
Abstract
In tissue engineering, stem cells have become an ideal cell source that can differentiate into most human cell types. Among the stem cells, bone marrow-derived stem cells (BMSCs) have been widely studied, and there is strong evidence that these cells can be differentiated into cells of the osteogenic lineage. Thus, BMSCs have become the gold standard for studies of tissue engineering in orthopedics. However, novel stem cell sources, such as amniotic fluid-derived stem cells (AFSCs) have been identified, and these have important and unique features that may lead to novel and successful applications toward the regeneration of bone tissue. This study was designed to originally compare the osteogenic potential of both BMSCs and AFSCs under distinct culture environments to determine whether the osteogenic differentiation process of both types of stem cells is related to the origin of the cells. Osteogenic differentiation was carried out in both two and three dimensions using a tissue culture plate and by means of seeding the cells onto microfibrous starch and poly(ɛ-caprolactone) scaffolds (a blend of starch and polycaprolactone), respectively. BMSCs and AFSCs were successfully differentiated into the osteogenic cell type, as cells derived from them produced a mineralized extracellular matrix. Nevertheless, the two types of cells presented different expression patterns of bone-related markers as well as different timing of differentiation, indicating that both cell origin and the culture environment have a significant impact on the differentiation into the osteogenic phenotype in AFSCs and BMSCs.
Introduction
Bone marrow-derived stem cells (BMSCs) are perhaps the most studied adult stem cell type. BMSCs are multipotent and can differentiate into many different lineages, including bone, as confirmed by several research groups.5,7–9 However, the process required to harvest BMSCs from a patient is an invasive and rather painful procedure. Furthermore, the number, proliferative capacity, and differentiation potential of BMSCs decline with age,10,11 suggesting that tissue-engineering strategies based on these cells might not be feasible in older patients. Some studies suggest that BMSCs are also sensitive to subculturing methodologies, and their stability in long-term in vitro culture is controversial.12,13 Nevertheless, BMSCs represent a promising cell source for cell-based therapeutics with high osteogenic potential in various tissue-engineered applications.14,15 Moreover, several studies have described that BMSCs show increased calcium mineralization levels when compared to other stem cell sources, such as adipose-derived stem cells. 16 BMSCs have also shown great promise in animal studies and even a few clinical trials.1,17–19
Amniotic fluid-derived stem cells (AFSCs) were described in 2007, 2 and while their behavior and characteristics have not been investigated in as much detail as BMSCs, it has been shown that AFSCs have a high proliferative capacity, 12 and that they can differentiate into cells from all the three embryonic germ layers.1,2 Differentiation into several specific cell lineages, including an osteogenic lineage, has been reported.2,6,20,21 This indicates that AFSCs may be useful for the development of novel bone tissue-engineering strategies. These cells are especially interesting, as they appear to be derived from embryonic and extraembryonic tissues during the processes of fetal development and growth. It has been suggested that AFSCs represent an intermediate stage between embryonic stem cells and adult stem cells. AFSCs are easy to obtain, and they represent a nearly inexhaustible stem cell source that does not raise the ethical concerns that are associated with human embryonic stem cell research.6,20 For these reasons, AFSCs have great potential in future regenerative strategies.
Several studies have compared human BMSCs and AFSCs to determine whether the body site of origin could influence the osteogenic differentiation potential of these stem cells.12,22,23 In mice, mesenchymal stem cells (MSC) derived from different sites are known to vary in clonogenic capacity, surface markers, and differentiation potential. 22 Studies with human cells indicate that MSC isolated from fetal tissues are more plastic and grow faster than adult MSC, even though they have similar immunophenotypic characteristics and maintain long and stable telomeres. 12
Culture conditions and the characteristics of the substrate or scaffold on which stem cells are grown may also deeply affect cell behaviors,24–27 as cells are inherently sensitive to their surroundings, 28 and the environmental niche surrounding the cells is of key importance in stem cell fate. 24 A previous study on nanofiber scaffolds 29 made of poly(ɛ-caprolactone) (PCL) produced by electrospinning showed that AFSCs deposited significantly more mineralized matrix than BMSCs by 4 weeks in an osteogenic culture medium. In another study, 30 AFSCs showed similar values of mineralization after 2 weeks in culture when compared to BMSCs in response to dexamethasone, but significantly increased levels when stimulated with BMP-7 in two-dimensional (2D) cultures. Also, studies performed by Peister et al. 31 on PCL scaffolds made by fused deposition modeling with a collagen network compared BMSCs and AFSCs for up to 15 weeks in culture on these scaffolds. AFSCs had higher calcium content in the surrounding matrix in 2D cultures than MSC at both 2 and 4 weeks. The same trend was observed in three-dimensional (3D) scaffolds after 15 weeks in osteogenic culture. Although some comparative analyses of the behavior of AFSCs and BMSCs were performed, the differences in the structural and chemical features between previously studied scaffolds and those studied in this article are quite considerable and may stimulate different cellular responses in either AFSCs or BMSCs, or both.
Scaffold architecture has been shown to affect cell binding and spreading, 28 and the scale of topographic features alone can also elicit diverse cell behaviors, such as changes in cell adhesion, surface antigen display, and modulation of cellular signaling pathways. 28 Thus, it is necessary to study different materials with different features to improve tissue-engineering strategies, as the scaffold characteristics may interfere with stem cell fate and/or promote the interaction between the implanted construct and the native environment.
In this study, our goal was to compare the behavior of human AFSCs and BMSCs in 2D and 3D cultures to determine which stem cell type is most suitable for bone tissue-engineering applications. A 2D culture was created by seeding and culturing these cells on tissue culture plates. The 3D culture consisted of stem cells grown on a well-characterized microfibrous mesh scaffold composed of a blend of starch and poly(ɛ-caprolactone) (SPCL). The SPCL scaffolds were obtained using melt-spinning and fiber-bonding processes. 32 This scaffold type was selected as an appropriate 3D environment for osteogenic differentiation, because previous studies suggest that it is suitable for bone tissue-engineering strategies.21,33–36 Human BMSCs and AFSCs were cultured in both 2D and 3D formats in the presence of an osteogenic medium for up to 3 weeks. At various time points, the cellular viability in each culture type was measured, and the expression of osteogenic markers and the formation of extracellular matrix (ECM) were assessed.
Materials and Methods
Scaffold production
SPCL fiber mesh scaffolds were produced by a melt-spinning technique as previously described, 32 where fibers ranging from 120–500 μm in diameter were produced. Next, 3D fiber mesh scaffolds were prepared by a fiber-bonding process that consisted of cutting and sintering the fibers obtained by the melt-spinning method. 21 SPCL scaffolds were cut into cylinders (4-mm diameter and 7-mm length) and sterilized with ethylene oxide prior cell culture.
