
Abstract
A multifaceted therapeutic approach involving biomaterial scaffolds, neurotrophic factors, exogenous cells, and antagonists to axon growth inhibitors may ultimately prove necessary for the treatment of defects resulting from spinal cord injury (SCI). The objective of this study was to begin to lay the groundwork for such strategies by implanting type I collagen scaffolds alone and incorporating individually a soluble Nogo receptor, chondroitinase ABC (ChABC), and mesenchymal stem cells (MSCs) into a standardized 3-mm-long hemiresection defect in the rat spinal cord. Statistically significant improvement in hindlimb motor function between the first and fourth weeks post-SCI was recorded for the scaffold-alone group and for the ChABC and MSC groups, but not the control group. Four weeks post-SCI, the scaffolds appeared intact with open pores, which were infiltrated with host cells. Of note is that in some cases, a few growth-associated protein 43 (GAP-43)-positive axons were seen reaching the center of the scaffold in the scaffold-alone and ChABC groups, but not in control animals. Angiogenic cells were prevalent in the scaffolds; however, the number of both macrophages and angiogenic cells in the scaffolds was significantly less than in the control lesion at 4 weeks. The results lay the foundation for future dose–response studies and to further investigate a range of therapeutic agents to enhance the regenerative response in SCI.
Introduction
The objective of this study was to investigate the use of collagen sponge-like scaffolds alone and for the delivery of select therapeutic agents to enhance a regenerative response in a standardized hemiresection defect created in the rat spinal cord. The agents included the scaffold alone, soluble Nogo receptor (sNgR), chondroitinase ABC (ChABC), and bone marrow-derived mesenchymal stem cells (MSCs). This investigation provided an initial qualitative and quantitative contemporaneous comparison of the effects of these therapeutic agents, delivered by collagen scaffolds, on certain features of the reparative response in a standardized defect in the rat spinal cord: ingrowth into the scaffold-filled lesion of neurogenic, angiogenic, and regulatory cells (viz., macrophages and astrocytes).
Prior studies have employed collagen in various formulations for the treatment of defects in the spinal cord: gel22–24 ; filaments 6 ; and preformed sponge-like scaffolds.25,26 Even though type I collagen is not a principal component of the extracellular matrix (ECM) of the central nervous system, it has been widely shown to support the growth and differentiation of neurons in vitro and has been applied as a gel scaffold in vivo.27–29 Collagen can also inhibit glial proliferation, and thus may decrease glial scar post-SCI. 30
The benefit to axon regeneration of neutralizing or blocking the activity of myelin proteins—Nogo, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein—or their signaling pathways has been demonstrated over the past decade.31–34 One specific approach is the use of the recombinant sNgR molecule aimed at preventing myelin proteins from reaching NgR1 on the cell surface. 20 sNgR has the advantage of targeting all three myelin proteins simultaneously while avoiding cytotoxicity and other problems associated with intracellular agents that much cross the cell membrane. Several studies have demonstrated the potential therapeutic benefits of sNgR33,35,36 for the treatment of SCI lesions.
Other inhibitory molecules present after SCI include CSPGs, which are components of the glial scar. Enzymatic degradation of CSPGs using ChABC has been shown to facilitate enhanced axon regeneration.21,37 In most studies, the method used to deliver agents to neutralize inhibitory molecules has been by intrathecal injection. No studies have yet examined delivery of antagonists to inhibitors within a biodegradable scaffold.
Various stem cell approaches have been studied for the treatment of SCI, including the implantation of neural stem and progenitor cells into spinal cord lesions38–40 and implantation of cells that were first genetically encoded ex vivo to express specific neurotrophic factors.41–44 The roles of MSCs as therapeutic agents for SCI relate to their potential for transdifferentiation into neural cell types and as sources of regulatory molecules. While there are conflicting data regarding these roles in treating SCI, 43 the ease with which autologous MSCs can be obtained and their putative benefits in other applications commended their investigation in this study.
Materials and Methods
Experimental design
A 3-mm-long hemiresection defect was created in the adult rat thoracic spinal cord, and a collagen scaffold was immediately implanted into the defect alone or as a delivery vehicle for cells or select therapeutic agents. The scaffold was implanted in combination with a collagen membrane overlying the defect area acting as a dorsal barrier. All animals were sacrificed at 4 weeks after injury. The experimental groups were as follows:
(1) Control group—3-mm hemiresection SCI with no implant (n=6);
(2) Dehydrothermally (DHT) crosslinked type I/III collagen scaffold (n=7);
(3) Carbodiimide (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide [EDAC])-crosslinked collagen scaffold (n=7);
(4) EDAC-crosslinked collagen scaffold delivering sNgR (n=6);
(5) EDAC-crosslinked collagen scaffold delivering ChABC (n=8); and
(6) EDAC-crosslinked collagen scaffold delivering MSCs (n=8).
The standardized hemiresection defect was created using a modification of procedures previously reported in the literature as a dorsal 24 or lateral10,45 hemisection; however, because the term, hemisection, has other medical meanings, we adopted the description of the defect as a hemiresection.
Hindlimb motor function was evaluated between 1 and 4 weeks postinjury. These time points were chosen to illustrate the difference in mean scores over an important time period. At 1 week following injury, the animals have stabilized from the surgical procedure, and by 4 weeks postinjury differences in the recovery profiles can be seen among the groups. The reparative tissue obtained at sacrifice was histomorphometrically evaluated.
Collagen scaffold fabrication with longitudinal pore orientation
Collagen scaffolds were fabricated using type I/III porcine collagen (Geistlich Biomaterials, Wolhusen, Switzerland). Collagen suspensions containing 2% weight/volume collagen were prepared by mixing the collagen into a solution of hydrochloric acid and water, at a pH of 3.0. The suspension was blended at 14,000 RPM for 2 h, with periodic checking and adjustment of the pH to 3.0. The suspension was then centrifuged for 15 min at 2000 RPM to remove air bubbles.
Longitudinal pore orientation was achieved by injecting a collagen suspension into cylindrical channels in a polyethylene mold and freezing along a longitudinal temperature gradient. 46 A temperature gradient was created along the length of the channel by inserting a copper plug into one end of the channel and placing the copper plug in contact with the freeze dryer shelf. The temperature of the freeze dryer shelf was set at −15°C. After freezing, the collagen suspension was sublimated by creating a vacuum of 200 mTorr and raising the temperature to 0°C as described previously. 47 This removes the ice content and produces a porous, sponge-like scaffold with oriented channels. The average pore diameter was found to be 100 μm. 48
DHT and carbodiimide crosslinking of scaffolds
Porous collagen scaffolds with longitudinally oriented pores were fabricated as described above, after which two cross-linking processes, a mild physical and a chemical process, were used to increase the cross-link density of the implants. The minimal, physical cross-linking was induced by DHT treatment. This process is normally applied to collagen scaffolds to impart a sufficient amount of mechanical stability for the sponge-like materials to withstand subsequent processing, including chemical cross-linking procedures, without collapse of the pores. The more substantial control over cross-link density and the attendant mechanical and degradation properties of collagen scaffolds comes with chemical cross-linking procedures, including carbodiimide treatment. 49
DHT cross-linking was performed by placing collagen scaffolds in a vacuum oven for 24 h at 105°C and 50 mTorr as done previously. 50 The use of a high vacuum (50 mTorr) and temperature (90°C–120°C) results in severe dehydration of collagen, leading to the formation of covalent cross-links between the polypeptide chains of the collagen fibers without converting the collagen into gelatin.
The chemical cross-linking method is based on the use of a carbodiimide, EDAC, to form zero-length cross-links between the collagen fibers. Unlike bifunctional reagents like glutaraldehyde, EDAC acts solely as a catalyst (i.e., it is not incorporated into the material) in the cross-linking reaction by activating the carboxylic acid groups of glutamic or aspartic acid residues, allowing the cytotoxic carbodiimide to be completely rinsed from the matrix after the cross-linking reaction. 51 Therefore, results obtained with carbodiimide-cross-linked collagen scaffolds are not likely to be due to toxicity of the material, as may be the case with glutaraldehyde-cross-linked collagen matrices. A solution of EDAC (Cat.# E-7750; Sigma-Aldrich; St. Louis, MO) and N-hydroxysuccinimide (NHS; Cat.#H-7377 Sigma-Aldrich) was prepared at an EDAC:NHS molar ratio of 5:2. Scaffolds were incubated at room temperature for 3.5 h in the EDAC-NHS solution, and then the scaffolds were rinsed twice in sterile phosphate-buffered saline (PBS) for 1 h. The scaffolds were then stored in fresh PBS at 4°C.
Collagen membrane to cover defect
In the current in vivo studies, a collagen membrane (BioGide®; Geistlich Biomaterials) was used to cover the defect. It is a collagen type I and III membrane with a bilayer design that combines 2 distinct layers to form the membrane. One is a smooth dense layer, which is less permeable, whereas the second is a porous layer that favors tissue integration. The layers were separated using fine forceps, and only the smooth layer was used to cover the defect.
