
Abstract
Damage of cartilage structures in the head and neck region as well as in orthopedic sites are frequently caused by trauma, tumor resection, or congenital defects. Despite a high demand in many clinical fields, until today, no adequate cartilage replacement matrix is available for these fields of application. Materials that are clinically applied for joint cartilage repair still need optimization due to difficult intraoperative handling and risk of early mechanical damage. We have developed and applied a novel chemical process to completely decellularize and sterilize human and porcine cartilage tissues (meniscus cartilage and nasal septum) to generate a new type of bioimplant matrix. To characterize this matrix and to determine the effect of the decellularization process, the content of denatured collagen (wD) and the content of glycosaminoglycans (GAGs) (wG) were determined. Possible cytotoxic effects and cellular compatibility of the matrix in vitro have been examined by seeding processed cartilage biomatrices with human primary chondrocytes as well as murine fibroblasts (L929). Vitality and state of metabolism of cells were measured using MTS assays. Both cell types adhered to scaffold surfaces and proliferated. No areas of growth inhibition or cytotoxic effects were detected. New synthesis of cartilage-specific extracellular matrix was observed. By histological staining, electron microscopy, and μCT analysis, an increase of matrix porosity, complete cell elimination, and high GAG removal were demonstrated. Being from natural-origin, processed xenogenic and allogeneic cartilage biomatrices are highly versatile with regard to shape, size, and biomechanics, making them promising candidates for various biomedical applications.
Introduction
The gold standard for reconstructive head and neck surgery is the application of autologous cartilage from rib, nasal septum, 2 or auricle. 3 Because these techniques frequently require multistage surgical procedures, several difficulties, like additional donor-site morbidity, insufficient cosmetic results at explantation, as well as implantation site, and postoperative dysfunctions, or pain, might occur as complications.3,4
Several medical conditions like meniscal cysts and traumatic or degenerative meniscal lesions are treated by total or partial meniscectomy. This may elicit degenerative changes in the affected joint that often increases the degenerative progress and can lead to osteoarthritis, requiring cartilage repair in joints.5–7
As availability of appropriate allogeneic and autologous cartilage donor tissues is limited,8–12 the worldwide interest in cartilage tissue replacement has become a main target in the tissue-engineering research.9,12 Although several xenogenic extracellular matrix (ECM) scaffolds have successfully been used in both preclinical animal studies and in human clinical studies for the repair or replacement of numerous defects, body tissues like musculoskeletal structures, lower urinary tract reconstruction, and integumentary structures,13,14 the extensive use of porcine or bovine cartilage tissues is not established as an alternative source for allogeneic or autogenic tissue transplantations. Until today, many attempts have been made to prepare xenogenic cartilage for use as cartilage implants.15,16
It had been speculated that because of its limited vascular, lymphatic, and neural supply, cartilage might be immunologically privileged. 17 Nevertheless, transplantation of discordant xenograft tissues exposes patients to the danger of antibody-mediated hyperacute and chronic rejection responses. These immune reactions are evoked by natural human serum polyclonal antibodies (anti-Gal) that bind specifically to mammalian carbohydrate α-galactosyl epitopes (Galα1-3Galβ1-4GlcNac-R) expressed on all cell types in porcine and bovine tissues.17–19
Because xenogenic and allogeneic cartilage tissues contain the risk of infection and disease transmission, extensive sterilization methods are necessary when using these tissues as bioimplant matrices. Today, several sterilization methods like autoclaving, dry heat, exposure to UV or gamma radiation, as well as the use of ethylene oxide, or antiseptic techniques using immersions in 70% ethanol aqueous solution are commonly used in bioimplant production. 20 The current gold standard, which is treating tissue by autoclaving, is not suitable for many tissues, especially as in natural tissues like cartilage, and it leads to denaturation of collagen, influencing tissue biomechanics.
After sterilization with ethylene oxide gas, the amino acid analysis demonstrated intensive reactions of ethylene oxide with amino acids of the collagen matrix. These changes lead to a decreasing helix stability, reduced shrinkage, and denaturation temperature, also influencing biomechanical stability. 21 Remaining deposits of ethylene oxide in tissue act cytotoxically and are able to provoke, for example, synovitis in case of meniscus replacement.21,22
Sterilization methods like gamma irradiation with radiation doses of ≥25 kGy can completely destroy HI virus, but result in numerous significantly negative changes with regard to tissue integrity and mechanical characteristics. 21 Furthermore, pathogenic agents like prions causing, for example, scrapies and Creutzfeld-Jakob-disease demonstrate a high radiation resistance and even remain active after tissue radiation with >0.5 MGy. 23
These causative organisms, which lead to a whole group of transmissible spongiform encephalopathies (TSEs), a group of fatal neurodegenerative diseases, are generally resistant to a multitude of pathogen inactivation and sterilization techniques. Therefore, the application of strong chemical reagents is necessary to ensure effective decontamination and inactivation in the source material. 24
In recent studies, it could be proved that NaOH is effective to inactivate aberrantly folded cellular proteins (PrPsc, prions), causative agents for TSEs.24–26 In consequence, PrPsc are eliminated and cross contaminations minimized. Furthermore, NaOH not only destroys PrPsc but also removes HIV, HAV, HBV and HCV, and DNA/RNA.27–29 Additionally, treatment with NaOH facilitates solvent penetration by tissue swelling.
H2O2 is used for tissue sterilization to inactivate and eliminate a broad spectrum of microorganisms like yeasts, fungi, and spores. It provides a high efficiency against a multitude of viruses like HIV-1, HAV, RSV, vaccinia, and herpes simplex virus type I. 30
The specific problem underlying this study was to provide a method that excludes the risk of infection and disease transmission and guarantees exhaustive decellularization for elimination of antigenicity and avoidance of immunoreactions.31,32 On the other hand, processing of the cartilage tissue should not affect biomechanical integrity of cartilage transplant matrix, matrix recellularization capability, or chondroconductive properties. 31
Material properties, biochemical stability, and integrity, as well as collagen composition and three-dimensional (3D) network formation of collagen fibers and fibrils, should be maintained to a large extent as in natural tissue. The created implant matrices should serve as scaffolds for the migration of cells and allow cell adhesion, recellularization, and proliferation. Thus, the aim of this work was to develop and evaluate a suitable method for processing cartilage meeting the requirements mentioned above.
Materials and Methods
Porcine cartilage
Porcine knee joints and nasal septal cartilage (NSC) were obtained fresh from young adult (7–8 months old) animals at a local abattoir. Cartilage was harvested under sterile conditions, and adherent noncartilaginous tissues, like perichondrium and olfactory epithelium, were dissected.
From all cartilage tissues, cylindrical discs (d=5 mm, h=1 mm) were produced to assure reproducible experimental settings and stored in a sterile NaCl solution (0.9%) at −24°C until further use.
Human nasal cartilage
Human NSC (hNSC) was obtained during routine surgeries, like septoplasties or septorhinoplasties, in the Department of Otorhinolaryngology, University Medical Center Ulm. Donor age ranged from 18 till 74 years, with an average age of 30.85±12.33 (n=143). Cartilage harvesting was approved by the University of Ulm Ethics Committee (No. 152/08), and all patients enrolled in this research have responded to an Informed Consent. All hNSC samples were first rinsed in the culture medium DMEM/HAM's-F12 (Biochrom), supplemented with fetal bovine serum (FBS, 10%; Biochrom) and gentamicin (0.5%, PAA Laboratories), under sterile conditions. Adherent noncartilaginous tissues, like perichondrium and epithelium, were dissected. Cylindrical discs (d=5 mm, h=1 mm) were punched out of hNSC samples for decellularization and sterilization process (DP), while smaller pieces were used for harvesting human primary nasal chondrocytes (hPChs).
Cartilage tissue DP
All steps of DP were performed as published in the patent applications33,34 and under continuous shaking. All intermediate washing steps were carried out for 24 h at room temperature (RT), whereas water (H2Odeion) was changed several times.
First, cartilage tissue samples were osmotically pretreated with H2Odeion for 24 h at RT. Samples were transferred to NaOH (1 N) solution, and processed for 3 h (porcine meniscus [pMC]), respectively, 1.25 h (porcine NSC [pNSC], hNSC), at RT for pathogen inactivation and denaturation of DNA/RNA as well as removal of cells and debris. After a washing step, samples were defatted with ethanol (70%) for 3 h at 40°C. To denature and remove noncollagenous components like glycosaminoglycan (GAG), samples were transferred to guanidine hydrochloride (1 M, GndHCl) solution containing sodium acetate (0.05 N) solved in an incubation buffer and incubated at +4°C for 96 h. Subsequently to another washing step, samples were treated with H2O2 solution (5%) for 48 h and shaded at 4°C for additional pathogen inactivation and protein denaturation. After a last washing step, pNSC und hNSC scaffolds were stored in a sterile NaCl solution (0.9%) until further use. Only pMC samples were treated again with NaOH solution (1 N) for 3 h and subsequently washed under the same conditions as previously performed.