Cell culture
Human BMSCs were purchased from Lonza®, where routine characterization of hMSC includes testing for surface antigen expression and functional testing for in vitro differentiation into adipogenic, chondrogenic, and osteogenic lineages as indicated by the manufacturer. BMSCs were also tested for expression of several cell surface markers via flow cytometry, and these cells were shown to be CD105+, CD166+, CD29+, CD44+, CD14−, CD34−, and CD45−. BMSCs were expanded in a basal BMSC medium composed of α-MEM (HyClone Laboratories, Inc.) supplemented with 15% embryonic stem-screened fetal bovine serum (ES-FBS; HyClone Laboratories, Inc.) and 1% penicillin/streptavidin solution.
Human AFSCs were isolated as previously described. 2 Briefly, AFSCs were obtained from human amniotic fluid specimens collected from amniocentesis procedures, using backup cells that would otherwise be discarded. AFSCs were immunoselected for c-Kit expression using magnetic microspheres. C-kit (or CD117) has been described to be the receptor for stem cell factor.
AFSCs were cultured in a basic amniotic fluid cell medium composed of an α-MEM (HyClone Laboratories, Inc.) with 18% Chang B (Irvine Scientific) and 1% Chang C (Irvine Scientific) medium as well as 2%
For 2D cultures, BMSCs (passage 5) and AFSCs (passage 24) were seeded onto tissue culture-treated six-well plates at a concentration of 30,000 cells/well. Cells were cultured for 3 days with a basal medium, and then on day 4 (day 0 of the osteogenic culture), the medium was replaced with an osteogenic medium consisting of the DMEM (HyClone Laboratories, Inc.) supplemented with 10% FBS (HyClone Laboratories, Inc.) and 100 nM dexamethasone (Sigma), 50 μM
For the 3D experiments, BMSCs and AFSCs were seeded onto SPCL scaffolds (7-×4-mm cylinders) at a concentration of 1.2×106 cells/scaffold. As in the 2D cultures, the seeded scaffolds were cultured in a basal medium for 3 days in nonadherent multiwell plates, and then in an osteogenic medium for 0, 7, 14, or 21 additional days. SPCL scaffold, a blend of SPCL, 30:70 (wt%) (Novamont), was produced by a fiber-bonding technique as previously described. 32 Before osteogenic characterization, cells were briefly rinsed in phosphate-buffered saline (PBS; HyClone Laboratories, Inc.) and fixed in 10% neutral buffered formalin (Surgipath Medical Ind., Inc.).
These time points were defined to achieve an osteoblastic-like state in the stem cells in the shortest possible period of time. The osteoblastic-like state was defined as when osteogenic-induced stem cells were able to synthesize a mineralized ECM. The 3-week time point was established in previous studies as this point for AFSCs. 21
Samples were retrieved after every 7 days in culture to be characterized for cellular viability with Calcein AM and for assessing the presence of osteogenic markers and matrix formation by alkaline phosphatase (ALP) and alizarin red staining, as well as the presence of RunX-2 and collagen I in the matrix by immunofluorescence. Cell morphology and matrix formation were also assessed by scanning electron microscopy (SEM) in the 3D environment. Characterization assays were made in triplicates.
Cell viability
At each time point, cellular viability in each 2D and 3D culture was measured. AFSC- and BMSC-containing wells and constructs were rinsed in PBS (HyClone Laboratories, Inc.) and incubated in a cell culture medium containing 3 μM Calcein AM (Molecular Probes, Invitrogen) for 30 min at 37°C in a 5% CO2 environment. Samples were rinsed in PBS and fixed in 10% buffered formalin (Surgipath Medical Ind., Inc.) overnight at 4°C. Cellular viability was observed under a confocal microscope (Axiovert 100 M; Zeiss) equipped with argon/He-Ne laser sources. DAPI (Molecular Probes) served as a nuclear marker in these cells (live or dead).
ALP staining
ALP staining was performed in 2D cultures to evaluate the osteogenic differentiation process of AFSCs and BMSCs in the well plates. The medium was removed from each well, and the cells were gently rinsed with PBS. Cells were then fixed in with 10% buffered formalin solution overnight at 4°C. Fixed cells were rinsed and kept in PBS until needed. To stain for ALP, the cells were incubated in a staining solution containing 0.25% Napthol AS-MX phosphate alkaline solution (Sigma-Aldrich) and Fast Violet B salt (Sigma) for 30 min at room temperature. After incubation, samples were rinsed in PBS to remove nonspecific staining and observed under an inverted microscope (Leica; DMI4000B). Images were acquired using a Q-Imaging (Retiga-2000RV) camera system.
Alizarin red staining
Alizarin red staining was performed in both 2D and 3D osteogenic constructs after fixing the constructs as described above. For this purpose, 2% alizarin red solution (Sigma-Aldrich) was prepared (pH adjusted to 4.1–4.3), and samples were stained by immersion for 2 min. After image acquisition of the stained cells and cell–scaffold constructs, alizarin red staining was solubilized in cetylpyridinium chloride (Sigma) at pH 7.0 for 15 min under mild agitation and quantified at 562 nm, using a plate reader (SpectraMax MS; Molecular Devices). In the case of 2D cultures, alizarin red staining was performed in the same samples following the Calcein AM assay.
Immunofluorescence
Immunofluorescence analysis was performed in samples retrieved of both 2D and 3D conditions to assess the presence of the bone-associated proteins RunX-2 and Collagen type I. Samples were rinsed and permeabilized with Triton X-100/PBS (Sigma) before incubation with a protein-blocking solution (Dako). After blocking, samples were incubated with antibodies rabbit anti-RunX-2 (ab23981; Abcam) and goat anti-type I Collagen (1310-01; Southern Biotech) (1:100 and 1:20 dilution, respectively) in an antibody diluent with background reducing components from Dako. Antibody binding was assessed using Alexa-Fluor-488 goat anti-rabbit IgG (A-11008) or Alexa-Fluor-594 donkey anti-goat IgG (A-11058), both from Molecular Probes (1:200 dilution), as a secondary antibody.
Scanning electron microscopy
To further characterize BMSCs and AFSCs grown in 3D culture, the cell-seeded scaffolds were analyzed using SEM (S-2600N; Hitachi Science Systems, Ltd.). SEM was performed with fixed constructs. After dehydrating the samples in a graded series of ethanol concentrations, the samples were dried in a critical point dryer (EMS850X; Electron Microscopy Sciences) and gold sputtered (Hummer 6.2 sputtering system; Anatech Ltd.).