Rat bone marrow MSC harvesting and culture
Rat MSCs were harvested from the bone marrow of the femurs and tibias of 5–7-week-old male Lewis rats. Under sterile conditions, the tibias and femurs were removed from the animals, and the bones were thoroughly cleaned with a knife to remove attached muscle, periosteum, cartilage, and ligaments. Using a 20-gauge needle attached to a 5-mL syringe, a small hole was made at each end of the bones, and the bone marrow was flushed out with the culture medium, collected, and filtered with a 70-μm cell filter. The culture medium consisted of low-glucose Dulbecco's modified Eagle's medium, 20% fetal bovine serum, and 1% antibiotics. The culture medium was supplemented with 10 ng/mL of fibroblast growth factor 2 (FGF-2), which has been reported to promote increased MSC proliferation and maintain the cells in a multipotent state while enhancing subsequent lineage-specific differentiation.52–57 We have also observed that FGF-2-treated MSCs have a smaller size, as has been previously reported, 54 which allows the cells to be more readily seeded into porous scaffolds. The cells were centrifuged, rinsed with PBS, and resuspended in the culture medium. Cells from one rat were plated in one 75-cm2 cell culture flask and cultured at 37°C with 5% CO2. After 24 h, the nonadherent cells were removed (by medium change), and the attached cells were cultured until they grew to ∼75% confluence. The culture medium was changed every other day. Once confluent, the primary culture cells were washed with PBS and detached by incubation with trypsin and ethylenediaminetetraacetic acid (EDTA). Detached cells were counted and frozen for future use in aliquots of 0.5 to 0.7 mL at a cell concentration of 1 million cells per mL.
Frozen aliquots of MSCs harvested from one of the rats, whose cells were found to grow well based on our prior experience, were used for implantation into the rat spinal cord. The cells were thawed, grown to passage 2 in monolayer, and then seeded into the collagen scaffolds.
Seeding of scaffolds with rat bone marrow MSCs
Scaffolds were first prewet in a series of 100% reagent alcohol, sterile distilled water, and sterile PBS. The scaffolds were placed on a sterile filter paper to absorb excess PBS and then were submerged in 25 μL cell suspension within a microcentrifuge tube containing 300,000 cells. The scaffolds were then moved to an agarose-coated cell culture dish and allowed to sit for 10 min at 37°C before adding the culture medium. The objective of this procedure, based on our prior work seeding MSCs into collagen scaffolds, was to incorporate as many MSCs as possible into the scaffold.
Incorporation of sNgR into collagen scaffolds
Prewet carbodiimide-cross-linked scaffolds were briefly dried on filter paper, and then 25 μL of a 9.8 mg/mL stock solution of sNgR (Biogen Idec, Cambridge, MA) was added to each scaffold. The scaffolds were then freeze-dried, and again 25 μL of a 9.8 mg/mL solution of sNgR was added to each scaffold. The concentration of sNgR in the stock solution was as high as possible without jeopardizing the degradation of the protein. The objective was to load the scaffold with as much of the protein as possible, a part of which was immobilized by the freeze-drying process, and a freely releasable amount added to rehydrate the samples.
Incorporation of ChABC into collagen scaffolds
Prewet carbodiimide-cross-linked scaffolds were briefly dried on filter paper, and then 25 μL of a 10-Units/mL ChABC (#C3667; Sigma-Aldrich) solution was added to each scaffold just before implantation. This dose was based in part on a previous study in which six 6-μL aliquots of 10 U/mL ChABC were injected into lesions in rats over a 10-day period. 21
Surgical procedure and animal care
Adult female Lewis rats (Taconic, Germantown, NY; Charles River Laboratories, Wilmington, MA) weighing 150–175 grams were used in this study. All animal care and surgical procedures were performed at the Veterans Affairs medical center Animal Research Facility (Boston, MA). All surgical and postoperative procedures were approved by the VA Boston Healthcare System Institutional Animal Care and Use Committee (IACUC).
Rats were anesthetized by an intraperitoneal injection with sodium pentobarbital (Nembutal solution, 50 mg/mL; Abbott Laboratories, North Chicago, IL) at a dosage of 45 mg/kg. The hair on the back of the anesthetized rat was shaved, and the skin was cleaned with Betadine. Surgery was performed under sterile conditions. A continuous oxygen supply was supplied to the rat during surgery. The rat was placed in a prone position on a flat operating board, on top of a water-circulating heating pad, and the rat's limbs were gently constrained with rubber bands in an extended position. A longitudinal incision, 2 inches in length, was made through the skin above the thoracic spine. The back musculature was incised along the midline and dissected away from the vertebral column. A dorsal laminectomy was performed at T8 and T9 using small bone rongeurs and microscissors. To provide enough space to create the 3-mm-long defect, two vertebrae were removed as has been done by others.10,58–60 The dura was opened with a surgical blade to expose about 7 mm of spinal cord, and the dorsal spinal artery was coagulated with a bipolar cautery. A small plastic template was placed on the exposed spinal cord to outline the area for incision. A lateral hemiresection of the spinal cord was created by making two lateral cuts (3-mm apart) on the left side of the spinal cord with an ultrafine surgical blade using the template as a guide. The template was removed once the cuts were made, and a midline incision was made between the two lateral cuts. A 3-mm-lateral gap was created by removing the tissue between the two lateral cuts with a forceps, followed by aspiration using a low-power surgical aspirator with a 16-gauge tip to remove blood and any remaining fragments of spinal cord tissue within the gap. Gelfoam was then temporarily placed in the gap to induce hemostasis. Once hemostasis was achieved, the defect was carefully examined with fine needle-tipped forceps to ensure that a complete hemiresection of the spinal cord was created. In the treatment groups, semicylindrical scaffolds (alone or as a delivery vehicle for therapeutic agents or cells) were placed within the defect, and a collagen membrane was placed extradurally over the dorsal aspect of the wound site and extended 2 mm past the spinal cord stumps on both sides of the defect to serve as a dorsal barrier. Following placement of the respective implants, the overlying musculature was closed with 4–0 vicryl sutures (Johnson and Johnson, Somerville, NJ), and the skin was closed with wound clips.
Immediate postoperative care of the animals included placement of the rat on a heating pad to maintain body temperature, subcutaneous injection of 1–2 mL of lactated Ringer's solution to compensate for blood loss during surgery, subcutaneous injection of antibiotics (cefazolin sodium 100 mg, Abbot Laboratories, North Chicago, IL), and a continuous supply of oxygen until the animal regained consciousness 4–6 h later. After regaining consciousness, the rats were transferred to plastic cages with fresh wood chip bedding and free access to food and water. An analgesic, ketofen (dose 5 mg/kg), was administered once a day for 4 days after surgery by subcutaneous injection. Subcutaneous injection of lactated Ringer's solution (2 mL per day) was continued for 4 days after surgery to prevent dehydration. Administration of antibiotics (cefazolin sodium) was continued for 1 week, to prevent bladder infection, at a dosage of 35 mg/kg twice a day. Postoperatively, animals lacked normal micturition reflex, and their bladders had to be emptied manually every 12 h using Crede's maneuver until the bladder function was recovered.
Hindlimb functional evaluation
Rats were removed from their cages and videotaped walking in an open space weekly until sacrifice. Hindlimb motor function was evaluated using a 0–5 motor scoring system (Table 1) similar to one previously described for rats by Wrathall, et al., 61 which itself was a modification of the 0–4 Tarlov scale, one of the first motor function grading systems—employed for a study of contusive SCI in dogs. 62 Scores from 0 to 5 were given by an evaluator blinded to the treatment group assignment. The scoring system used for the present study is shown in Table 1 along with the Wrathall motor score for comparison. This simplified hindlimb function scoring system was used instead of the more-extensive Basso-Beattie-Bresnahan (BBB) 63 scoring system because of the desire to primarily detect major functional differences among groups.
Transcardial perfusion and tissue removal at 4 weeks
At 4 weeks postinjury, the rats were given a dose of 150 mg/kg of sodium pentobarbital administered intraperitoneally, and secured to a surgical board. A thoracotomy was performed to expose the heart, after which a needle attached to a peristaltic pump was inserted into the left ventricle. Heparinized saline was circulated through the animal, followed by 4% paraformaldehyde. Following transcardial perfusion, the thoracic spinal column was removed using a scalpel and surgical scissors. The extracted spine was placed in 4% paraformaldehyde at 4°C for a minimum of 72 h. The extracted spine was trimmed using bone rongeurs and surgical scissors; the spinal column surrounding the defect area was left intact, and the tissue specimens were then placed in a solution of EDTA (Cat.#S311-10; Fisher Scientific, Waltham, MA) for 10 to 14 days to decalcify the bone surrounding the defect. The EDTA solution was changed every other day.
Histology, immunohistochemistry, and histomorphometry
The entire decalcified spines were embedded in paraffin using a tissue processor (HypercenterXP, Tissue Processor; Thermo Shandon, Houston, TX). Serial longitudinal sections of the defect were cut at 6 μm with a microtome, and mounted on glass slides (Cat.#15-188-48; Fisher Scientific).
Adjacent tissue sections were stained with hematoxylin and eosin for general observation of cellular and ECM features and Masson's trichrome, which was used to identify the presence of collagenous tissue within the defect.
Antibodies to the proteins listed in Table 2 were used in the immunohistochemical staining of paraffin-embedded tissue sections containing the defect area.