Electron microscopy and microcomputed tomography analysis
To visualize increasing porosity as a consequence of DP, comparative scanning electron microscopy (SEM), transmission electron microscopy (TEM), and microcomputed tomography (μCT) analysis of native and processed pMC as well as pNSC matrices were performed.
Standard primary fixation for all three examination methods was performed with 0.1 M phosphate buffer (pH 7.3), containing 2.5% glutaraldehyde and 1% saccharose for 2 h at RT, then rinsed in phosphate-buffered saline (PBS), and postfixed in 2% osmium tetroxide for 2 h at RT. Before dehydration in a graded series of ethanol, samples were washed in PBS.
For SEM examination, dehydrated samples (each n=3) were critical point-dried, sputtered with gold–palladium (Au-Pd, 20 nm), and studied using a Zeiss DSM 962 SEM.
After dehydration, samples for TEM analysis (each n=3) were embedded in EPON. Ultrathin sections (70 nm) were poststained with lead citrate and examined in a Zeiss EM 10 (Zeiss) at 80 kV. Image acquisition and processing were conducted with EM-Menu 4 software (TVIPS).
Critical point-dried specimen of native (n=10) and processed pNSC (n=10) underwent a μCT analysis (SkyScan 1172; SkyScan) applying 40 kV and 250 μA, at 5-μm resolution in all spatial directions. After reconstruction, the scans were analyzed using analysis software CTAn (SkyScan). Scaffold porosity was obtained by using binary masks at a threshold of −384 HU. The ratio of pore volume (PV) to scaffold volume yields the percentage of total porosity (Ptot). Ptot in native pNSC discs was then compared with Ptot in processed pNSC scaffolds.
Amount of denatured collagen wD assessed by the hydroxyproline assay
The amount of denatured collagen wD was determined according to the modified method of Bank et al. 35 The hydroxyproline content in both fractions was measured according to Stegemann and Stalder 36 by a modified method of Berginski. 37 Absorption was measured at 565 nm within 1 h. Samples were extrapolated against hydroxyproline standards (data not shown).
Quantification of sulfated GAGs—the DMMB assay
The amount of sulfated GAGs was determined using the DMMB assay as described previously by Barbosa et al. 38 Absorbance was measured at 656 nm. Sulfated GAG quantities were determined by comparison with calibration curves of standard chondroitin sulfate solutions (data not shown).
Assessment of Galα1,3-Galβ1-4GlcNAc-R (α-Gal epitope) in processed and native pNSC
For assessment of remaining Galα1,3-Galβ1-4GlcNAc-R (α-Gal epitope) in decellularized pNSC, an enzyme-linked immunosorbent assay (ELISA) was performed slightly modified according to Galili et al. 39 and Stone et al. 32 Processed and native pNSC was homogenized in PBS (each 400 mg mL−1). Aliquots (100 μL) of homogenized samples were diluted with a blocking solution (1% bovine serum albumin in PBS) in different concentrations. Different cell concentrations of the fibroblast-like fibrosarcoma cell line L929 (DMSZ No. ACC 2) (see Cell culture for in vitro cytotoxicity assessment section) resuspended in PBS were used as control. Each aliquot was mixed with an equal volume of M86 anti-Gal antibody (Enzo; dilution 1:100) and incubated overnight at 4°C with a constant rotation. Samples were centrifuged at 14.000 g for 5 min, and the pellet was discarded, whereas the supernatants (sample solutions) were used for further experiments.
Microtiter plates (Nunc Maxisorb) were coated with 50 μL of α-Gal epitope (alpha-Gal; Dextra; 5 μg mL−1 in bicarbonate/carbonate buffer [200 mM]) each well and incubated at 4°C overnight. Subsequently, plates were washed twice with PBS containing Tween20 (0.01%). Remaining protein-binding sites were blocked by adding the blocking solution (200 μL). After 2-h incubation, at 37°C, plates were washed twice, and the sample solution (50 μL) was added to each well. The sealed plates were incubated for another 1 h at RT under continuous shaking. Coated plates were washed three times, whereupon the secondary antibody solution (Goat anti-mouse IgM, 1:1000, HRP-conjugate; Enzo) was added and an incubation step (1 h, RT) enclosed. To detect antibody binding, a TMB substrate reagent (BioLegend) was added and incubated for 20 min at RT. The reaction was stopped by adding H2SO4 (50 μL, 2 M), and absorbance was measured at 450 nm.
Microbiological evaluation
To verify the bactericide and fungicide effect of the developed DP, microbiological analysis according to ISO 11737-1:2006+AC: 200940 was established. The presence of germs or fungi on native as well as processed scaffolds was determined by using three different types of culture media. Blood agar plates (Merck) were used for detection of fastidious organism and differentiation of bacteria based on their hemolytic properties, Standard-I agar plates (Merck) for exigent germs, and Sabouraud agar plates (4%, Merck) for detection of fungi.
Ten grams of each cartilage type (pMC, pNSC, native, and processed, each n=4) was minced in pieces and incubated in 10 mL sterile 0.9% NaCl solution for 10 min on an orbital shaker, to rinse off and collect the adherent germs. Serial dilution (100, 10−2, and 10−4) of the NaCl solution was performed, and 100 μL of each dilution were plated in triplets on the different culture medium plates. All plates were incubated for 48 h at 37°C. After incubation, the number of colony-forming units per gram cartilage (cfu g−1) was calculated.
Cell isolation and culture
hPChs were isolated from hNSC samples. The cartilage was rinsed in a culture medium, minced, transferred to a digestion medium, culture medium containing collagenase type II (0.3%; Worthington), and incubated for 16 h at 37°C in a shaking water bath. Cells were pelleted by centrifugation, and the total cell number and vitality were determined. hPChs were seeded for amplification with a density of 0.5×104 cells cm2.
When reaching 80%–90% confluence, cells were detached, counted, and cryopreserved to ensure that all hPChs harvested from different patients were treated equally, and the differentiation status of cells was maintained. Cells were not passaged during the expansion period.
Cytotoxicity testing
Cell culture for in vitro cytotoxicity assessment
hPChs and the murine fibrosarcoma cell line L929 were used to determine cytotoxic effects.
For in vitro cytotoxicity assessment, hPChs were thawed, seeded with a density of 0.5×104 cells cm2, and cultured in a culture medium until 80%–90% confluence was reached.
Cryopreserved L929 cells were seeded with a density of 1.0×104 cells cm2 in cell culture flasks and cultured in an RPMI 1640 medium, supplemented with FBS (10%), penicillin (100 U mL−1), and streptomycin (100 U mL−1), until 80%–90% confluence at 37°C and 5% CO2. The medium was changed three times a week.
Preparation of liquid extracts of processed cartilage matrix
To determine cytotoxic effects due to DP, the matrix was examined using a test method for in vitro cytotoxicity for biological evaluation of medical devices, according to the international standard ISO 10993-5:2009 (E). 41 Liquid extracts of processed pMC, pNSC, and hNSC matrices were prepared for each cell type using the respective culture medium as an extraction medium. Sterile scaffolds were incubated in the extraction medium for 24 h at 37°C and 5% CO2 under aseptic conditions using sterile, chemically inert cell culture plates. The culture medium was used as an extraction vehicle because of its potential to extract polar and nonpolar substances.
Cytotoxicity test using L929 cells and hPChs
Except for the respectively used cultivation medium, the test procedures were performed identically for both cell types. Amplified cells were detached, seeded in 96-well cell culture plates with a density of 1×104 cells per well, and incubated for 24 h at 37°C and 5% CO2 to allow cell adherence. The medium was removed, and cells were incubated in extract or control solutions (100 μL) for 24 h at 37°C and 5% CO2. DMSO (10%) in the cultivation medium served as positive control while using ThinCert™ PET membranes (3 μm; Greiner Bio-one) as negative control.
The medium was changed completely, and the MTS solution (20 μL; Promega) was added to each well. Cells were cultured for 2 h under standard culture conditions, and absorbance was measured photometrically at a wavelength of 490 nm.
Seeding and 3D culture on processed pNSC
For scaffold seeding, cells were thawed and cultured until 80%–90% confluence in a monolayer culture for 4 days. Before seeding, pNSC was sterilized in 80% ethanol for up to 2 h at RT to exclude contaminations caused by shipping and thawing. To adjust the pH value and rehydrate the scaffold matrix, pNSC scaffolds were transferred into 24-well plates and incubated in an expansion medium for 24 h at 37°C and 5% CO2. The medium was removed, and scaffolds were seeded with 1×106 cells per scaffold in an expansion medium. To enable cell adhesion, seeded scaffolds were incubated for 1 h at standard culture conditions. The 3D culture was performed by using a chondrocyte differentiation medium (NH Chondro Diff medium; Miltenyi) supplemented with 0.5% gentamicin. Seeded scaffolds were transferred into new wells and were cultured at 37°C and 5% CO2. The medium was changed three times a week. Scaffolds were analyzed on day 7 and 21. For all seeding experiments, cells in passage 1 were used.