Statistical analysis
All quantified results are expressed as the mean±standard deviation. Two-way ANOVA followed by Bonferroni's Multiple Comparison test was also used to determine whether differences between sample groups were significant. Differences were considered significant when the p-value was<0.05.
Results
Osteogenic differentiation of AFSCs and BMSCs (2D environment)
Cellular viability assays (Calcein and DAPI) showed that both AFSCs and BMSCs were able to attach to the tissue culture plates and proliferate for up to 3 weeks in 2D cultures (Fig. 1). This assay suggested that there was an increase in AFSC viability after 2 weeks in an osteogenic medium, while BMSCs maintained high viability levels during the 3-week period. Alizarin red staining, which indicates calcium deposition, was detected in both cell types after 14 days in an osteogenic medium. The calcium deposition during osteogenic differentiation appeared to become more intense in AFSCs by the end of week 3 in an osteogenic medium. However, an increase in the staining intensity was not evident in cultures of BMSCs at this time. In addition, ALP staining, which is associated with ECM maturation, was highest in culture of BMSCs after 2 weeks in an osteogenic medium, suggesting that this may be an active phase for the development of ECM by these cells. In contrast, AFSCs did not express significant amounts of ALP at any time during the 2D experimental study (Fig. 2).

Viability assay (green) and alizarin red (red) staining of amniotic fluid-derived stem cells (AFSCs) and bone marrow-derived stem cells (BMSCs) cultured in a two-dimensional (2D) environment for 0, 7, 14, or 21 days in an osteogenic medium. Scale represents 100 μm. Color images available online at www.liebertpub.com/tea
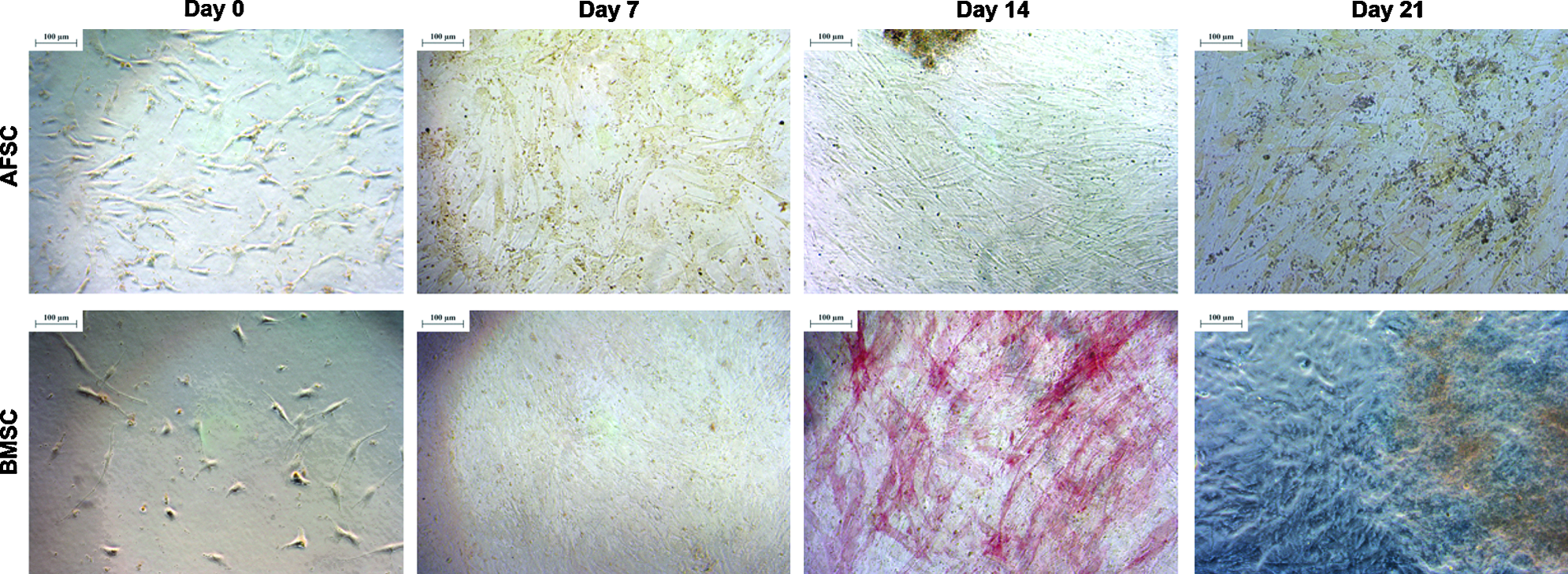
Alkaline phosphatase staining of AFSCs and BMSCs cultured in a 2D environment for 0, 7, 14, or 21 days in an osteogenic medium. Scale represents 100 μm. Color images available online at www.liebertpub.com/tea
RunX-2 is an important transcription factor involved in osteogenic development. In our 2D cultures, RunX-2 was expressed by both AFSCs and BMSCs at day 0, and a weaker signal was also detected at day 7. At this time point, the RunX-2 signal was more evident in BMSC culture. After 14 days in culture, the presence of RunX-2 was below the detection limit of this assay (data not shown). In addition, collagen I, which is an important protein in the bone ECM, was not detected in either AFSCs or BMSCs during the 3 weeks in 2D culture.
Calcium levels were quantified using a microplate reader after solubilizing the alizarin red staining described above. For the first 2 weeks in 2D osteogenic culture, calcium deposition by both cell types followed a similar pattern, but was relatively low during the first 14 days in an osteogenic medium. At 21 days of culture, calcium levels deposited by osteogenically differentiated AFSCs increased dramatically (Fig. 3), suggesting that AFSCs produce a greater amount of mineralized ECM in the 2D osteogenic culture environment when compared to BMSCs.