The numbers of cells within the defect area staining for von Willebrand Factor (vWF) and for CD68 were counted, and the results expressed as an areal density by dividing by the area. For the vWF staining, equal weight was given to vessels and individual cells. This was done because the vessels were nearly always small with only 1 or 2 endothelial cells lining the vessel in a cross-section. Also, the proportion of individual cells to identifiable vessels was similar among the groups.
Statistics
Numerical values are reported as mean±standard error. Statview (version 5.0.01; SAS Institute Inc., Cary, NC) software was used for statistical analysis. Fisher's exact test was employed to determine if there were significant differences among groups with respect to the percentage of animals recovering their bladder function. One-way analysis of variance (ANOVA) was employed to determine the significance of the effect of group on select outcome measures. Fisher's protected least squares difference (PLSD) post hoc testing was then performed to compare specific groups. The paired-sample sign test was used to determine the significance of improvement in the hindlimb function of the animals in the respective groups from 1 to 4 weeks post-SCI. The Kruskal–Wallis test was employed to reveal statistically significant differences among groups for the change in hindlimb score from 1 to 4 weeks. The criterion for significance was set at p≤0.05.
Results
Return of bladder function
All of the animals lost their ability to empty their bladders immediately after SCI. The number of animals per group that recovered bladder function is shown in Figure 1. The percentage of animals in each of the groups recovering their bladder function before sacrifice at 4 weeks ranged from 55% to 85% of the animals. Fisher's exact test showed no significant difference among groups. The time to bladder function return ranged from just a few days in some animals to up to 2 weeks in other animals (Fig. 2). However, ANOVA showed no significant effect of treatment on the time to bladder function return (p=0.37; power=0.33).
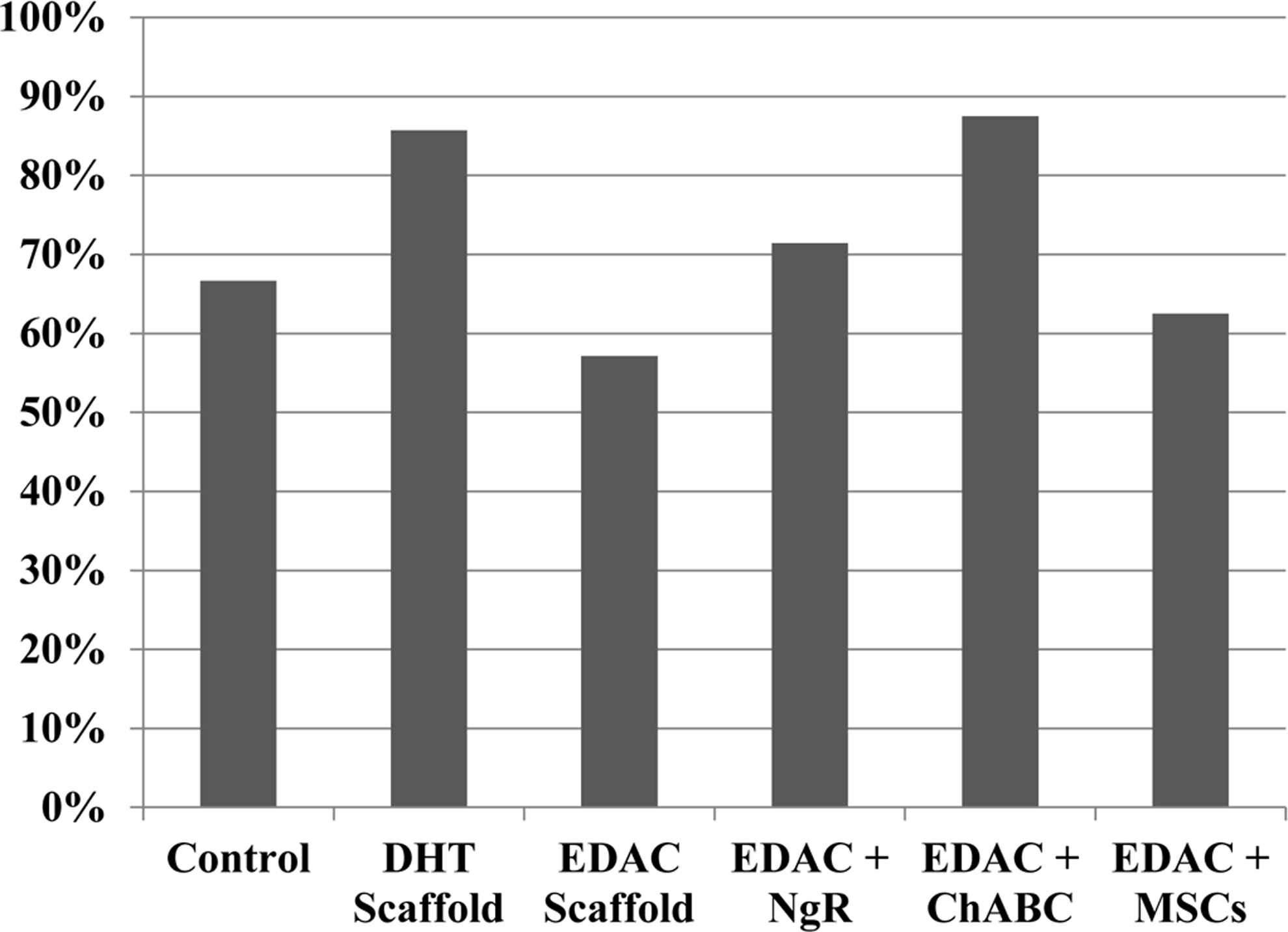
Percentage of animals that recovered bladder function before sacrifice at 4 weeks. DHT, dehydrothermal; EDAC, 1-ethyl-3-(3-dimethylaminopropyl) carbodimide; ChABC, chrondroitinase, ABC; NgR, nogo receptor; MSC, mesenchymal stem cells.

Time to bladder function return. Mean±SEM. Statistical analysis was performed using one-way analysis of variance (ANOVA) (p=0.37; power=0.33).
Hindlimb function
Immediately after the hemiresection, all animals lost control of both hindlimbs. Statistical analysis using the paired-sample sign test showed a significant improvement in left and right hindlimb function from week 1 to week 4 after SCI in the following groups (Table 3 and Figs. 3 and 4): DHT (p=0.02 left and right); EDAC+ChABC (p=0.04 left, p=0.02 right); and EDAC+MSCs (p=0.02 left, p=0.04 right). Statistically significant improvement was not seen in the EDAC scaffold-alone group (p=0.13 left, p=0.13 right), EDAC+NgR group (p=0.07 left, p=0.13 right), or control group (p=0.48 left and right; Table 3). Kruskal–Wallis ANOVA revealed no significant difference among groups for hindlimb function when comparing the mean scores of all the groups at a single time point (at 1 week following injury, p=0.44 for left hindlimb and p=0.75 for right hindlimb, and at 4 weeks, p=0.57 for left hindlimb and p=0.56 for right hindlimb). As expected, there was a tendency for the right (spared side) hindlimb functional score of each animal to be higher than the left (lesion side); however, statistical analysis using the paired-sample sign test showed no significant difference between left and right hindlimb function for any of the groups.
