
Abstract
This work describes the development of a three-dimensional (3D) model of osteogenesis using mouse preosteoblastic MC3T3-E1 cells and a soft synthetic matrix made out of self-assembling peptide nanofibers. By adjusting the matrix stiffness to very low values (around 120 Pa), cells were found to migrate within the matrix, interact forming a cell–cell network, and create a contracted and stiffer structure. Interestingly, during this process, cells spontaneously upregulate the expression of bone-related proteins such as collagen type I, bone sialoprotein, and osteocalcin, indicating that the 3D environment enhances their osteogenic potential. However, unlike MC3T3-E1 cultures in 2D, the addition of dexamethasone is required to acquire a final mature phenotype characterized by features such as matrix mineralization. Moreover, a slight increase in the hydrogel stiffness (threefold) or the addition of a cell contractility inhibitor (Rho kinase inhibitor) abrogates cell elongation, migration, and 3D culture contraction. However, this mechanical inhibition does not seem to noticeably affect the osteogenic process, at least at early culture times. This 3D bone model intends to emphasize cell–cell interactions, which have a critical role during tissue formation, by using a compliant unrestricted synthetic matrix.
Introduction
Research during the past 15 years has shown that matrix compliance plays a critical role in cellular functions such as spreading, migration, proliferation, differentiation, or abnormal phenotype. 7 In the particular case of osteogenesis, numerous studies on 2D substrates have indicated that stiffness favors the osteogenic differentiation of progenitor cells, although soluble factors are required to synergistically induce a fully developed phenotype.8–10 The translation of these results to a 3D context is more challenging due to the difficulty to find 3D models that allow practical encapsulation, as well as an independent control of the mechanical and biochemical properties. However, recent reports have correlated matrix stiffness and osteogenic potential in 3D matrices, which proved that osteogenesis is enhanced by stiffness in the tested hydrogel systems.11–13
Besides matrix stiffness, cell–cell interaction and communication play a critical role in the proper tissue development and function. In the particular case of bone, cell–cell coupling has been found to be important in mature bone functions, such as bone remodeling. 14 In addition, during bone development, cells aggregate forming highly condensed networks in a process known as mesenchymal condensation, which is mainly controlled by cell–cell interactions. 15 However, in most synthetic hydrogels, cells remain physically entrapped such that spreading, migration, and cellular interconnections are hindered. One usual approach to enable cell adhesion and spreading has been the functionalization of hydrogels with adhesive motifs, such as the integrin-binding RGD peptide.16–18 Nevertheless, the presence of the RGD peptide does not fulfill the expectations in promoting good cell adhesion and spreading in 3D as reported in 2D systems. 19 Progress has been made using more complex strategies such as combining RGD sequences with peptides sensitive to matrix metalloproteinases (MMP) 20 or the incorporation of cell–cell communications cues. 21 In spite of these biochemical modifications, cellular migration and matrix remodeling in 3D are still not optimal.22,23
In the present study, a new strategy was evaluated to develop bone-like structures based on allowing cell–cell interactions in 3D in an effort to recreate the environment during the first stages of bone formation in vivo. Therefore, the working hypothesis was that certain hydrogels—at extremely low compliance—might be able to provide an unrestricted environment, where cells could spread, create cellular connections, condense the matrix, and develop into a bone-like tissue. For this purpose, MC3T3-E1 cells were encapsulated in a soft self-assembling peptide hydrogel—known as RAD16-I and commercially available as BD™ PuraMatrix™—that structurally resembles the extracellular matrix. 24 The sequence of the peptide (AcN-(RADA)4-CONH2) does not provide any recognizable residue for the cells and, thus, it is considered noninstructive. Once in salt-containing solutions, these peptides rapidly self-assemble, thereby creating a nanofiber network with pores around 50–200 nm. Furthermore, this biomaterial has already been proven to support growth and proliferation of a wide variety of cells: chondrocytes, 25 neuronal cells, 26 osteoblasts, 27 endothelial cells,28–30 and hepatocytes,31,32 as well as differentiation of progenitor cells to osteogenic,33,34 neural, 35 and hepatic 36 lineages.
Materials and Methods
Cell expansion
MC3T3-E1 cells (subclone 4) were acquired from ATCC (cat # CRL-2593) and expanded under standard conditions, as indicated below. Reagents were purchased from Invitrogen, unless otherwise stated. Cells were grown on traditional culture flasks (75 cm2 of growth area) with the supplier's recommended medium (control medium), consisting of the alpha minimum essential medium (α-MEM) with ribonucleosides and deoxyribonucleosides, supplemented with 10% (w/v) fetal bovine serum (FBS, Lonza cat # DE14-801F), 2 mM L-glutamine, 1 mM sodium pyruvate, and 1% (w/v) penicillin/streptomycin. Cultures were maintained at 37°C in a humidified incubator equilibrated with 5% carbon dioxide. Cells were passaged every 3–4 days at approximately 80% confluence using 0.05%/0.02% trypsin–EDTA.