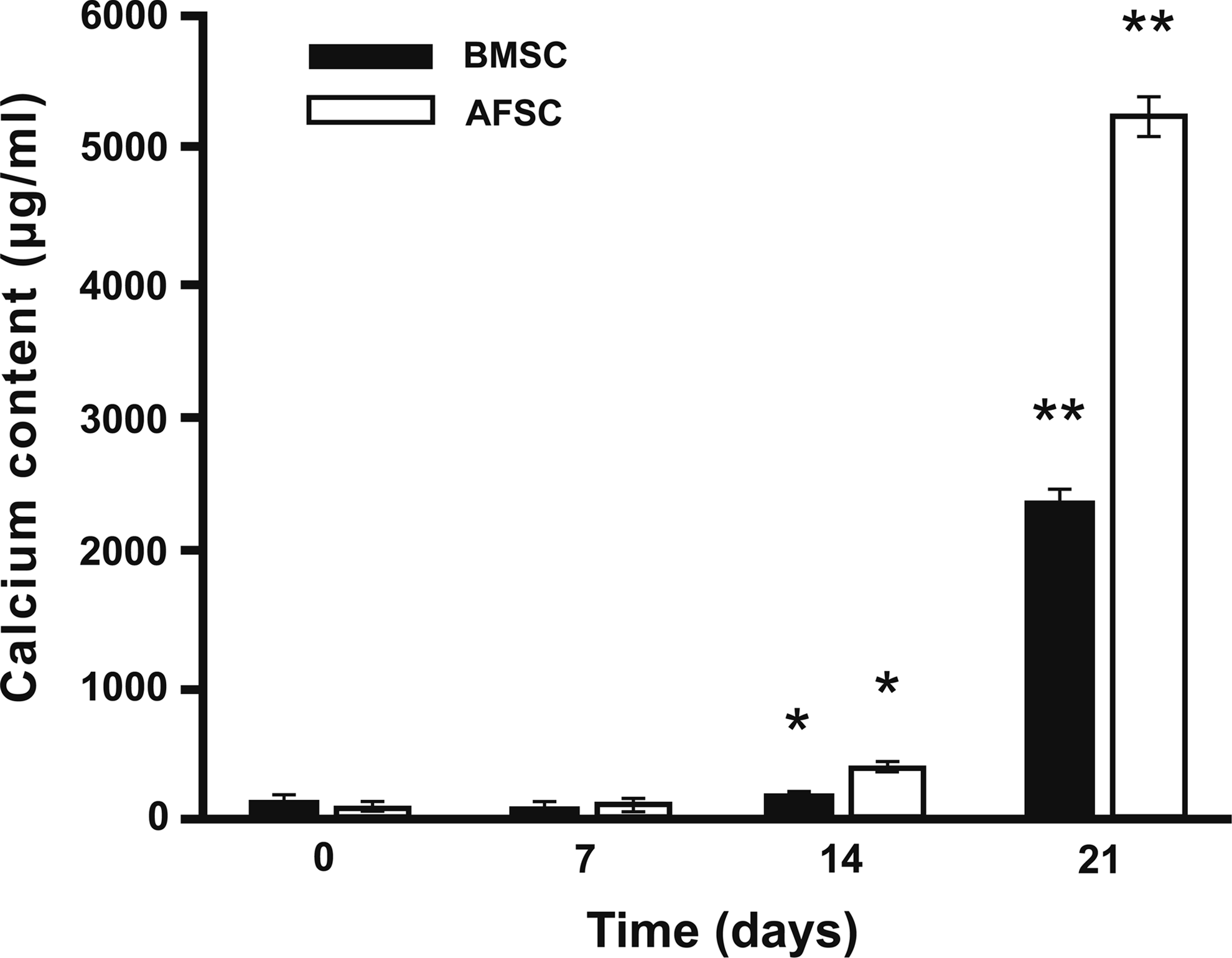
Calcium content (μg/mL/well) of AFSCs and BMSCs in a 2D culture environment in an osteogenic medium for 0, 7, 14, or 21 days (symbols * and ** denote p<0.05).
Osteogenic differentiation of AFSCs and BMSCs in SPCL scaffolds (3D environment)
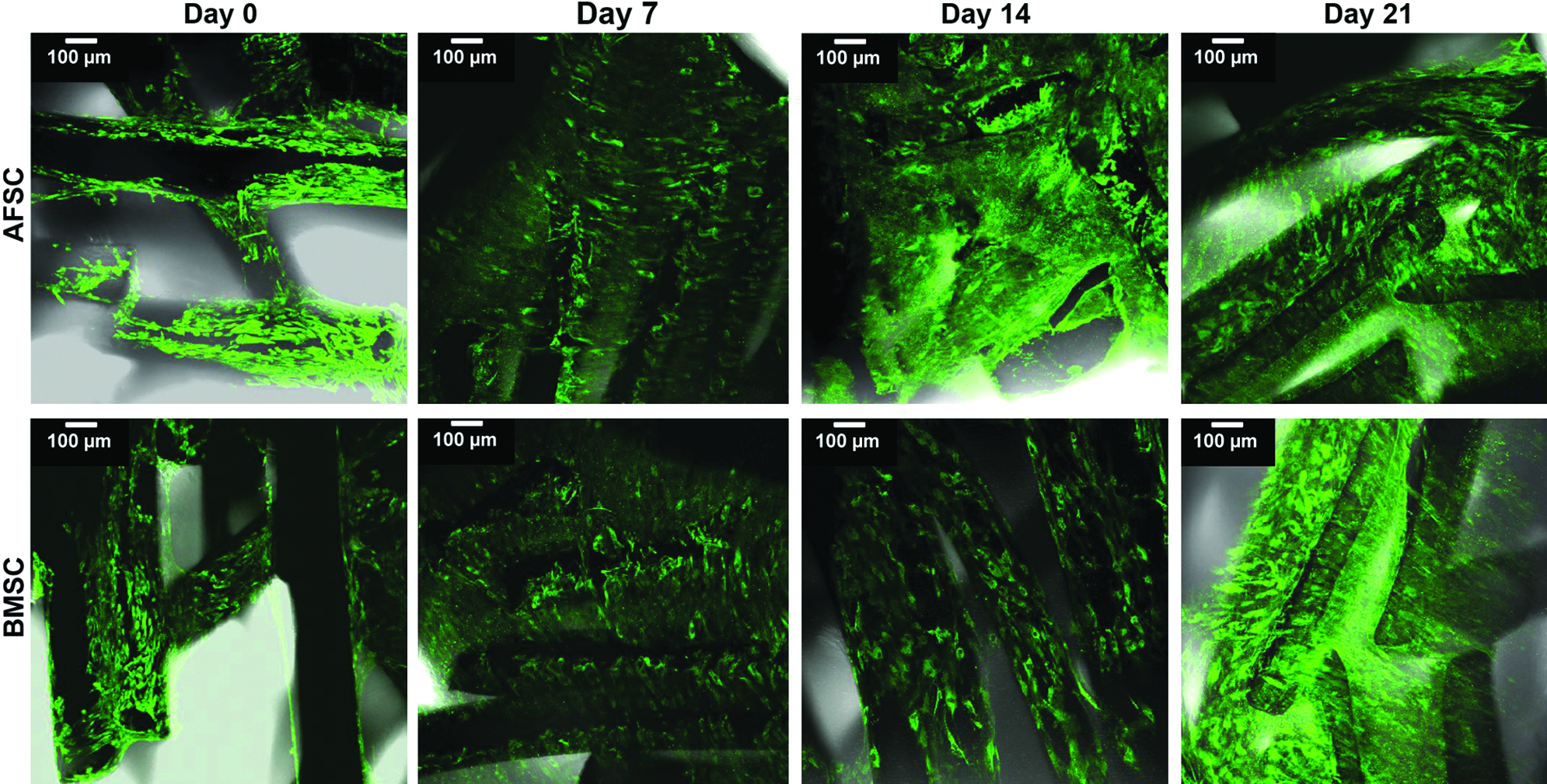
According to immunofluorescence observations, both stem cell types remained viable when cultured onto SPCL scaffolds (3D culture) for up to 3 weeks (Fig. 4). SEM images revealed that both AFSCs and BMSCs proliferated on the scaffold, and they were distributed throughout the scaffolds as clearly evidenced by thick layers of cells seen throughout the scaffold (Fig. 5). The SEM images also suggest the presence of the newly synthesized ECM in BMSC-SPCL constructs beginning at 2 weeks in an osteogenic medium. Some mineralization nodules (white dots in SEM images) could be seen as well, and these nodules had increased in number by 3 weeks in an osteogenic medium. In contrast, AFSC-SPCL constructs did not develop these nodules until week 3 (Fig. 5).