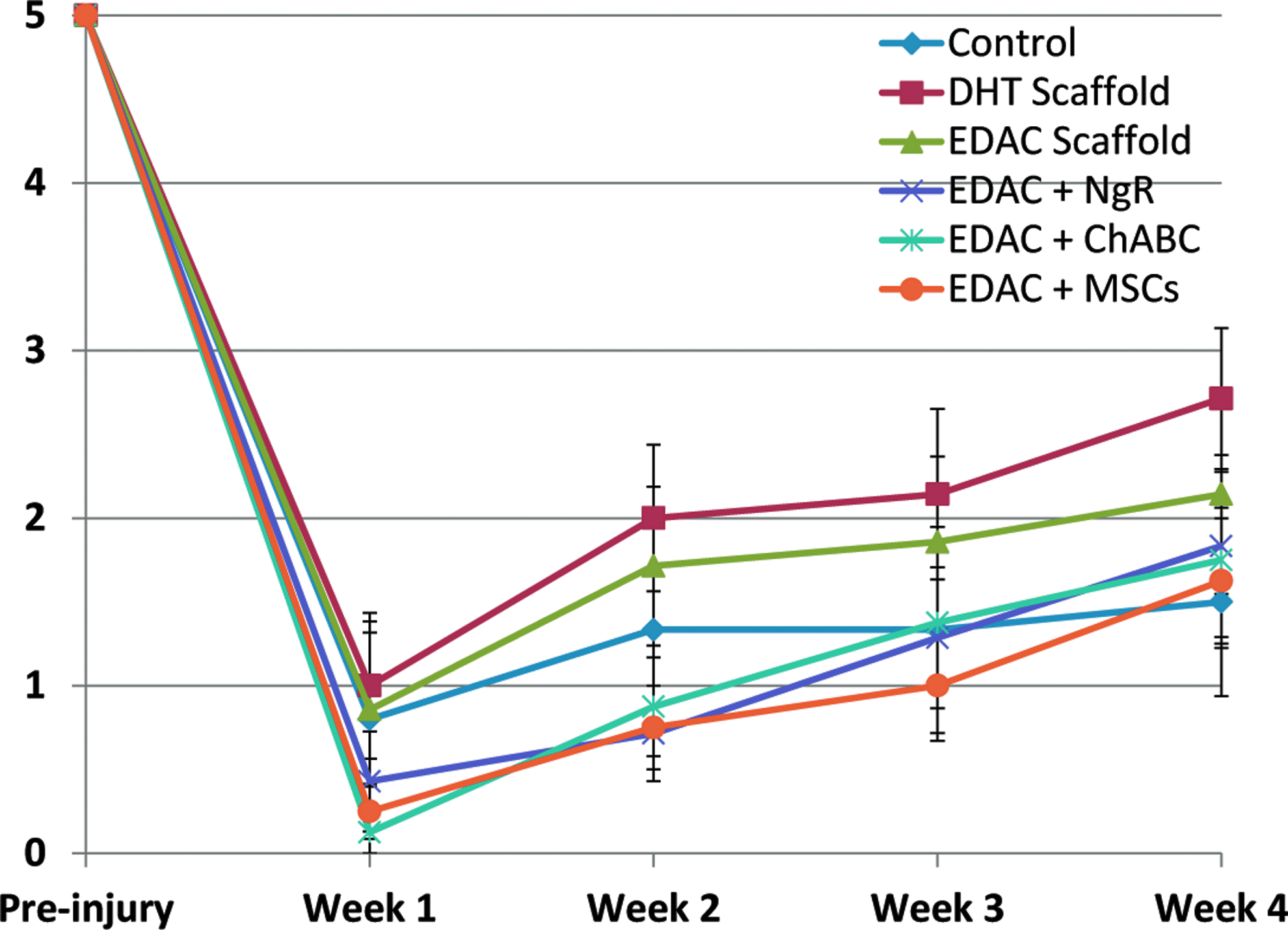
Left hindlimb function; lesion side. Mean±SEM. Grades on the ordinate are described in the text. Color images available online at www.liebertpub.com/tea
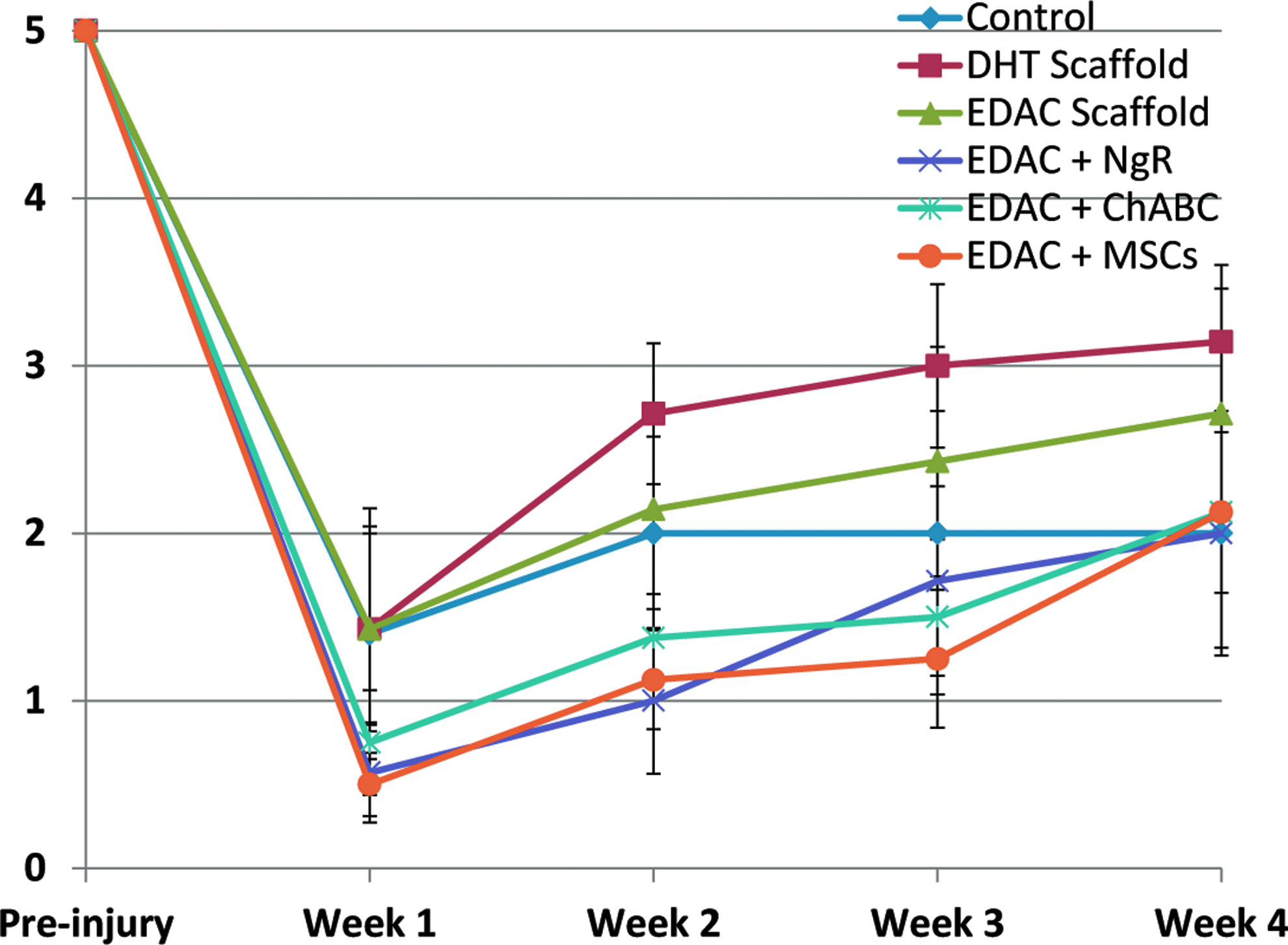
Right hindlimb function; spared side. Grades on the ordinate are described in the text. Mean±SEM. Color images available online at www.liebertpub.com/tea
DHT, Dehydrothermal; EDAC, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; NgR, Nogo receptor; ChABC, chondroitinase ABC; MSCs, mesenchymal stem cells.
Axon regeneration
Axons in and around the lesion site were identified by neurofilament staining at 4 weeks postinjury (Fig. 5). A band of spared tissue on the contralateral side of the defect was visible in all of the groups, whereas a few regenerating axons could be seen bordering the rostral and caudal ends of the defect. To help identify axons in the process of regeneration, growth-associated protein 43 (GAP-43; Fig. 6) was immunostained. Qualitative assessment of the staining revealed GAP-43-positive axons at the rostral and caudal boundaries of the defect in animals from all groups, including some animals in the control group (GAP-43 staining of spinal cords from uninjured rats was negative–not shown). In some animals, GAP-43-positive axons were seen entering the periphery of the scaffolds in the DHT, EDAC+ChABC, and EDAC+MSC groups. Of note was that in some cases, a few GAP-43-positive axons were seen reaching the center of the scaffold in the DHT and EDAC+ChABC groups, but not in control animals (Fig. 6).