Histological and immunohistochemical analysis
Cryo-, as well as paraffin, sections (3–5 μm) were performed for histological and immunohistochemical evaluation of native, processed, and seeded tissue scaffolds. Before staining, scaffolds were rehydrated using a decreasing alcohol series, starting with xylol. To assess cell morphology, cell distribution, as well as presence of GAGs, hematoxylin and eosin (H&E) and Alcian blue (AB) staining were performed.
Immunohistochemical detection of newly synthesized aggrecan in seeded pNSC scaffolds was performed by using LSAB+System-HRP (Dako), according to the manufacturer's instructions. The primary antibody for aggrecan (Serotec) was added to the tissue sections and incubated for 2 h in a humified box. Subsequently, slides were rinsed, and the secondary antibody was added for 20 min.
Mechanical testing
To assess biomechanical properties of native and processed (each n=12) pNSC scaffolds, a uniaxial confined compression test was performed. The linear modulus of pNSC samples was examined comparatively biomechanically. Before the confined compression test, the scaffolds were equilibrated in 0.9% NaCl up to RT. The exact scaffold height measurement and the confined compression test were carried out in a standard material testing machine (Z010; Zwick GmbH) using a 40 N load cell. Samples were placed between two porous aluminum oxide (Al2O3) ceramic cylinders and transferred to the confining compression chamber containing 0.9% NaCl. The loading speed was set to 100% compression/30 min, whereas the preload was set to 0.1 N. The test was aborted when a maximum load of 40 N or 50% height compression of the samples was reached. The linear modulus was determined using testXpert V12.2 (Zwick GmbH).
Statistical evaluation
Statistical analysis was conducted using SigmaPlot® 11.2 software (Systat Software GmbH). The relative percentage of wD and wG of all examined samples is reported as mean±standard deviation (SD), with 0.95 confidence interval given in brackets. Student's t-test and Mann–Whitney U test were performed to determine significance (level of significance α=0.05) of the influence of DP on content of denatured collagen, GAG content, matrix porosity Ptot, D-periodicity of collagen fibrils, as well as on biomechanical properties. The Kruskal–Wallis one-way analysis of variance on ranks was used for evaluation of significance (level of significance α=0.05) for evaluation of in vitro cytotoxicity tests.
Results
Successful decellularization and sterilization
The aim of the DP was to gain highly pure, 3D chondroconductive collagen scaffolds. The tissue samples were derived from different species and types of cartilage tissue (Fig. 1). Starting DP, tissue samples were osmotically pretreated with H2Odeion and subsequently underwent alkaline hydrolysis to remove all lipids as well as proteins and inactivate pathogenic agents like PrPsc or HI virus. By using GndHCl, the 3D structure of macromolecules, for example, proteins, DNA or RNA, was denatured, and GAG extracted. At least, one further inactivation step was performed by using H2O2. pMC was additionally treated a second time with NaOH. Several tissues (pMC, pNSC, and hNSC) (Fig. 1) were decellularized and sterilized by the described DP. Macroscopically, a whitening of all cartilage tissues as a result of the DP was visible. This change of color is most likely due to the removal of all noncollagenous tissue components, blood, and cells.
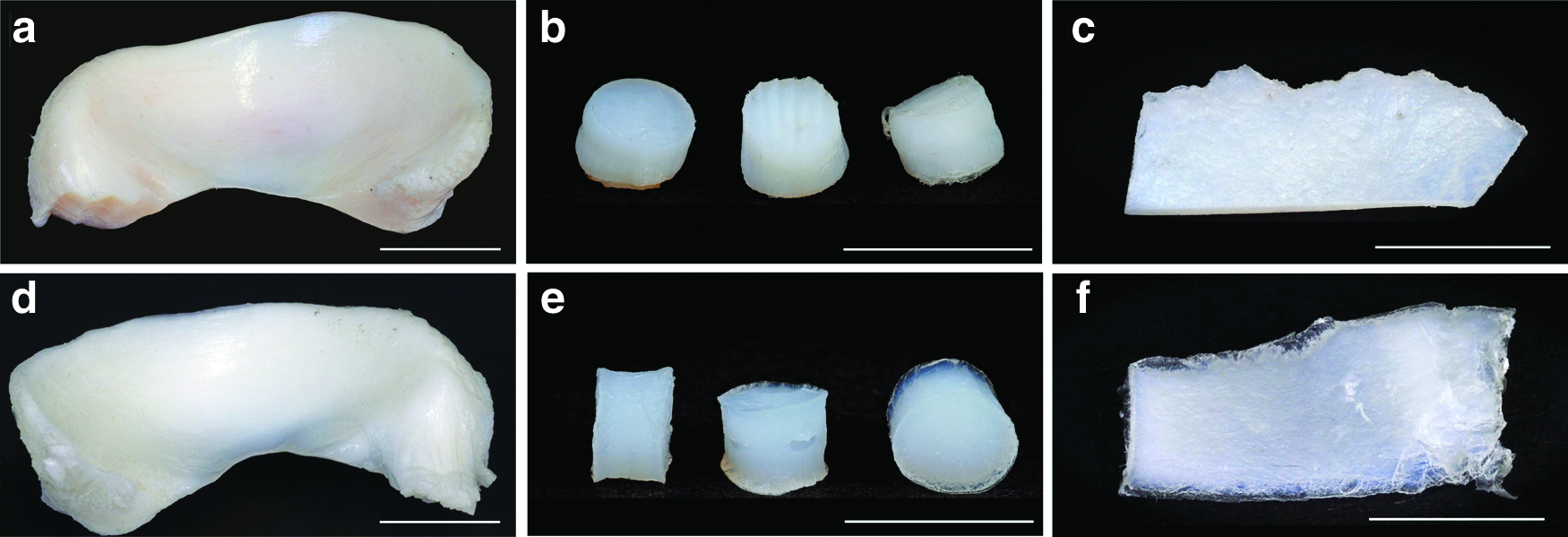
Tissue samples were derived from different species and types of cartilage tissue. Porcine meniscus
On native pNSC, 8.6×102±1.1×102cfu g−1 gamma-hemolytic and 1.91×103±0.59×103cfu g−1 exigent microorganisms, as well as 3.34×102±0.21×102cfu g−1 fungi, were detectable. About 0.67×102±0.25×102cfu g−1 gamma-hemolytic and 0.86×102±0.22×102cfu g−1 exigent microorganisms, but no germs belonging to the class of fungi, were found on native pMC. In contrast, no microorganisms or fungi were detected on processed pMC or pNSC.
Microscopic morphology
H&E staining was performed to investigate the cartilage structure and cell distribution within the different cartilage tissues. These stainings demonstrated that cells were removed completely from cartilage tissue by the DP (Fig. 2). Lacunae of all treated cartilage samples were empty, and no cell nuclei were left. In native pMC, pNSC, and hNSC, cell nuclei are stained in dark blue and are easily recognizable in the red-stained collagen matrix. AB is a cationic phthalocyanine dye that contains copper and binds stoichiometrically to acidic, sulfated GAGs like keratin and chondroitin sulfate. 42 Therefore, the intensity of staining allows an estimation of the amount of present GAGs in the matrix. As seen in Figure 3, the histological staining of native, unprocessed cartilage demonstrates a very intense blue staining of the matrix of all examined cartilage types, pMC, pNSC, as well as hNSC. Tissue sections of processed cartilage provide only a slight light blue staining. The matrix in fact is nearly clear, evidencing almost complete removal of GAGs. In electron microscopy and microcomputed tomography analysis section, quantitative results obtained by DMMB assay further detail these findings.