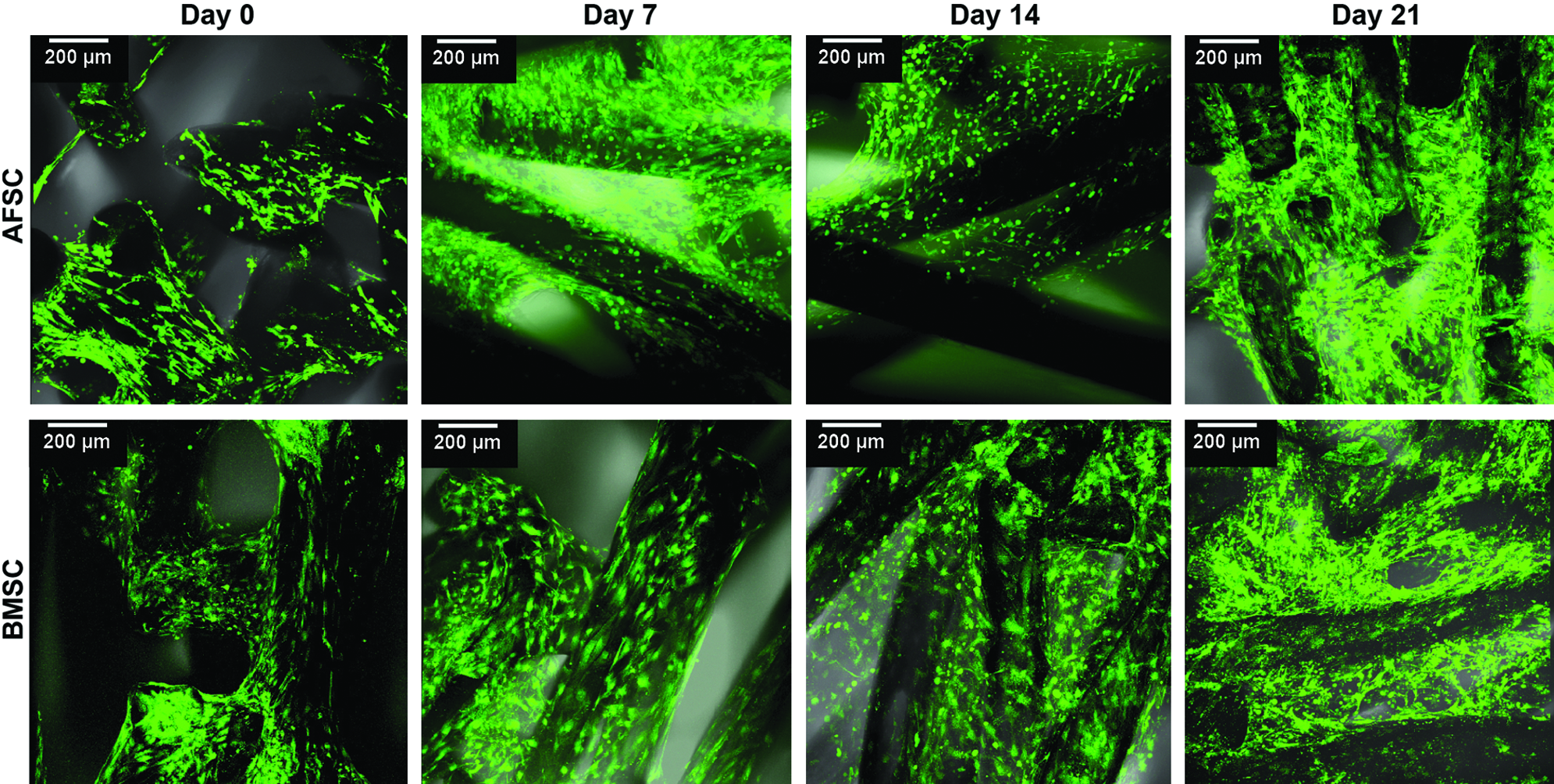
Viability assay of AFSCs and BMSCs seeded onto starch and poly(ɛ-caprolactone) (SPCL) scaffolds and cultured in an osteogenic medium for 0, 7, 14, and 21 days. Scale represents 200 μm. Color images available online at www.liebertpub.com/tea
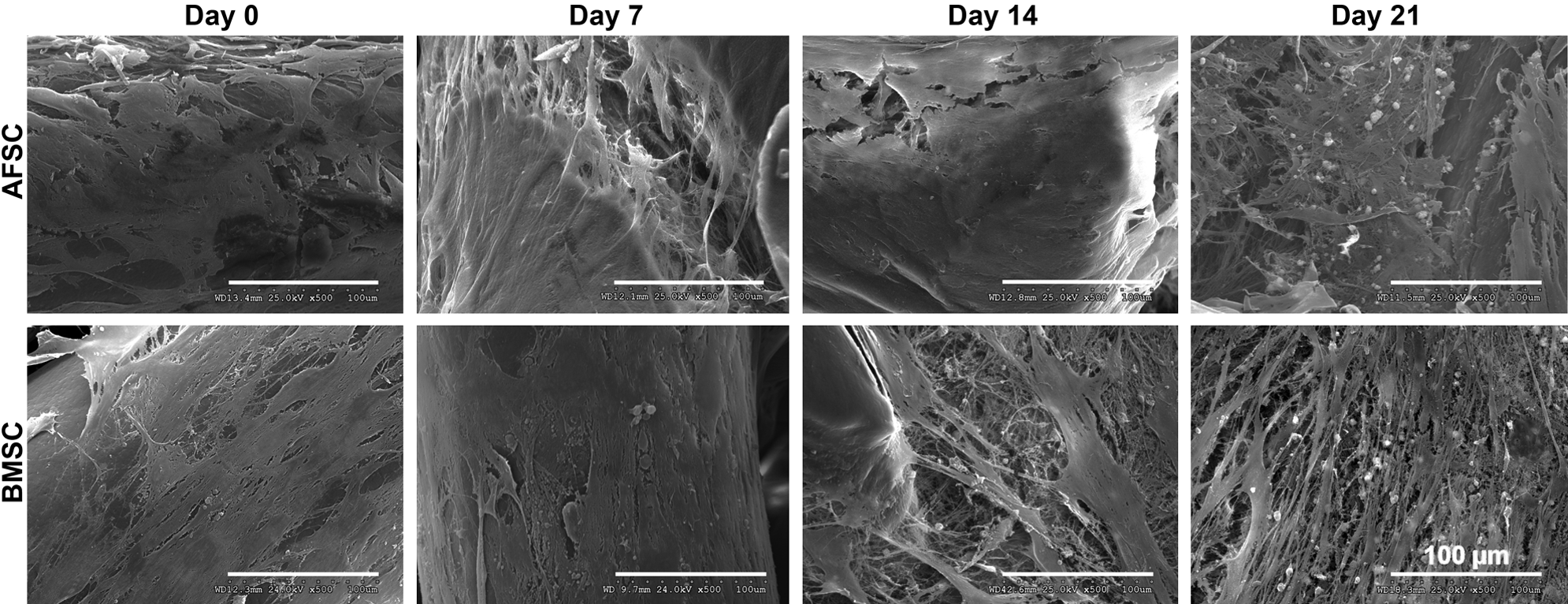
Scanning electron microscopy images of AFSCs and BMSCs seeded onto SPCL scaffolds in an osteogenic medium for 0, 7, 14, or 21 days. Scale represents 100 μm.
The formation of mineralization nodules was also evaluated in BMSC-SPCL and AFSC-SPCL constructs by alizarin red staining (Fig. 6) to determine