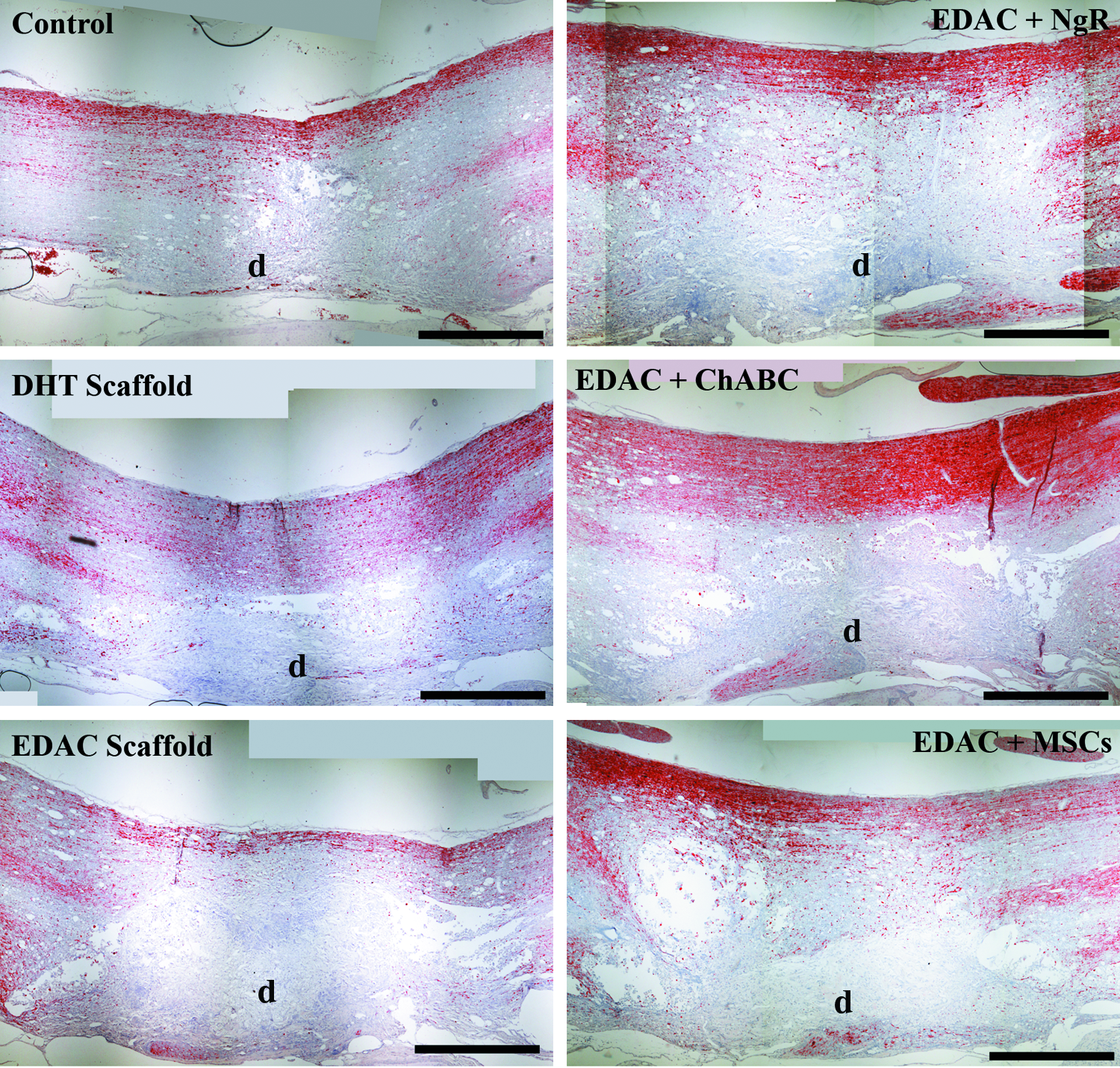
Neurofilament staining. Positive stain, red chromogen. d, defect. Scale bar 1 mm. Color images available online at www.liebertpub.com/tea
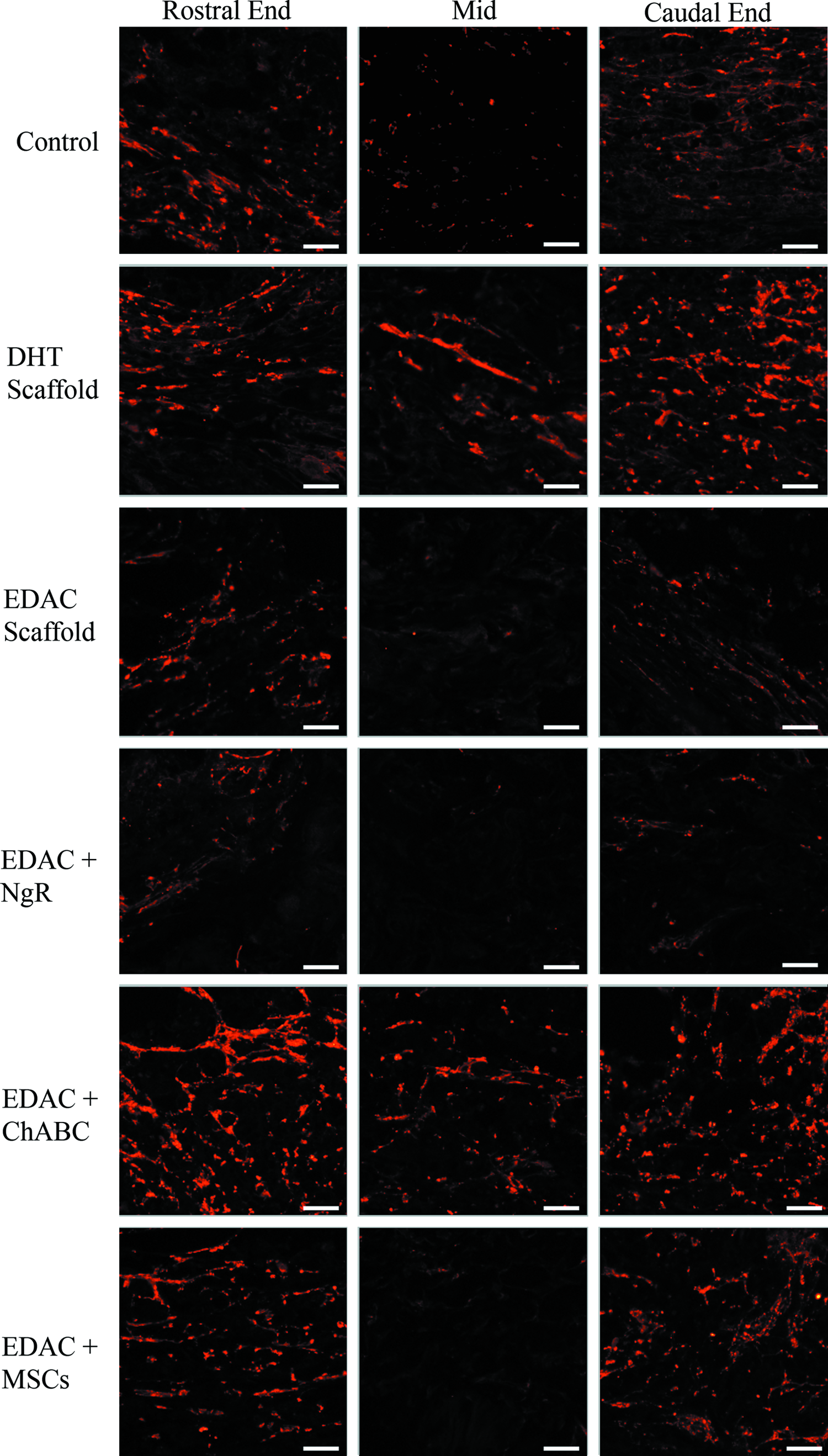
Growth-associated protein 43 (GAP-43) staining (red fluorescence). Representative images of the middle and the boundaries (rostral and caudal ends) of the defect area. Scale bar 50 μm. Color images available online at www.liebertpub.com/tea
Angiogenesis
Staining for vWF revealed new blood vessel formation in the defects of all groups 4 weeks after injury (Fig. 7). The implanted scaffolds were permissive to the ingrowth of vasculature; however, the control group had considerably more (∼2-fold higher vessel density) blood vessels and vWF-positive cells in the defect (Fig. 8). The areal density of blood vessels in the scaffold-implanted defects was similar to the vessel density determined for normal rat spinal cord tissue. One-way ANOVA showed that there was a significant effect of treatment on the areal density of blood vessels and vWF-positive cells within the defect (p=0.003, power=0.95). Analysis between individual groups using Fisher's PLSD revealed significantly more blood vessels in the defect of the control group compared to all of the treatment groups, but showed no significant differences between treatment groups.