To assess morphology and distribution of chondrocytes of native and processed cartilage tissues [pMC
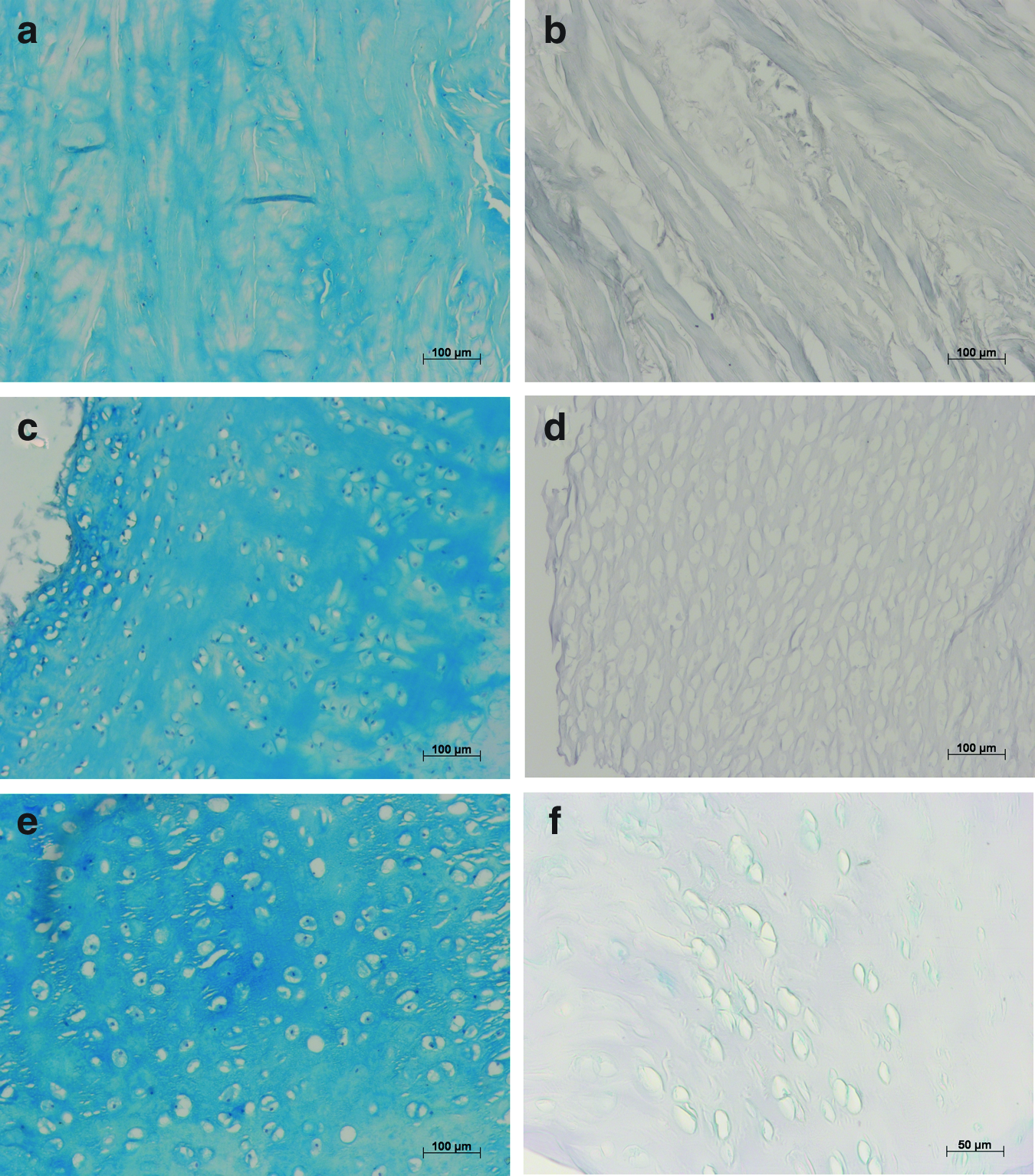
Presence of glycosaminoglycans (GAGs) in native, unprocessed pMC
DP increases scaffold porosity, but maintains collagen structure
SEM analysis of native pMC reflects the unique organization of the collagen fibers in meniscus cartilage. The fiber orientation varies according to the location within the meniscus. The examined vertical sections from native pMC demonstrate cross-sections of densely parallel-arranged and circumferentially orientated large fiber bundles and a small number of radial collagen fibers (Fig. 4a).
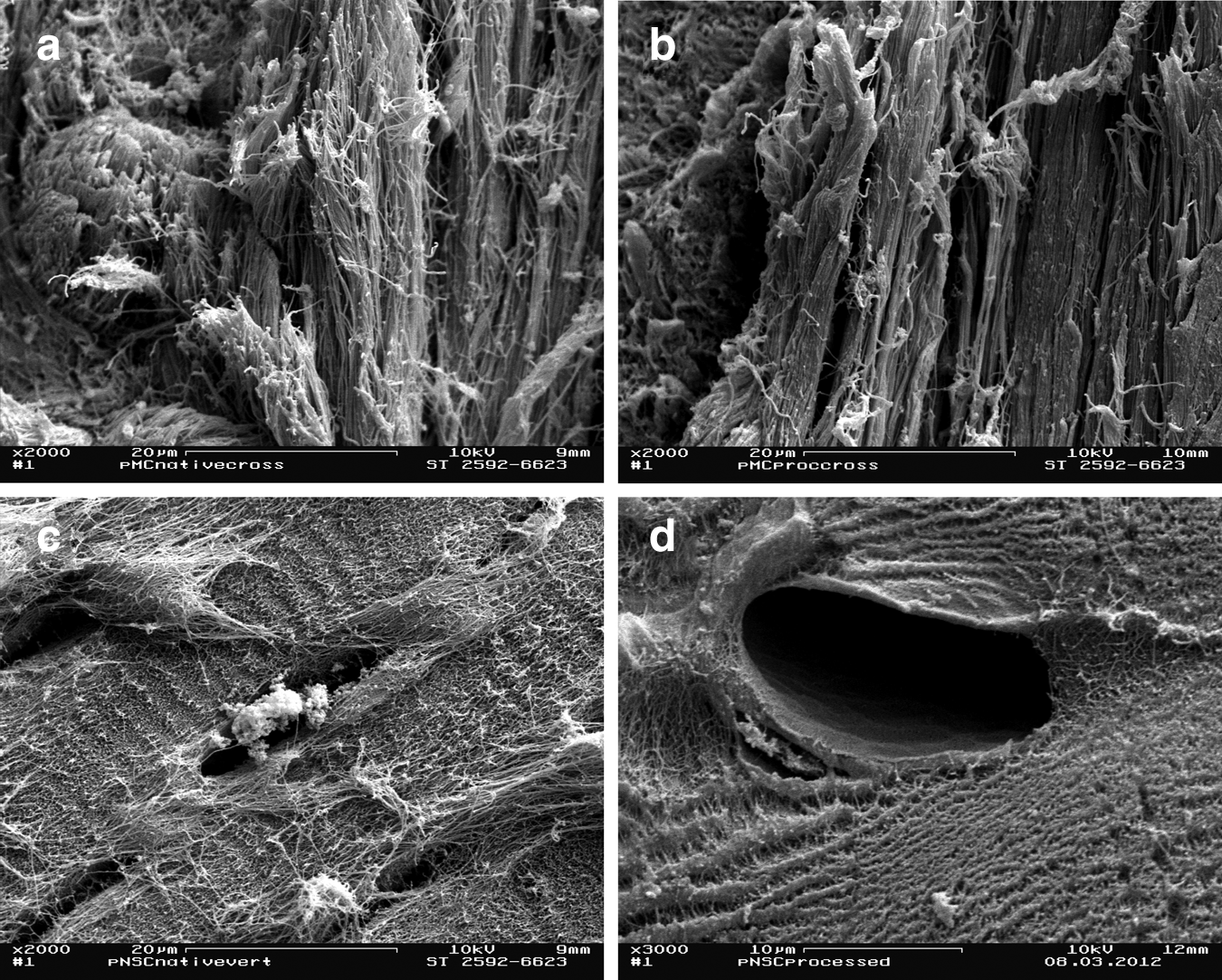
Scanning electron microscopy analyses of pMC
SEM images of native pNSC demonstrate a fine, interwoven network of collagen type II fibers. In native pNSC, single chondrocytes occupy all lacunae (Fig. 4c).
SEM analyses of processed pMC (Fig. 4b) and pNSC (Fig. 4d) visualize the capability of DP, to maintain the 3D arrangement of collagen fiber organization and to remove all cells. All empty lacunae in processed pNSC demonstrated a diameter of 10–40 μm.
The most characteristic feature of both, collagen type I, as well as type II, and the D-periodic, banded structure of collagen fibrils was observed in TEM for native and processed pMC, as well as pNSC. In native pMC (Fig. 5a), the collagen fibrils demonstrated a periodicity with D=63.96±6.42 nm, and in processed pMC (Fig. 5b) with D=64.29±3.75 nm. Comparison of the results of native (D=65.10±6.08 nm) (Fig. 5c) and processed (D=65.55±2.66 nm) (Fig. 5d) pNSC reveals that there is no significant difference between the D-periodicity of native and processed collagenous tissue (pMC p=0.979; pNSC p=0.781). The results of the TEM analysis confirm that, applying the chemical DP, no changes in D-periodic structure of collagen fibrils in pMC or pNSC occur. The respective gap/overlap ratio of collagen type I and collagen type II fibers was maintained.

The characteristic D-periodic, banded structure of native collagen type I and type II fibrils with D=67 nm in hydrated or native collagen,57 and 64–65-nm long in dehydrated, fixed samples,58 examined by transmission electron microscopy (TEM) for native and processed cartilage tissues. No statistical significant differences of D-periodicity in collagen type I fibrils of native pMC
As a consequence of DP, total matrix porosity Ptot of pNSC increases significantly. In native pNSC, the basic Ptot amount was 22.20%±10.83%. In consequence of cell removal and reduction of GAGs, caused by the applied chemical treatment, processed pNSC possessed a Ptot of 44.14%±6.84% (p≤0.001). In pMC, the porosity increased clearly from 19.01%±12.18% in native samples to 28.93%±12.67% in processed scaffolds.