the presence of calcium in these aggregates. In addition, the alizarin red stain determined that AFSCs required more calcium during the first days of culture for metabolic processes, but this tendency changed after 2 weeks in an osteogenic medium. By 3 weeks in culture, the calcium concentration produced by BMSCs on SPCL scaffolds was dramatically higher than that produced by AFSC-SPCL constructs (Fig. 6).
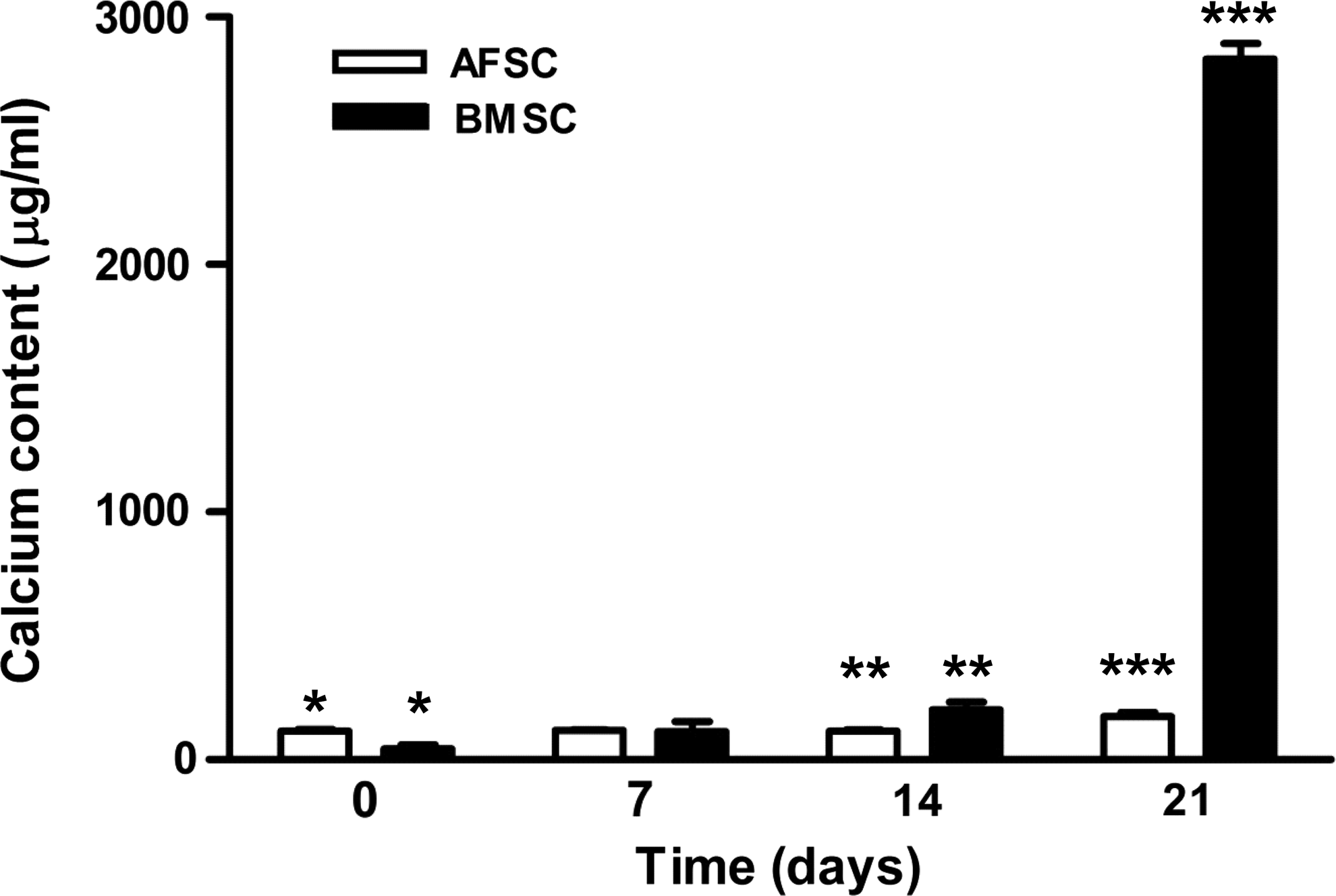
Calcium content (μg/mL/construct) of AFSCs and BMSCs seeded onto SPCL scaffolds in an osteogenic medium for 0, 7, 14, or 21 days (symbols *, **, and *** denote p<0.05).