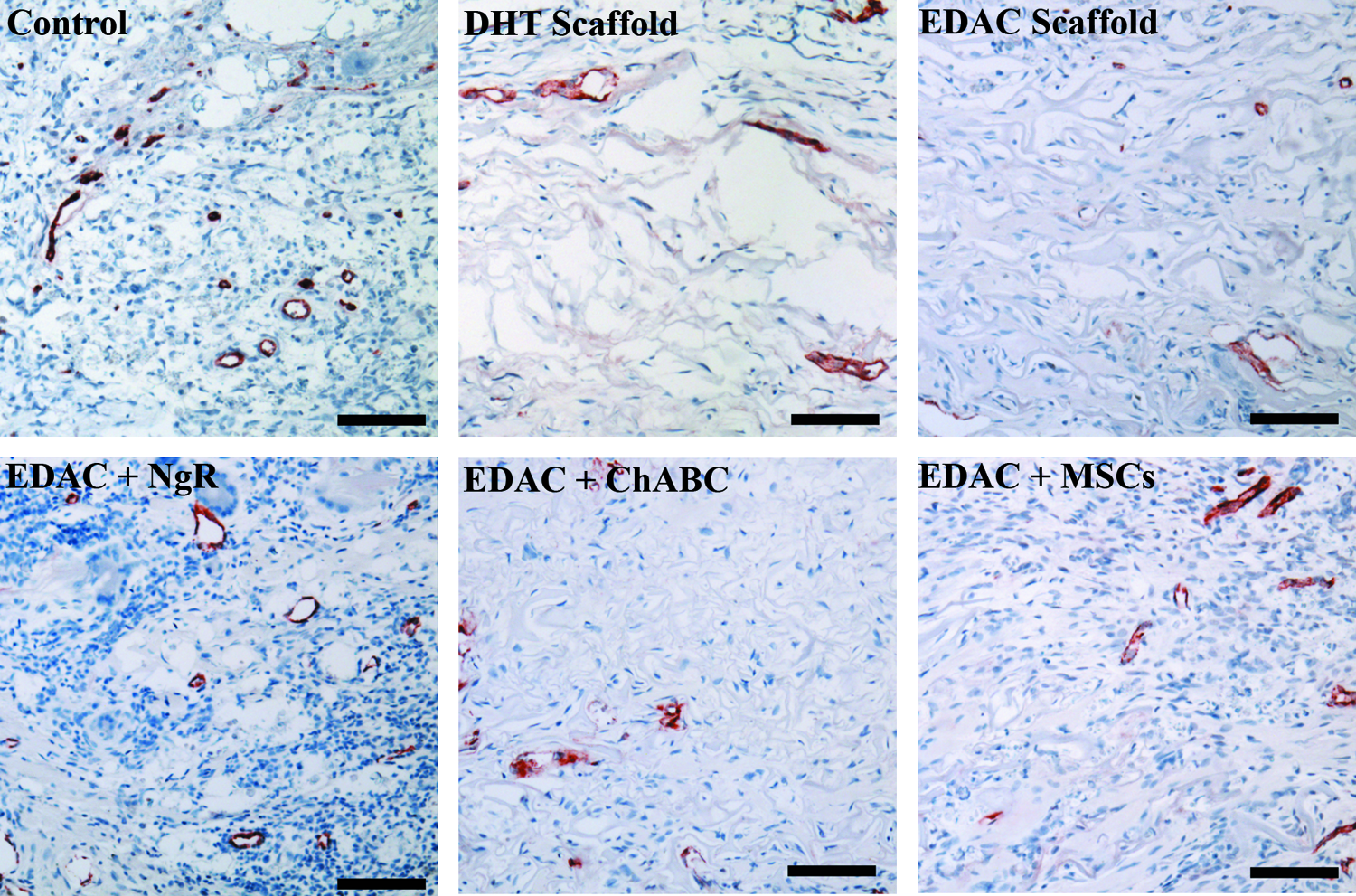
von Willebrand factor staining. Positive stain red. Scale bar 100 μm. Color images available online at www.liebertpub.com/tea
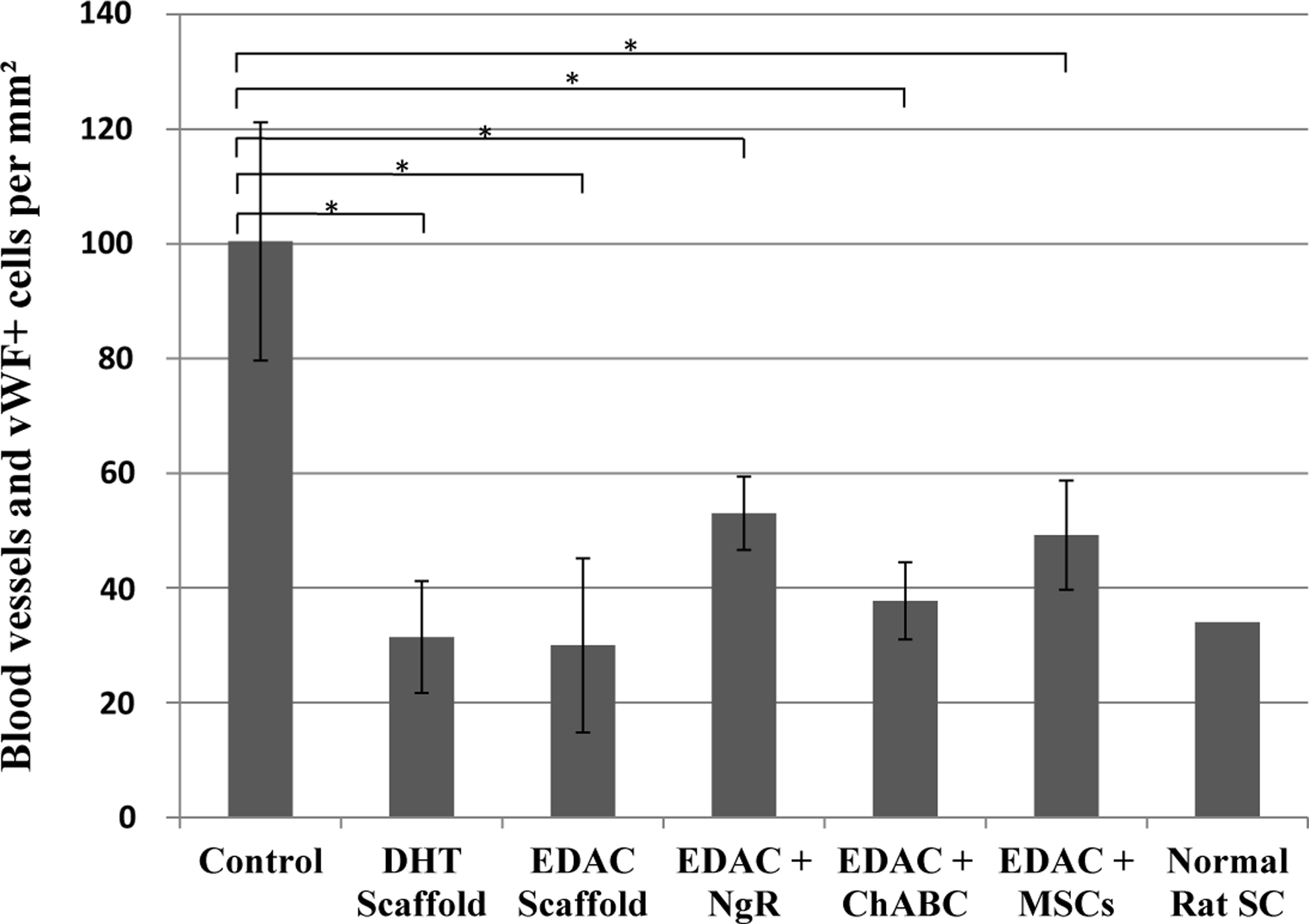
Number of formed blood vessels and individual von Willebrand Factor (vWF)-positive cells in the defect. Mean±SEM. (*p<0.01, Fisher's protected least squares difference [PLSD]).
Inflammatory response
Staining for CD68, a marker for macrophages, revealed extensive presence of macrophages within the defect in the control group, and immediately surrounding the defect of all groups (Fig. 9). Considerable numbers of CD68-positive cells were also seen within the undamaged cord up to several millimeters away from the defect in both the rostral and caudal directions of all groups. Areas of necrotic debris had an especially high number of macrophages present. The number of CD68-positive cells was quantified at the center of the defect (corresponding to the center of the scaffold for the treatment groups) as shown in Figure 10. Many more macrophages were found in the defect center of the control group than any of the other groups (Fig. 11), and one-way ANOVA showed a statistically significant effect of treatment on the areal density of macrophages (p<0.001, power=1). Comparison between individual groups using Fisher's PLSD post hoc testing revealed a significant difference between the control group and each of the treatment groups (p<0.0001). There was also a significant difference between the EDAC and EDAC+NgR groups (p=0.01) as well as between the EDAC+NgR and EDAC+MSC groups (p=0.03). The difference between the DHT scaffold and EDAC scaffold-alone groups was not significant (p=0.19) nor was the difference between the EDAC and EDAC+ChABC groups (p=0.07).

CD68 staining. Positive stain red. d, defect. Scale bar 1 mm. Color images available online at www.liebertpub.com/tea

CD68 staining at the defect center. Positive stain red. Scale bar 100 μm. Color images available online at www.liebertpub.com/tea

Number of CD68-positive cells in the defect. Mean±SEM
Fibrous tissue
The tissue filling the defects of all groups at 4 weeks postinjury contained a large amount of collagen as was seen by staining with Masson's trichrome (Fig. 12). Closer examination of the tissue in the defect showed a loose organization of fibrous tissue in all of the groups (Fig. 13) with many fibroblast-like cells within the tissue. In the treatment groups, much of the implanted scaffold was present.