The amount of denatured collagen wD increases during DP
One of the major aims of the DP was to maintain in pMC, pNSC, and hNSC the 3D collagen structure and to avoid collagen denaturation to preserve the original mechanical matrix properties and the stability of the tissue as far as possible. Thus, it was essential to determine wD as a basic quality criterion of the scaffolds. wD of the scaffolds was compared to native tissue as reference. Each group consisted of 14–19 samples.
One interesting result was that native cartilage tissue had relatively high basic values for wD. In native hNSC (donor age ranged from 16 to 55 years), the basic wD amount was 18.56%±4.39% (16.02; 21.10) (Fig. 6), whereas the wD values for pNSC had even a higher levels of wD, around 25.29%±8.05% (20.64; 29.93) (Fig. 6). The results of the hydroxyproline assay reveal a significant increase of wD due to chemical treatment of the cartilage tissues in pNSC as well as in hNSC (Fig. 6). After DP, 52.8%±9.9% (47.73; 57.91) of the pNSC collagen network was affected, whereas in hNSC, wD demonstrated a damage of 42.98%±6.76% (39.38; 46.59). The comparison of wD values of processed (4.89%±1.66% (4.10; 5.70) and native (4.06%±0.98% [3.56; 4.57]) pMC reveals that in 3D ECM scaffolds of fibrous pMC, the effect of chemical decellularization treatment is not so drastic. The denaturation level of the collagen network in this case seems to be of minor importance, as no significant increase in content of wD of pMC could be detected.
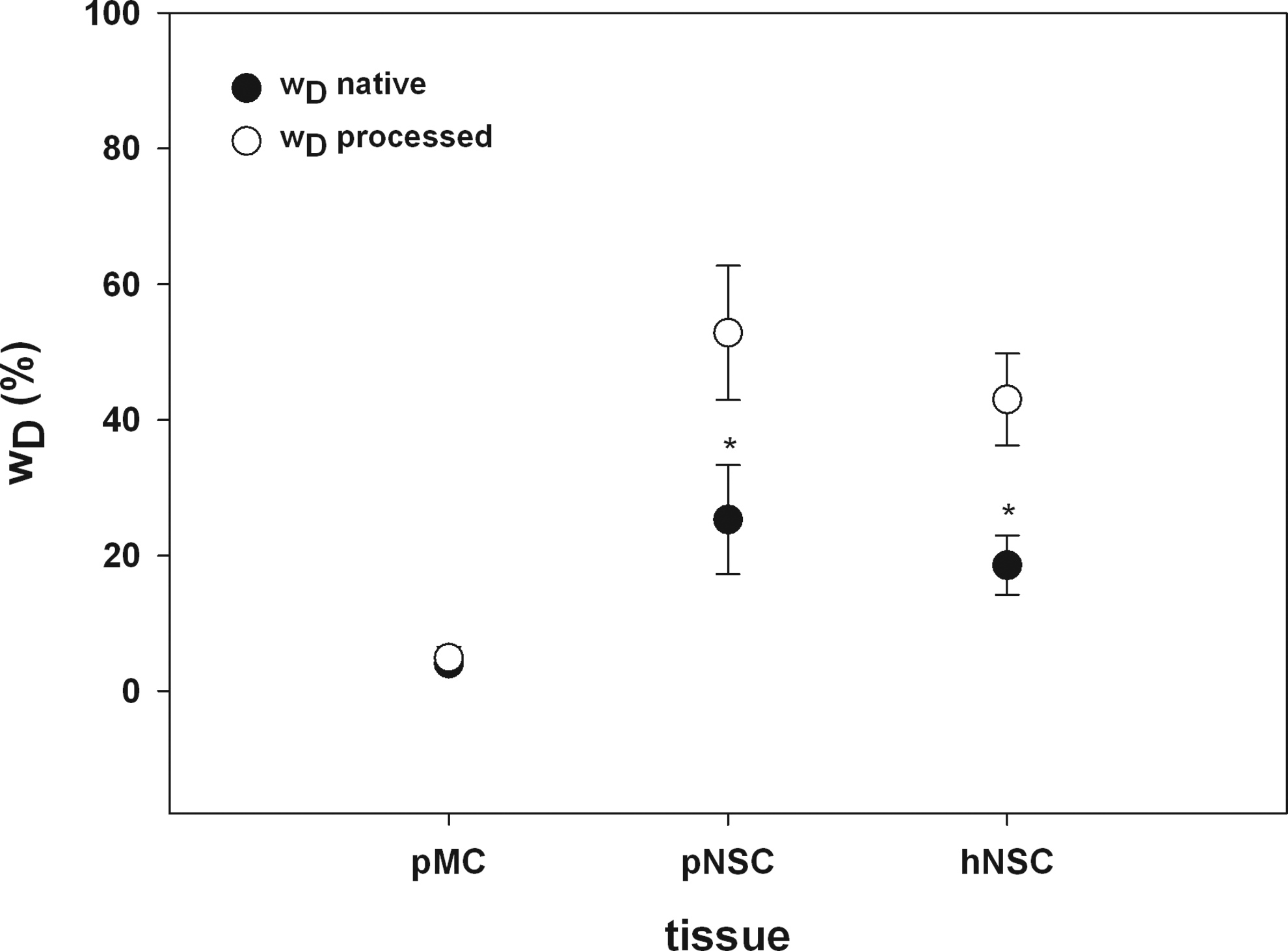
Determination of wD as a basic quality criterion of DP-treated collagenous scaffolds. The amount of denatured collagen wD increases significantly in processed pNSC and hNSC (white circles) compared to native cartilage tissues (black circles). No changes of wD were found for pMC. For preserving the mechanical properties of the biomatrices, maintenance of the 3D collagen structure and minimizing collagen denaturation are essential (*p<0.001; Student's t-test; n=14–19).
Reduction of sulfated GAGs due to DP
Reduction of sulfated GAGs was an important step to increase porosity and to enable migration of cells into the scaffolds. Each sample group contained 16–23 samples.
Figure 7 demonstrates the results of the DMMB assay to quantify GAG content of processed pMC, pNSC, and hNSC in comparison with the native cartilage tissues. In the case of pMC, the average wG in native tissue accounts for 0.92%±0.64% (0.59; 1.25) of the dry weight. The high SD can be traced back to the fact of heterogeneous allocation of GAGs in native pMC and all other cartilage tissues. After chemical treatment of the porcine collagenous ECM meniscus matrix, wG was decreased to 0.42%±0.06% (0.39; 0.46). In native pNSC, a GAG content of 4.82%±0.94% (4.31; 5.32) per dry weight was measured and was significantly reduced to 1.50%±0.43% (1.315; 1.68) during the production process. As a consequence of DP, the content of GAG in hNSC decreased significantly from 7.17%±0.89% (6.73; 7.61) to 2.44%±0.89% (2.00; 2.89). For all examined cartilage tissues, the wG was reduced about 55%–69%.