SEM data are in agreement with results obtained from the calcium quantification assay in AFSC-SPCL and BMSC-SPCL constructs, showing that BMSCs are faster in achieving a calcified matrix in 3D microfibrous SPCL scaffolds. When cultured on the SPCL scaffolds, both BMSCs and AFSCs expressed similar levels of RunX-2 for the first week of osteogenic differentiation (Fig. 7). The amount of this transcription factor decreased from day 0 to day 7 in an osteogenic medium, but then increased again after 2 weeks, especially in AFSC cultures. By the end of week 3, RunX-2 expression remained high, suggesting that the osteogenic process continues in the 3D constructs throughout the 3-week timeline even when significant mineralized matrix was present.
Immunofluorescence of RunX-2 expression in AFSCs and BMSCs seeded onto SPCL scaffolds in an osteogenic medium for 0, 7, 14, or 21 days. Scale represents 100 μm. Color images available online at www.liebertpub.com/tea
Immunofluorescence for collagen type I showed that this protein was expressed in cell–SPCL constructs during the experimental time. At day 0, which represents an undifferentiated stage, similar levels of this protein were observed for both AFSCs and BMSCs (Fig. 8). After 7 and 14 days of culture in an osteogenic medium, a more intense fluorescence was observed in AFSC-SPCL constructs, but this tended to decrease by week 3. However, in BMSC-SPCL constructs, collagen I fluorescence increased incrementally over the study period. Furthermore, in the BMSC-SPCL constructs, the collagen fibers seemed to be oriented in along the SPCL scaffold fibers, forming a dense ECM.
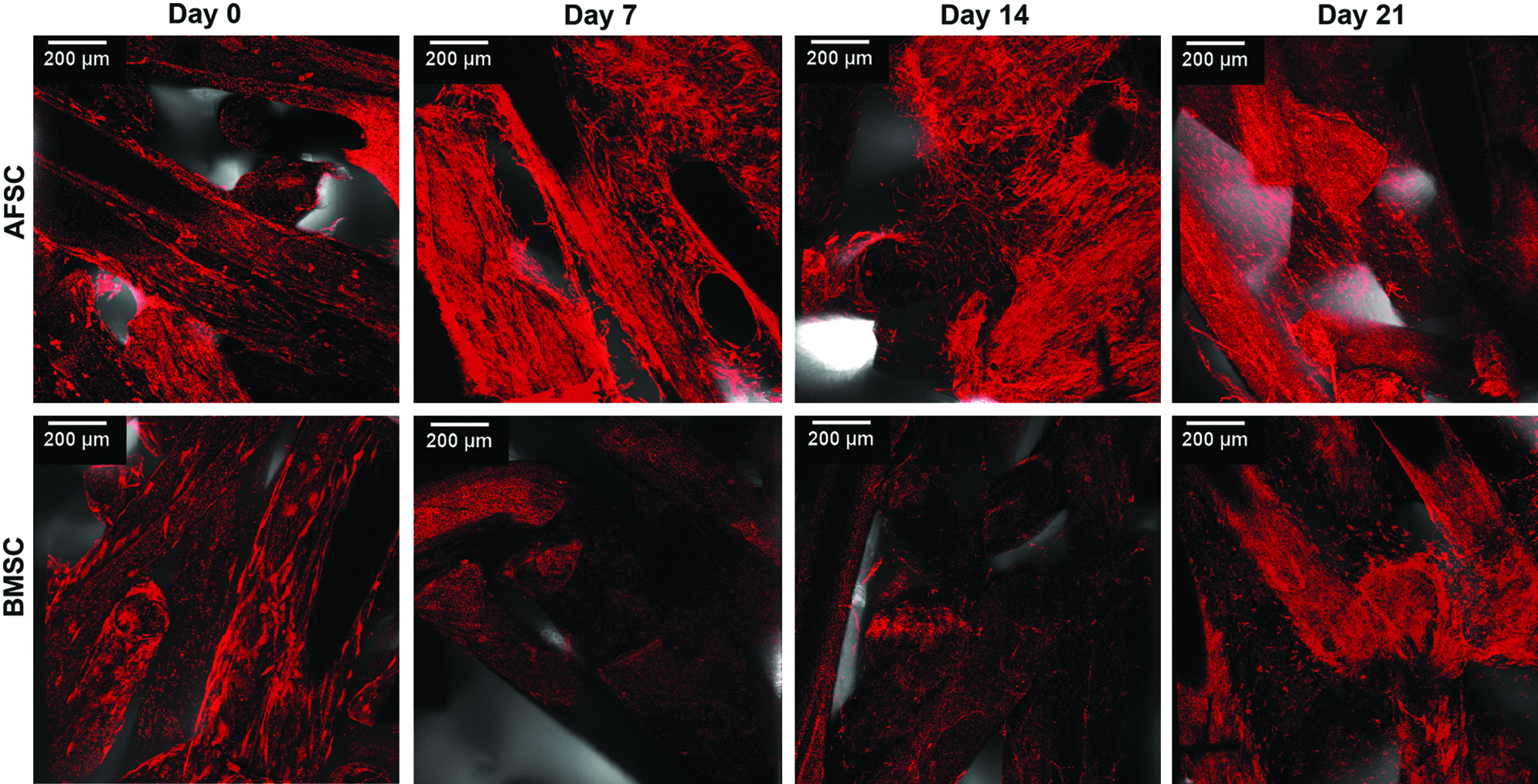
Immunofluorescence of collagen type I expression in AFSCs and BMSCs seeded onto SPCL scaffolds in an osteogenic medium for 0, 7, 14, or 21 days. Scale represents 200 μm. Color images available online at www.liebertpub.com/tea
Discussion
In a 2D culturing environment, both AFSCs and BMSCs were able to differentiate into the osteogenic lineage and produce mineralized ECM, even in the absence of collagen I and RunX-2 expression. Although collagen I is the most abundant protein in the organic matrix of bone tissues, collagen I cannot initiate tissue mineralization, since it is not responsible for nucleation and the postnucleation growth of apatite crystallites. 37 However, it has been shown that osteogenic cells can produce a layer of noncollagenous matrix in vitro, at the surface of nonbiological substrata, before the development of a calcium- and phosphorus-rich ECM. This layer has been shown to be similar to the cement line in vivo. 38 Interestingly, AFSCs showed higher proliferation rates and enhanced mineralization of the ECM in 2D cultures when compared to BMSCs, but this mineralization process seems to compromise the viability of AFSCs in 2D culture. In the 3D environment, the two types of stem cells were seeded on SPCL scaffolds and cultured them in an osteogenic medium. In these conditions, mineralization of the ECM was observed at 14 days for BMSCs and at 21 days for AFSCs, and did not seem to influence cell viability. AFSCs and BMSCs also showed changes in the expression of osteogenic markers depending on whether they were cultured in 2D or 3D culture, indicating that culture environments play an important role in guiding cellular behavior during the osteogenic differentiation process.