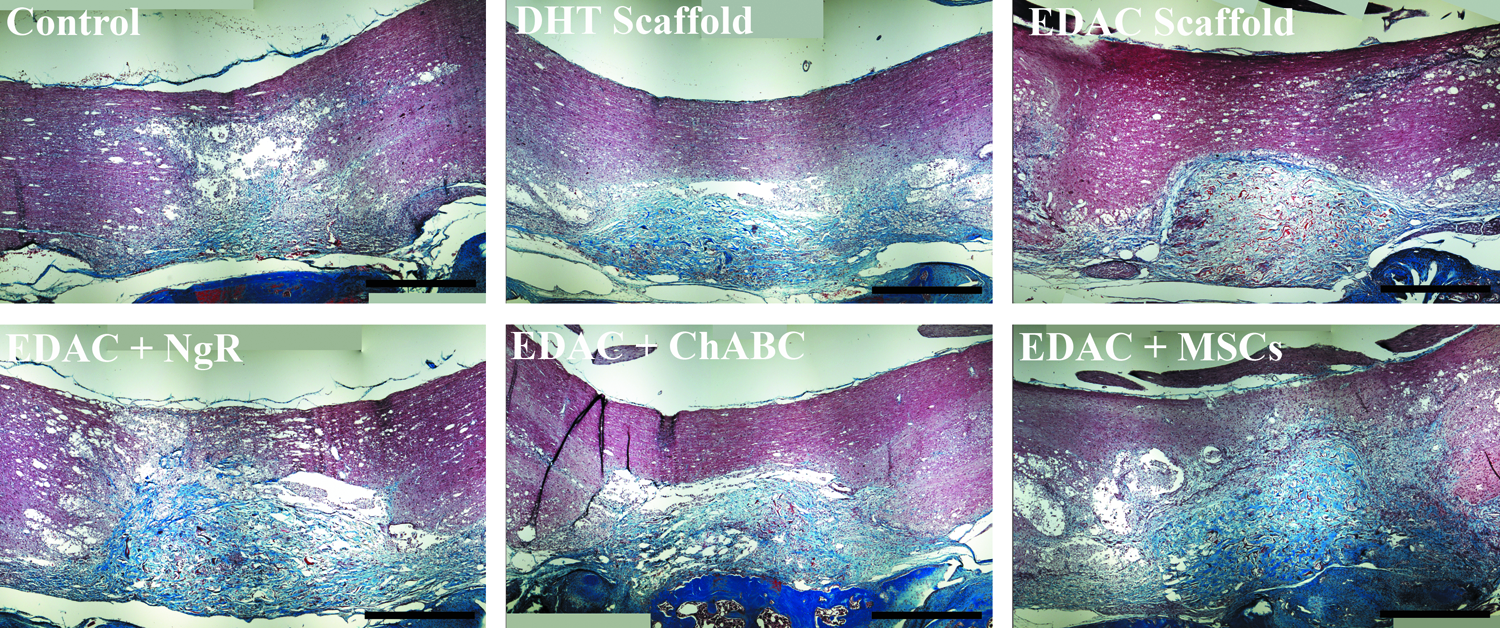
Masson's trichrome staining. Collagen stained blue/green and red. Scale bar 1 mm. Color images available online at www.liebertpub.com/tea
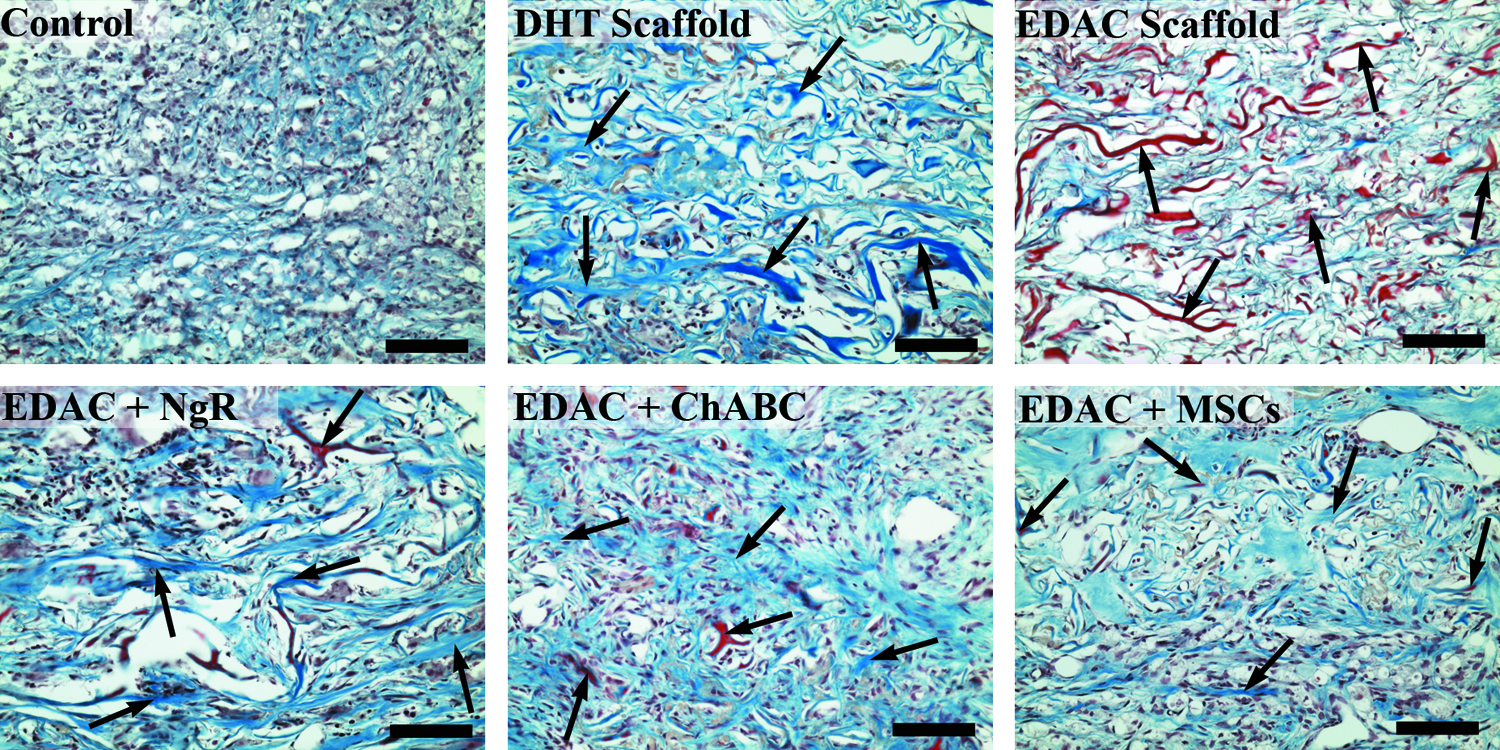
Masson's trichrome staining at the defect center. Collagen stained blue/green and red. Arrows indicate scaffold struts. Scale bar 100 μm. Color images available online at www.liebertpub.com/tea
Astrocyte response
The astrocyte response to injury was assessed by glial fibrillary acidic protein (GFAP) staining at 4 weeks following injury. There were few-to-no GFAP-positive astrocytes seen within the defect areas of any group (Fig. 14). Astrocytes were seen immediately bordering the lesion, and the intensity of the GFAP staining diminished gradually at a distance from the injury site.

GFAP staining. Positive stain red. Defect area outlined by black line. Scale bar 1 mm. GFAP, glial fibrillary acidic protein. Color images available online at www.liebertpub.com/tea
α-smooth muscle actin
A few tissue sections from the control and EDAC scaffold groups were stained for α-smooth muscle actin (α-SMA) to identify cells within and around the defect expressing the myofibroblast phenotype. Although many of the cells in the defect appeared to be fibroblasts, there were no fibroblast-like cells that stained positive for α-SMA within the tissue sections stained at 4 weeks postinjury. The only cells that were seen to stain positive for α-SMA in and around the defect were smooth muscle cells and pericytes lining the blood vessels (Fig. 15). An interesting, but unexplained, finding was the positive α-SMA staining of cells with motor neuron morphology in the gray matter a few millimeters away from the defect (not shown). Positive α-SMA staining was not seen in motor neurons found in sections of uninjured rat spinal cord.
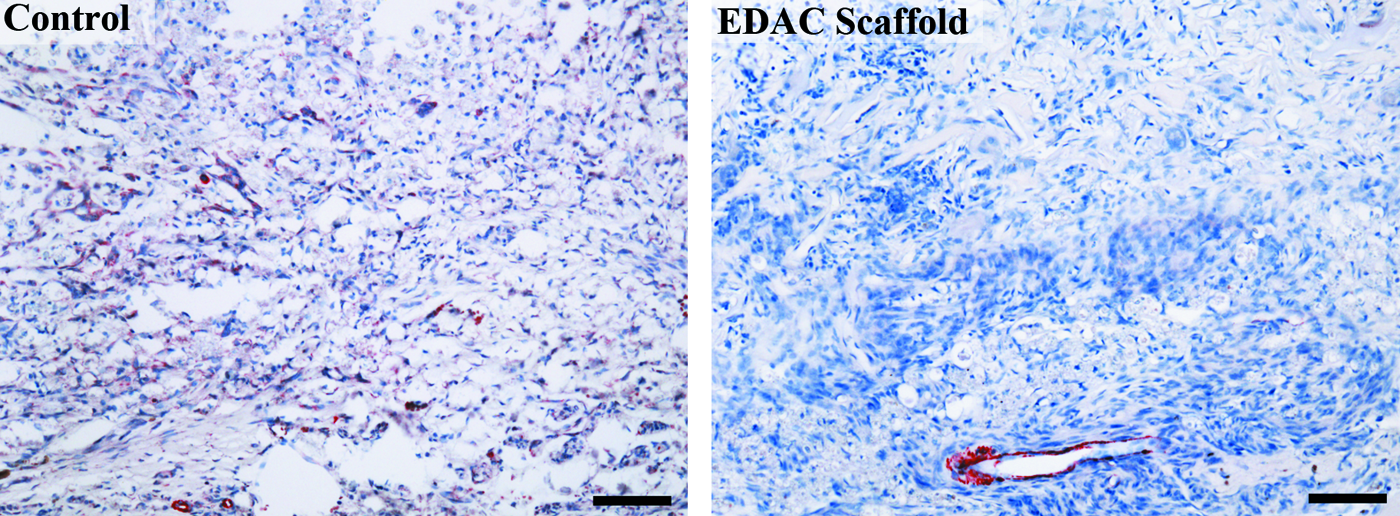
α-smooth muscle actin staining within the defect. Positive stain red. Scale bar 100 μm. Color images available online at www.liebertpub.com/tea
Discussion
This investigation provided an initial qualitative and quantitative comparison of the effects of several collagen scaffold-based implants on select features of the reparative response in a hemiresection in the rat spinal cord. After 4 weeks, the scaffold material was present in the treatment groups, indicating that at the site of implantation, the collagen scaffolds had not yet fully degraded. Notable findings were that host cells were able to migrate across the implant–tissue interface to gain access to the pores of the scaffold, and that the pores of the scaffold remained open and interconnecting through 4 weeks, postimplantation.
