively high mRNA levels of CD39, although they persistently exhibited clear antithrombotic properties, demonstrated by high TF levels, undetectable NO production after acetylcholine-induction, extremely little PGI2 production, and high levels of platelet adhesion, even under unstimulated conditions. Although CD39 reduces platelet aggregation in EC, the relatively high CD39 levels in MSC-EC did not result in low platelet adhesion. Because CD39 plays, in addition to a thromboregulatory role in EC, an important immunomodulatory role in neutrophils, lymphocytes, and monocytes, 30 we believe that this again points toward the MSC-EC as being partially differentiated hematopoietic stem cells. Therefore, these cells seem unsuited for seeding the luminal side of synthetic vascular grafts. Thus, we believe that bone marrow–derived MSCs are not appropriate for creation of the endothelial layer of small-diameter TEVGs.
On the other hand, we and others8,52 have shown that MSC seem perfectly able to differentiate into cells with a vascular SMC phenotype. Gong et al. 32 and we show that the designated MSC medium (MSCGM) induces this differentiation, even though the particular medium was designed to keep MSC in an undifferentiated state. The fact that, at high confluence, MSC are prone to differentiate rather than stay in a proliferative, undifferentiated state could partially explain this, 53 although medium supplemented with vascular SMC-specific growth factors (SmGM-2, Lonza) does not induce vascular SMC phenotype, even at higher passages (>7; data not shown), also at high confluence. In addition, several other culture media that were tested (Dulbecco's modified Eagle medium/10% FBS, stem cell expansion medium (Millipore), cardiomyocyte differentiation medium (Millipore) or MSC-chemically defined medium (Lonza); data not shown), induced proper proliferation rates and variable SMC differentiation, albeit none superior to the MSC medium of Lonza. Also, no EC differentiation occurred under these culture conditions. In contrast to the decrease in EC markers over time in MSC cultured in EC medium, expression of SMC markers increased over time when cultured in MSC medium (up till passage 14; data not shown). Unfortunately, the composition of the MSC medium was not disclosed, making it difficult to speculate on the mechanisms taking place during the in vitro differentiation steps. It is known that this culture medium contains 10% FBS. Most likely, TGF-β and PDGF-BB present in serum largely contribute to the differentiation process.8,54 Because the SMC medium contains only 5% FBS, this might be insufficient for proper SMC differentiation, although Gong et al. found no differences between culturing with 10% or 2% FBS. 32 The fact that we observed stronger SMC differentiation of MSC cultured in MSCGM and possibly a stronger effect of FBS than Gong et al. 32 could be because of species differences or differences between alternate batches of serum.
When MSC are cultured in MSC medium, not only their phenotype, but also their functional properties largely overlap with those of vascular SMC. In contrast to MSC cultured in SMC medium, protein expression of SMC markers is not merely present, but can also be localized to the actin stress fibers within the cell, suggesting the presence of a contractile apparatus. 28 In addition, for use in the medial part of small-diameter tissue engineered grafts, it is important that cells produce sufficient amounts of collagen to provide for the ultimate strength of the graft. We show, at the mRNA and protein levels and according to the QuickZyme collagen-production assay, that these cells are perfectly able to produce collagen in an amount comparable with that of vascular SMC. We also show that these cells are sensitive to PDGF-BB, because they migrate in vitro toward a PDGF-BB gradient in a pattern comparable with that of vascular SMC. It has been published that correct interaction between intimal and medial cells is dependent on PDGF-BB. 55 Thus, we consider that these cells are capable of such interaction, which is probably required for a fully functional vascular wall of a small-caliber artery.
In conclusion, we show that porcine bone marrow–derived mononuclear cells (MSC) can reproducibly be isolated and cultured. In vitro, these cells are unable to gain sufficient endothelial properties to be considered useful in small-diameter tissue engineered vascular grafts, but they can acquire an adequate vascular smooth muscle phenotype and as such should seriously be considered for use as medial cells in small-diameter tissue engineered vascular grafts. It is important to further translate these findings to the human situation8,32 and to continue the search for an autologous source for intimal cells. For the latter, the usefulness of blood-derived endothelial (progenitor) cells 56 or of microvascular EC that can be isolated out of, for example, adipose tissue 57 or skin biopsies can be explored. 58
Footnotes
Acknowledgments
This project was executed by the Institute for the Technical Development of Autologous Cell Therapy consortium. DGM Molin is supported by the Euregional Meuse-Rhine Interreg IVA grant BioMIMedics. The authors would like to thank Marion Feijge, Rik Olde Engberink, Mat Rousch, and Geertje Swennen for technical assistance.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.