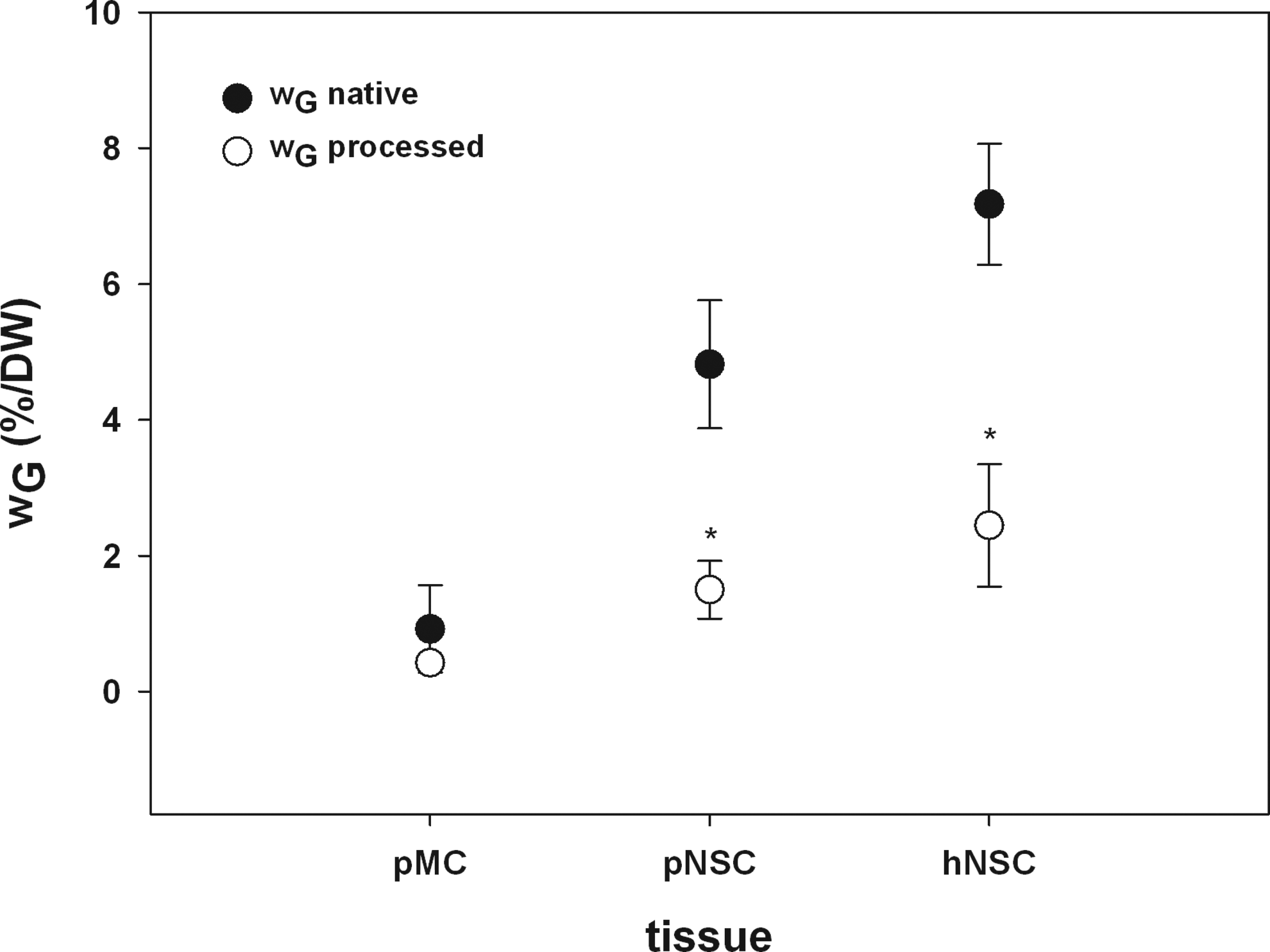
Results for quantification of sulfated GAGs (wG) using the DMMB assay of native pMC, pNSC, and hNSC (black circles) compared to the processed cartilage tissues (white circles). The results of DMMB assay support the TEM and microcomputed tomography analysis and demonstrated a significant reduction of wG in pNSC and hNSC. In pMC, no significant enhancement of wG was detectable. The high standard deviation can be traced back to the fact of heterogeneous allocation of GAGs in native pMC and all other cartilage tissues (*p<0.001; Mann–Whitney U test; n=16–23).
DP eliminates Galα1,3-Galβ1-4GlcNAc-R (α-Gal epitope)
Detection and quantification of Galα1,3-Galβ1-4GlcNAc-R (α-Gal epitope) in native (black circles, Fig. 8) and processed (white circles, Fig. 8) pNSC were compared to different cell concentrations of the murine fibrosarcoma cell line L929 (black squares). Data of ELISA inhibition assay are shown as % inhibition of M86 binding to α-Gal epitope. As shown in Figure 9, M86 is bound to L929, whereas 50% inhibition is demonstrated by a cell number of ∼2.5×105 cells. Furthermore, M86 binding and a similar inhibition could be detected in native pNSC at a tissue concentration of 250 mg mL−. Decellularization and sterilization of pNSC result in complete elimination of α-Gal epitopes, indicated by the lack of inhibition reactions. No interaction between M86 and homogenized tissue was detectable even at higher cartilage concentrations like 200 and 400 mg mL−1 (Fig. 8). These findings imply that α-Gal epitopes are removed completely from the porcine cartilage matrix.

Detection and quantification of Galα1,3-Galβ1-4GlcNAc-R (α-Gal epitope) in L929 (black squares) compared to native (black circles) and processed (white circles) pNSC. Data of enzyme-linked immunosorbent assay (ELISA) inhibition assay are shown as % inhibition of M86 binding to α-Gal epitope. About 50% inhibition is demonstrated by a cell number of ∼2.5×105 cells. A similar inhibition was detectable at a tissue concentration of 250 mg mL

In consequence of DP, the linear modulus of the scaffold matrix significantly decreased in processed pNSC tissue compared to native tissue (*p<0.001).
Biomechanical loading capacity decreases
The linear modulus of the scaffolds decreased significantly from 6.5 MPa±2.3 in native pNSC to 1.92±0.85 MPa in processed tissue (Fig. 9, p<0.05). Thus, the matrix stiffness is reduced about 69.5%.
Cytotoxicity
Processed pMC, pNSC, and hNSC matrices were examined to exclude any traces of cytotoxic components due to the chemical DP, possibly leading to changes in morphology, vitality, metabolism, and growth behavior of cells. Each sample group contained 12–32 samples.
For both hPChs and L929, no significant influence on cell growth and metabolism was detected, whether grown in an undiluted extract of pMC, pNSC, or hNSC (Fig. 10). Cell shape, growth, and metabolism of each cell type were unchanged compared to respective negative (noncytotoxic) controls ThinCert PET membranes. Cells incubated in a positive control, 10% DMSO solution in a respective culture medium, reflected the cytotoxic effects of DMSO on growth and vitality.
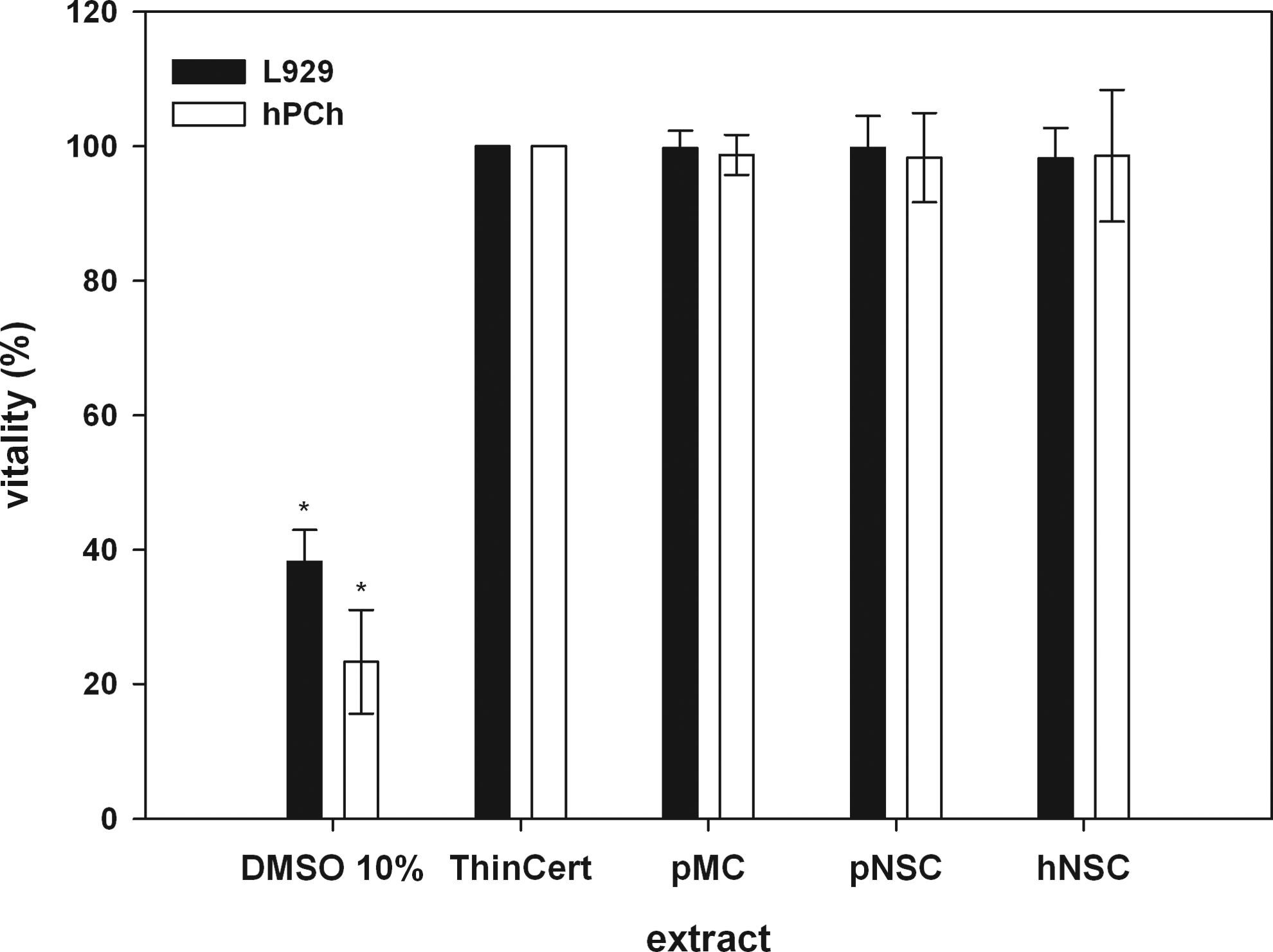
In vitro cytotoxicity test for biological evaluation of medical devices according to international standard ISO 10993-5:2009 (E) of processed pMC, pNSC, and hNSC. L929 (black) and human primary nasal chondrocytes (hPChs) (white) were used as indicator cells for determination of possible cytotoxic effects. Cells of both cell types, cultured in negative control extract as well as in undiluted sample extracts, demonstrate a high viability and reflect the noncytotoxic effect of the extracted processed xenogenic cartilage matrices. No significant differences between negative control and extracts were detectable. Compared to these results, the cytotoxic effect of the 10% DMSO solution, used as positive control, is significant (*p<0.001).