As regard to differences in the behavioral and histopathological findings among the treatment groups, there were several findings that are of importance in informing future investigations. One of the interesting observations of the current study was the significant improvement in the hindlimb function from 1 to 4 weeks postinjury in three of the treatment groups, which was not seen in the control group. A significant difference in hindlimb function was not seen when solely comparing the groups at a single time point; however, we found that there was a significant difference compared to control when considering the change of hindlimb function with time within each group for each animal individually, in a paired comparison analysis (which is represented by the slopes of the lines in Figs. 3 and 4). The three groups that showed statistically significant improvement of hindlimb function from 1 to 4 weeks postinjury were the DHT scaffold-only group, the EDAC+ChABC group, and the EDAC+MSC group. Interestingly, the EDAC-crosslinked scaffold-alone group did not show a statistically significant improvement in the hindlimb function; however, when used in combination with ChABC or to deliver MSCs, a functional benefit was seen. In future work, it will be interesting to compare the functional improvement in the treatment groups to the control over a longer time period to see if there is continued improvement in hindlimb function with increased time. Of note in this regard, however, were the prior findings 10 using a similar SCI defect (comparable location, but 4 mm instead of 3 mm in length in Sprague-Dawley rats) that demonstrated that most of the motor recovery was achieved at 4 weeks post-SCI in both untreated control groups and in groups implanted with a 50:50 poly(lactic-co-glycolic acid) (PLGA) scaffold seeded with murine neural stem cells (NSCs). In that study, the controls received a BBB score of about 5 at 4 weeks, compared to the BBB score of 10 for the treated group. While a direct comparison of those motor function findings with the present results is not possible because of the different scoring systems, the general trends in the recovery of function in the control and treatment groups are similar. The prior study also found no statistical difference in the motor function recovery between a scaffold-alone group and the group made up of animals that received NSC-seeded implants. In the current study, also, 2 groups of non-cell-seeded scaffolds yielded results comparable with an MSC-seeded scaffold group.
In the present investigation, we found GAP-43-positive axons entering the periphery of the implanted scaffold in some animals from three of the treatment groups: the DHT scaffold, EDAC+ChABC, and EDAC+MSC groups. Furthermore, a few GAP-43-positive regenerating axons were able to reach the center of the scaffold in the DHT scaffold and EDAC+ChABC groups. Interestingly, GAP-43-positive axons were seen at the rostral and caudal boundaries of the defect in animals from all groups, including some animals in the control group (staining of spinal cords from uninjured rats was negative). The positive GAP-43 staining in some control animals implies a continued spontaneous regenerative response at 4 weeks post-SCI in untreated Lewis rats. However, axons did not penetrate into the defect of the control animals. Despite seeing only a small number of axons entering the scaffold in the DHT scaffold, EDAC+ChABC, and EDAC+MSC groups, it is encouraging to find, by 4 weeks, the possibility of at least some axons to grow within the collagen scaffolds. Prior work with a similar hemiresection model 10 reported GAP-43 staining just rostral to the defect implanted with an NSC-seeded PLGA scaffold, 70 days post-SCI, but no staining within the defect and very few GAP-43-positive fibers rostral to the nontreated control site.
Blood vessel formation is an integral part of the body's healing response and an important factor to consider when employing tissue-engineering strategies for tissue regeneration. Similar to the wound-healing response seen in most tissues of the body, previous studies have shown a rapid increase in the number of new blood vessels after contusion SCI, with angiogenesis peaking at 7 days followed by a subsequent decrease in the vessel number by 14 days postinjury.64,65 Of interest is that the angiogenic response following SCI closely correlates with the dramatic increase of macrophages within the lesion site, 64 which are known to stimulate angiogenesis. 66 Bartholdi, et al. found that the expression by macrophages of the potent angiogenic inducer, vascular endothelial growth factor, corresponded temporally and spatially with the neovascularization of the lesion site after SCI. 67 In the current study, we found that the implanted collagen scaffolds (with or without NgR, ChABC, or MSCs) were permissive to the ingrowth of vasculature; however, there was a greater number of blood vessels in the lesion of the untreated control at 4 weeks postinjury compared to the scaffold-implanted groups. There were also significantly more macrophages present within the lesion of the control group compared to the treatment groups at 4 weeks. It is therefore possible that the persistent increase of blood vessels in the lesion of the control at 4 weeks is due to stimulation by macrophages—suggesting a continued inflammatory response, whereas when a scaffold is present, the inflammatory and remodeling processes within the lesion may resolve more quickly.
Macrophages play a complex role in the healing response of the injured spinal cord. They are key players in the clearing of necrotic debris at the site of injury and also as a part of the Wallerian degeneration, which occurs in the distal ends of severed axons. 68 Furthermore, they have been implicated in both tissue destructive (primarily through the release of inflammatory cytokines and reactive oxygen and nitrogen intermediates) and tissue regenerative/protective (by the release of growth-promoting and neuroprotective factors) processes after SCI.69–72 A striking demonstration of the seemingly dual role of macrophages after SCI is a study by Kostyk, et al. 73 that showed a paradoxical association between a marked increase in axon regeneration and an impaired functional recovery in MRL/MpJ mice, which have been shown to have scarless regeneration of ear-puncture injury and a substantially reduced macrophage response after SCI. Interestingly, in the newt, axon regeneration occurs in the presence of a significant inflammatory response that includes macrophage and lymphocyte infiltration; however, this response does not seem to be detrimental to axonal growth. 74 Still, others have shown the benefits of macrophage activation on axon regeneration in the mouse optic nerve 75 and the rat spinal cord. 76 The dichotomy between the protective and destructive effects of the macrophage response is likely the result of distinctive macrophage phenotypes resulting in a dramatically different behavior. 76
In the present study, at 4 weeks, we found a robust macrophage response in the tissue immediately surrounding the defect area as well as in the white matter tracts distant from the lesion site in all of the experimental groups. However, only a few macrophages were seen in the scaffolds of the treatment groups, whereas in the control group, there were a large number of macrophages filling the defect area. This suggests the continued presence of necrotic debris in the defect of the control group, to which macrophages are responding, and the absence of substantial stimulation of macrophages to migrate into the scaffold, at 4 weeks postinjury. Interestingly, the addition of the sNgR to the EDAC-treated scaffold resulted in a significant increase in the number of macrophages within the scaffold at 4 weeks, suggesting an inflammatory response to the presence of the NgR.
Astrocytes have also been shown to have both beneficial and detrimental effects on the healing response in the injured spinal cord. 77 In the acute stage of injury, astrocytes are activated, become hypertrophied, proliferate, and migrate to the site of injury. This response serves to isolate and contain the injury site, perhaps in an attempt to prevent further damage to the surrounding tissue.78,79 Astrocytes also respond to injury by synthesizing a scar tissue that is rich in CSPGs, which are inhibitory to axonal growth.80,81 Thus, at early time points after injury, astrocytes appear to have a protective effect; however, this benefit comes at the expense of impaired axon regeneration once a glial scar has been formed. In our present work, we observed a modest increase in the number of astrocytes present at the boundary of the defect area in all groups, with only a few entering into the implanted scaffolds. This seems to suggest the ability of the astrocytes to enter the scaffolds, but an absence of signaling to stimulate any considerable astrocyte migration within the scaffolds at 4 weeks.
Also of note in the implant groups and nonimplanted controls was the absence of contracture of the newly synthesized fibrocollagenous matrix by α-SMA-expressing myofibroblasts, as is a common feature in the reparative response in many other types of tissues. Few α-SMA-containing nonvascular cells were seen in any of the groups. There is the possibility that the transient expression of this contractile actin isoform 82 occurred at an earlier time point, and by 4 weeks, was so diminished as to be unnoticeable. However, that the fibrocollagenous bundles were not found densely packed and uniaxially aligned with a detectable crimp, as is often the result of contracture, raises doubt that in this particular hemiresection, SCI tissue contraction plays a meaningful role in the healing process.
The results of this study have provided the foundation to further investigate collagen scaffolds alone and as delivery vehicles for select therapeutic agents for the treatment of SCI. Of importance will be dose–response studies for each of the agents and combinatorial approaches in the same standardized SCI model.
Conclusion
This study showed the potential of a collagen scaffold alone and as a delivery vehicle of select therapeutic agents to modulate the healing and potentially enhance the regenerative response in SCI using a hemiresection rat model.
Footnotes
Acknowledgments
The research reported here was supported by the U.S. Department of Veterans Affairs, Veterans Health Administration, Rehabilitation Research and Development Service, and the Department of Defense. MS was supported by a VA Research Career Scientist Award, and RC received a fellowship from the Gates Millennium Scholars Program. We thank Dr. Daniel Lee and Dr. Paul Weinreb at Biogen Idec for generously providing the soluble Nogo receptor. The authors are grateful to Larry Benowitz, Ph.D., for supplying the GAP-43 antibody.
Disclosure Statement
No competing financial interests exist.