Other types of stem cells have been shown to be quite sensitive to various microenvironments and to respond to topographical patterns as well, suggesting a physical contribution to the cellular expansion and differentiation process.24,39,40 Some osteoblast responses also seem to be substrate dependent, further highlighting the importance of the culture surface in modulating cell proliferation, matrix deposition, and inflammatory factor expression. 41
Unlike the results obtained for 2D culture, collagen I is present before mineralization occurs in 3D SPCL scaffold-based culture with both AFSCs and BMSCs. This collagen provides the framework and spatial constraints for ordered crystal deposition, and thus assists the formation of calcium phosphate nodules. The collagen fibers covering the scaffolds seem to be aligned along the scaffold fibers, providing some level of organization, which may be important in the process, since natural regeneration typically involves an organized distribution of the ECM rather than a randomly dispersed matrix which, in the in vivo scenario, is associated with scar or fibrotic tissue formation.
Despite similar viability, collagen I and RunX-2 levels detected during these experiments, after 21 days in osteogenic culture, BMSCs and AFSCs behaved differently in terms of mineralization of the ECM. Mineralization occurs later in AFSC constructs, and these cells also produced less-mineralized ECM when compared to cultures of BMSCs when seeded onto SPCL scaffolds.
In addition to culture substrate–ECM interactions, other inductive factors also influence the programming of MSC by the matrix. 24 Multiple in vitro and in vivo studies have confirmed that osteogenic-specific growth factors exert autocrine and paracrine effects on the proliferation, differentiation, and maturation of osteoprogenitor cells. In this study, the participation of growth factors in the culture medium is quite evident, due to their pivotal role in achieving the osteogenic differentiation process. The continuous expression of RunX-2 in SPCL scaffolds also indicates that the osteoblast differentiation process is likely to continue in time, thus reinforcing the ECM production and maturation.
In both environments, AFSCs and BMSCs were able to proliferate and differentiate into the osteogenic phenotype with the production of a mineralized ECM, although differences in osteogenic marker expression, such as ALP activity, RunX-2, and collagen I, were observed. These differences may be related to different contributions from the external environment as well as the influence of the stem cell source in cellular responses toward the osteogenic differentiation process, which directly affects ECM production and composition. AFSCs have a higher proliferative capacity, and their ability to maintain their pluripotency at higher passages could be exploitable as a readily available source for large numbers of osteogenic progenitor cells. It is thought that AFSCs may have greater applicability for allogeneic treatment strategies than BMSCs, which may be limited to autologous interventions and by the donor's age. Despite these limitations, BMSCs represent a promising cell source for cell-based therapeutics, especially in orthopedics, since BMSCs are predisposed, to a certain extent, to follow the osteogenic differentiation pathway due to their natural commitment to the mesenchymal lineage.
The fact that BMSCs from passage 5 are compared with AFSCs from passage 24 in this study is based on the fact that BMSCs and AFSCs have different population-doubling times, which may be more relevant in predicting potential outcomes than the number of cell passages. The average number of population doublings for marrow-derived adult human MSCs was determined to be 38±4, at which time the cells finally became very broad and flattened before degenerating. 42 Also, studies performed on BMSCs suggest that the proliferation rates and other properties of BMSCs gradually change during expansion, and this led to the recommendation that hMSC should not be expanded beyond six passages, 43 supporting the importance of using low-passage BMSCs. On the other hand, AFSCs share characteristics with both embryonic stem cells and adult stem cells, and they have been described as genetically and phenotypically stable pluripotent cells.23,44 Furthermore, these cells can be extensively expanded while maintaining long telomeres and a normal karyotype for over 250 population doublings, and they are still able to differentiate into functional cells. 2 Studies that compared AFSCs to BMSCs 30 at different passages (P16–17 to P3–6) have highlighted the fact that the cell passage number may not be as significant as other stem cell properties in these types of comparisons.
The origin of stem cells may also influence the cellular response toward external signals, causing changes in ECM production and composition. Studies describe stem cell differences in proliferation 20 and differentiation3,20 potential, survival capacity, 20 cytokine expression, 6 and immunomodulatory properties, 22 and all of these appear to depend on the tissues from which the stem cells were obtained. This is also observed for lineage-specific markers such as ALP activity and RunX-2 expression. 20 Our data follow this trend, as AFSCs and BMSCs behave differently on different substrate matrices, affecting the timing of differentiation and bone-related protein synthesis. Animal studies are envisioned to understand the full potential and behavior of both AFSCs and BMSCs, analyzing the influence of the cell source and the 3D scaffold when implanted in vivo in a preclinical situation.
Conclusions
The application of stem cells to clinical practice is an exciting alternative for many treatments, since these cells can be used to enhance the body's own regenerative potential or to develop new therapies. The data presented here indicate that both AFSCs and BMSCs are able to proliferate, and subsequently colonize a substrate in both 2D and 3D cultures. Both cell types were able to cover the culture surface with adherent cells, forming a network that followed the osteogenic differentiation process for up to 3 weeks when an inductive osteogenic culture medium was used. Markers specific for osteogenic differentiation were detected in the cultures, and a mineralized matrix was formed in 2D as well as 3D cultures. Interestingly, cells from different origins express different osteogenic markers at different time points, which may be a result of both cellular origin and substrate properties. Specifically, the origin of the stem cells seems to have an important role in selecting pathways involved in ECM synthesis, resulting in different time frames for protein synthesis. The interplay of ECM molecules, cells, and culture surface results in a dynamic and balanced system with different possible outcomes. Thus, the selection of a particular stem cell type for a designated tissue-engineering strategy may not be a simple and direct process, and AFSCs might be an interesting alternative to BMSCs for bone tissue-engineering applications.
Footnotes
Acknowledgments
The authors wish to thank Dr. Jennifer Olson for editorial assistance with this article. M. T. Rodrigues thanks the Portuguese Foundation for Science and Technology (FCT) for providing a Ph.D. scholarship (SFRH/BD/30745/2006). This study was supported, in part, by the Telemedicine and Advanced Technology Research Center (TATRC, W81XWH-07-01-0718) at the U.S. Army Medical Research.
Disclosure Statement
The authors have no conflict of interest to declare.