A significant difference of cell vitality in comparison to negative controls as well as all cultures in extracts was detected. The viability of hPChs cultured in 10% DMSO solution decreased to 23.31%±7.72%, while the same cells cultured as negative control were 100% viable.
For L929, a higher percentage of viable cells were detected in the positive control (38.20%±4.72%). According to the ISO guidelines, cell viabilities in the range between 0% and 40% reflect a strong cytotoxic effect of the tested extract.
Vitality of hPChs seeded in a pNSC extract (98.30%±6.64%) was unchanged as compared to negative controls. The same observation was made for hPChs cultured in pMC and hNSC extracts. Cell viability of hPChs seeded in the pMC extract was 98.71%±2.98%, and in hNSC extract, 98.56%±9.79%.
In vitro cytotoxicity tests applying L929 in the pMC (99.77%±2.52%), pNSC (99.76%±4.75%), or hNSC (98.20%±4.51%) extract revealed no cytotoxic effects on cells. No significant difference in cell vitality of both groups compared to negative controls was detectable. Data analysis was performed following ISO guidelines, whereas the values for cell vitality of positive control, as well as tested extracts, were referred to negative control.41,43
For all in vitro cytotoxicity tests, a significant difference of cell viability compared to cytotoxic controls was detected (Fig. 10). The vitality of cells cultured in matrix extracts and the respective diluted solutions was above 90%. According to ISO guidelines, cell viabilities between 70% and 100% prove the absence of cytotoxic components in matrix extracts.
hPChs adhere to biomatrices and produce cartilage-specific ECM
Processed xenogenic pNSC scaffolds were seeded with hPChs and cultured for 7 and 21 days with the influence of chondrogenic growth factors. During amplification in the monolayer culture, hPChs assumed a branching, elongated, and spindle-shaped morphology, which has been described earlier. 44 After seeding, cells adhered to processed pNSC surfaces, and within 7 days, cells established a tight cell layer on the scaffold surface. No inhibition areas around the scaffolds were detectable.
AB staining of seeded pNSC scaffolds demonstrated an increasing GAG accumulation during cultivation by enhanced blue staining, first, in the hPCh layer on the construct surface and subsequently, within the superficial zones of the scaffold (Fig. 11). GAG accumulation increased visibly from day 7 (Fig. 11a) to day 21 (Fig. 11b) and spread to deeper construct zones. These results were confirmed by immunohistochemical staining for the cartilage-specific marker aggrecan (Fig. 11c). The distribution of counterstained cell nuclei evidenced that cells not only adhered to the biomatrix but also started to migrate into the matrix during cultivation.
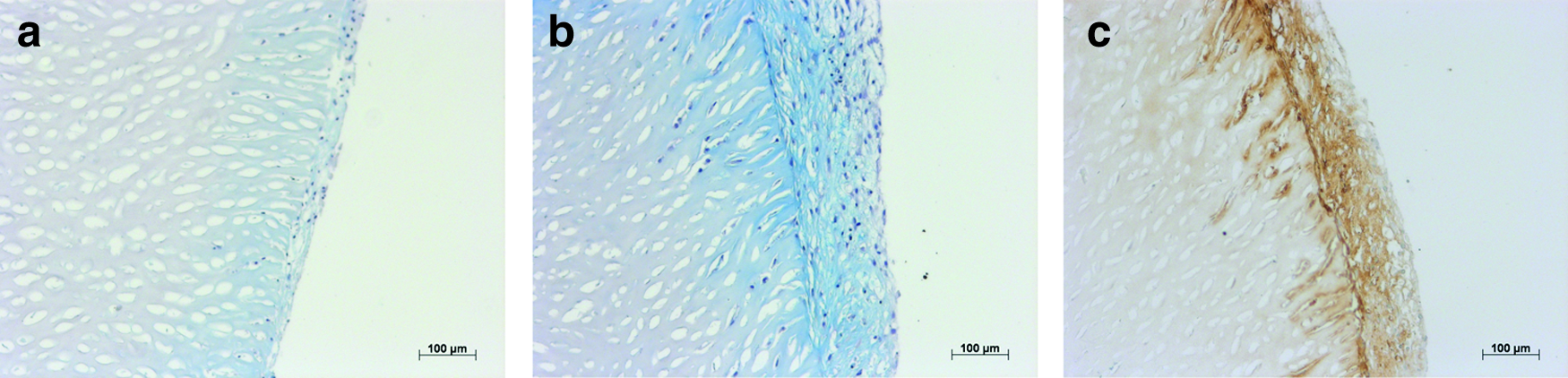
AB staining of seeded pNSC scaffolds after 7
Discussion
Today, in different clinical fields, especially in otorhinolaryngology and orthopedics, replacement and reconstruction of cartilage tissue defects are, due to the lack of healing capacity of cartilage and an increasingly aging population, of outstanding concern.1,9 Because there is a persistent demand for adequate replacement matrices, 45 the specific aim of this study was to create a new chondroconductive cartilage replacement matrix on the basis of xenogenic and allogeneic cartilage specimens. Material properties, biochemical stability and integrity, as well as composition of the native collagen network consisting of fibers and fibrils should be affected as little as possible. Furthermore, the created implant matrices should serve as scaffolds for cell adhesion, proliferation, and migration into the matrix, as well as neosynthesis of ECM components. The challenge of this project was the simultaneous elimination of all components possibly causing immune as well as rejection reactions and at the same time, minimizing the risk for disease transmission 31 while maintaining matrix integrity.
Shape and size of processed cartilage scaffolds were maintained during the purification process as evaluated by measuring pMC and pNSC discs, before and after DP (data not shown), but the fraction of denatured collagen is clearly increased in hNSC and pNSC mainly caused by 1 N NaOH solution, which is necessary for inactivation of PrP.Sc24–26 Additionally, treatment of cartilage with NaOH leads to a destruction of cellular components and hydrolysis of GAGs. 46 The degree of denaturation caused by NaOH treatment depends on the applied concentration of NaOH solution and time of exposure.47–49 Hydroxide ions form ionic and nonionic bonding to collagen and induce tissue swelling, as well as lyotropic effects, and subsequently, the hydrolysis of collagen due to the breakdown of intermolecular bonding and release of further hydroxide ions. 48 Applied chaotropic salt and hydrogen peroxide solutions have an inconsiderable influence on the amount of denatured collagen, as own unpublished examinations have shown. While most of the contained proteins of cartilage are destroyed hydrolytically by this treatment, the triple helical conformation of collagen is not disturbed because of the relatively short incubation period used in the process presented here. 47 The consequences of collagen fragmentation caused by alkaline treatment combined with the effects of H2O2 treatment result in the measured and demonstrated an increased percentage of denatured collagen in processed cartilage compared to native cartilage.
Regarding the amount of denatured collagen, the results show that fibrous cartilage is very stable, and drastic DP conditions seem to have no measurable effect on the collagen network. Compared to these findings, hyaline cartilage shows stronger impairment of the 3D collagenous matrix, whereas it must be considered that in the native state of pNSC and hNSC, the measurements demonstrated high basic wD values. From our point of view, the high wD values in native tissue may be a consequence of the pretreatment, according to Bank et al.,35,50,51 whereas the proteoglycans of native tissue have to be removed to guarantee optimal enzyme access to differentiate between intact and denatured collagen. Furthermore, in pNSC, the high wD may be additionally increased because of 3–4 min of scalding of the animal during slaughter. We assume that the different resistance to the used chemical agents during DP could be explained by different function and composition of collagen type I and II of processed tissues. Although both collagen types belong to the group of fibril-forming collagens, there is a huge difference between them. Type I collagen, as found in meniscus cartilage, forms strong and stable fibrils and fibers that are highly cross linked to fulfill the role as a stabilizing, load transmission, and attenuation structure in the knee joint (Fig. 4a).52–55 In contrast to meniscus, type II collagen in septal cartilage is much more delicate, with smaller fibrils and fibers (Fig. 4c). Here, the main function of collagen is to act as a stiff antagonist to the GAG–water gel, which is responsible for the elasticity of the tissue. 56 Based on these two differences in the collagen structure, we assume that the reason for the higher wD in septal cartilage is the more delicate type II fibrils. This structure results in a larger surface area, and therefore the NaOH solution can affect type II collagen to a larger extent than type I collagen with its bigger fibrils.
TEM images of collagen type I (pMC) and II (pNSC) fibrils from native and processed cartilage tissues demonstrate the D-periodic structure of both collagen types (Fig. 5). The Hodge-Petruska D-periodic fibril-packing structure, which is known to be 67-nm long in hydrated samples or native collagen 57 and 64–65-nm long in dehydrated, fixed samples, 58 was maintained during DP. The overlap/gap ratio of collagen type II and collagen type I was not affected by the chemical decellularization and sterilization. The overlap/gap ratio for type II collagen, previously suggested to be closer to 0.4D/0.6D than type I, 58 was also confirmed by our TEM data.
Despite the negative effects of the strong alkaline solution, NaOH solution is necessary not only for inactivation of PrPSc24–26 but also to guarantee complete decellularization to exclude possible immune reactions mediated by cell surface structures like the oligosaccharide α-Gal (Galα1,3-Galβ1-4GlcNAc-R) (α-Gal epitope) 31 in xenogenic tissue grafts.
For further quality control, it was necessary to determine the amount of sulfated GAGs in native and decellularized tissue with the assumption that not only GAGs are removed but also the complete proteoglycan network is removed. We were able to demonstrate that the main part of GAGs was removed from the cartilage matrix during DP, whereas in septal cartilage, wG was diminished about 70% and in the fibrous cartilage matrix of pMC about 50%. This, in addition to cell removal, leads to a higher porosity of the scaffold matrix. Regarding the results presented here, the amount of remaining GAGs has no cytotoxic effect (Fig. 10) or other negative influence on the matrix quality.
As location within the tissue depends on the age of the host and microenvironment, 59 this issue should be regarded during scaffold production using xenogenic and allogeneic cartilage tissues. Furthermore, several properties of GAGs, like promoting water retention, binding growth factors, and cytokines, as well as contributing to the gel properties of the ECM, lead to the assumption that the remaining part of GAGs after DP enhances biocompatibility,38,59,60 rather than having negative effects on scaffold properties. Possible remaining heparin-rich GAGs could positively affect cell growth because of heparin-binding properties of various growth factors and surface receptors. Hyaluronic acid, besides chondroitin sulfate, one of the most common GAGs present in cartilaginous ECM, has been used as a bioimplant matrix for tissue reconstruction and as a carrier for selected cell populations in therapeutic tissue-engineering applications. For example, orthopedic products like Synvisc® (Genzyme Corporation, Cambridge, MA) 59 are based on hyaluronic acid.
For in vitro production of functional neocartilage tissue, the induction of substantial neosynthesis of acidic, sulfated GAGs and collagen is required.12,61 The 3D in vitro culture demonstrated that hPChs adhered to the scaffold surface and were able to migrate into the matrix (Fig. 11). The capacity of hPChs to synthesize a new cartilage-specific ECM in a long-term 3D culture was demonstrated by AB and immunohistochemical staining for aggrecan (Fig. 11).
In the last years, a variety of mammalian tissues and organs like urinary bladder, pericardium, small intestines, dermis, and heart valves have been decellularized before their use as ECM bioscaffolds to support reconstruction of defective tissue.62,63 Furthermore, recent studies have shown that ECM bioscaffolds, including structural and functional proteins, as well as 3D organization comparable to the native tissue, provide constructive tissue remodeling instead of scar tissue formation.13,14
The new patented decellularization and sterilization method33,34 presented here is applicable for highly dense tissues, for example, for meniscus and facial cartilage, by maintaining the natural hydration status. Thus, rehydration procedures with the risk of incomplete hydration of the inner regions of scaffold matrix or matrix defect formation due to crystalline water content 64 are avoided. No initial homogenization steps for matrix disintegration and final cross-linking steps known for other cartilage replacement materials 65 or ECM scaffolds are necessary.33,34
An increase in matrix porosity, as a result of DP, takes place, as proved by a μCT analysis. The size of resulting pores in the consequence of cell removal was additionally estimated by light and electron microscopy and is consistent with the dimensions of natural lacunae from ∼20 to 30 μm. 66 The presented implant matrix offers the advantage of a 3D, naturally structured collagen network with an optimal pore size in the range between 15 and 50 μm,56,67 providing ideal cellular microenvironmental conditions to human donor cells. Thus, cell–cell interactions as well as 3D cell attachments, which play a major role in phenotype and morphology expression, 66 are close to the in vivo situation. Pore size and architecture of implant matrices are known to influence diffusivity of nutrients, oxygen, and serum into the matrix.61,68 This might be the reason why the natural alignment of collagen fibrils, fibers, and interspaces facilitate an apparently adequate nutrient supply of seeded and migrated hPChs within the pNSC matrix, as indicated by increased collagen type II and GAG secretion as well as proliferation rate. 69
The increase of matrix porosity, caused by DP, is accompanied by a significant reduction of the linear modulus of xenogenic cartilage matrices. More detailed characterizations of biomechanical properties of pNSC, described by Schwarz et al., 69 confirm that the decreased linear modulus in processed, compared to native pNSC scaffolds seems to provide adequate matrix stiffness, to support redifferentiation of hPChs. After 3D culture for up to 42 days, enhanced GAG secretion led to an increase of biomechanical stability of processed pNSC. 69
The newly developed scaffolds are easy to handle, individually shapeable, and useable as a cell delivery matrix. Encouraging results with various collagen matrices presented within the last years56,70–72 led to the development of our novel natural cartilage-based collagenous scaffolds. One outstanding characteristic is that we have no evidence of shrinkage of these scaffolds—neither during DP nor during storage and cell culture over at least 42 days (results not shown, report in progress)—as described frequently when using collagen type I or II gels or scaffolds,56,73 thus envisioning a successful in vivo application.
The presented method is carried out with standard chemicals and without any steps of enzymatic digestion, enabling a cost-effective and relatively simple production compared to other decellularization procedures using DNase/RNase, 31 pepsin, 65 or papain degradation. 62 No expensive recombinant enzymes have to be used, 74 and the risk of inhomogeneous enzyme diffusion or inappropriate preparation of digestion dilution during matrix processing is avoided.
The existence of α-Gal epitopes and their effects on immune responses were examined in biological implant matrices composed of xenogenic ECM, like standard decellularized bovine pericardium, 31 native xenograft meniscus grafts, 17 or ECM scaffold materials derived from porcine small intestine submucosal ECM, porcine bioprosthetic heart valves, 75 porcine anterior cruciate ligament, and cartilage.32,74,76,77 Thereby, the interaction of natural human anti-Gal antibody with these membrane-bound carbohydrate structures is presumed to be a major obstacle in xenotransplantation. 39 The risk of exposing patients to the danger of antibody-mediated hyperacute and chronic rejection responses known from transplantations of discordant xenograft transplantations17,32,78 is successfully excluded by complete removal of these membrane glycoproteins from porcine cartilage by applying DP (Fig. 8). Additionally, the developed DP provides sterility and complete removal of germs as evaluated according to the guidelines of ISO 11737-1:2006–2008. 40
The application of glutaraldehyde was avoided in our process to exclude cell toxic effects due to glutaraldehyde residues following washing steps and the risk of shrinkage as has been described for glutaraldehyde-preserved meniscus implants. 79 The matrix gained through DP is sterile, noncytotoxic, free of cells, retains a low GAG content, and provides pore sizes close to original pNSC cartilage The almost intact collagen demonstrates a high mechanical stability and durability while maintaining the original shape and size. Methods known to be mechanically harmful, applied, for example, for urinary bladder matrix and submucosa to delaminate adhesive tissue, 80 were not integrated into the DP to avoid an additional mechanical damage and weakening of the implant matrix.
Applying the presented DP could be the first step toward the use of xenogenic cartilage in larger amounts and would provide cartilage replacement scaffolds from human, porcine, or bovine origin in unlimited availability.
The newly developed DP successfully removes cells and cell membranes, and thus cell surface structures of xenogenic tissues that mediate inflammatory reactions. The risk of disease and germ transmission is eliminated by using solutions like NaOH, alcohol, and H2O2. The amount of denatured collagen is increased, but the remaining intact collagen still provides a suitable stability of the tissue in terms of gross mechanical stability. It was demonstrated that fibrous cartilage is less sensitive to the described DP compared to hyaline septal cartilage. By adaption of the DP to the respective tissue, wD and fraction of removed GAG can be optimized for each tissue type. Bioimplants produced with this newly developed DP can be used as resorbable, chondroconductive, and therefore adequate cartilage replacement materials derived of virtually any cartilage type and species. In particular, by the use of such decellularized xenogenic tissues, such well-known problems as limited availability, donor-site morbidity, encapsulation and extrusion, germ and disease transmission, and immune reactions might no longer be of relevance.
Footnotes
Acknowledgments
The authors wish to thank the DFG (German Research Foundation, BR 3817/1-1 and BU 461/21-1) for funding the project Production of a 3-dimensional chondroconductive collagen matrix made from porcine menisci. Furthermore, the authors thank S. Lessig, M. Jerg, K. Urlbauer, G. Cudek, R. Weih, and E. Schmid for their excellent technical support.
Disclosure Statement
No competing financial interests exist.