Cell encapsulation in self-assembling peptide hydrogel (3D cultures)
The general protocol for cell encapsulation into self-assembling peptide was previously described in detail. 37 Specifically, commercially available self-assembling peptide RAD16-I (1% (w/v), BD PuraMatrix, BD Biosciences) was diluted to obtain a final concentration between 0.3% and 0.6% in 10% (w/v) sucrose. In parallel, cell culture inserts (Millipore, cat # PICM01250) were placed into six-well plates and 500 μL of the control medium was added underneath to wet each membrane. Afterward, MC3T3-E1 cells were harvested from the culture flasks and suspended also in 10% (w/v) sucrose solution at a final concentration of 8×106 cells/mL. A volume of this suspension was gently mixed with an equal volume of the peptide solution, and 80 μL was immediately loaded into each cell culture insert. Since the insert membrane had already been wet with the medium, the peptide would instantly start the self-assembling process when loaded, due to medium diffusion. After several rinsing steps with a medium, 2.5 mL of the control medium was added into the well and 200 μL into the insert. Then, the 3D cultures (henceforth also named constructs) at different peptide concentrations (from 0.15% to 0.30%) were maintained in the incubator at 37°C and the medium was changed every day by removing 0.5 mL of the medium from the well and adding 0.5 mL of a fresh medium in the insert.
Cellular and construct morphology evaluation
The shape of final construct was daily monitored with a stereoscopic microscope (Nikon SMZ660) and the size was measured with image software. To calculate the contraction degree, the construct was ideally simplified to a disk with an average diameter and the contraction degree has been reported as the percentage of diameter decrease respect to the initial one. Cellular morphology into the constructs was routinely examined under phase-contrast microscopy using an inverted Nikon microscope (Nikon Eclipse TS100). For fluorescence staining, constructs were fixed in 1% (w/v) p-formaldehyde, permeabilized with 0.1% (w/v) Triton X-100, and incubated with phalloidin–tetramethylrhodamine B isothiocyanate (Phalloidin-TRITC, Sigma) and 4′,6-diamidino-2-phenylindole (DAPI, Sigma), which stain F-actin and nuclei, respectively. Finally, the constructs were inspected in a Zeiss Axiovert inverted microscope with a coupled ApoTome, which allows optical sectioning.
Field emission gun-scanning electron microscopy analysis
After 24 days of culture, constructs were fixed using 5% (w/v) gluteraldehyde. After fixation, 3D cultures were washed twice in phosphate-buffered saline (PBS) and subsequently dehydrated. The dehydration process included several immersions for 10 min in ethanolic solutions: once in 30% (v/v) ethanol, twice in 50% (v/v) ethanol, three times in 70% (v/v) ethanol, three times in 90% (v/v) ethanol, three times in 96% (v/v) ethanol, and three more times in 100% (v/v) ethanol. Once dehydrated, the cultures were dried using a CO2 critical point dryer (Polaron, CPD Jumbo E-3100). Then, the samples were sputter-coated with gold and platinum alloy using the equipment Emitech SC7620 (60 s, 18 mA, and chamber pressure 0.2 mbar). Samples were examined with an FEG-SEM (JEOL JSM-7100F) at 13 kV.
Cell viability assessment
Cell viability in the constructs was determined using the LIVE/DEAD® Viability/Cytotoxicity kit for mammalian cells (Invitrogen). Briefly, 3D constructs after 19 or 42 days of culture were washed three times with PBS and subsequently covered with a solution of ethidium homodimer-1 and calcein AM (calcein acetoxymethyl ester), both at optimized concentrations of 2 μM. The incubation time was set to 15 min. Finally, the samples were rinsed with PBS and fluorescence was visualized with a Zeiss ApoTome system (for optical sectioning and 3D reconstruction) coupled to the Zeiss Axiovert inverted microscope.
Cell culture on self-assembling peptide hydrogel layer and on plastic dishes (2D cultures)
As two-dimensional controls, cells were grown on traditional plastic plates, as well as on surfaces that were previously modified with a thin layer of self-assembling peptide RAD16-I. In the first case, cells were seeded in six-well plates at a density of 2.6×104 cells/cm2 and grown in the control medium. In the second case, substrates were prepared as described in a recent publication. 32 Briefly, PTFE membranes (Millipore) were treated by graft polymerization of pentafluorophenyl methacrylate (PFM) in a plasma reactor. Afterward, the membranes (0.3×0.3 cm2) were soaked overnight at 37°C in an aqueous solution of a peptide with the same sequence as RAD16-I peptide, but presenting a free amine (NH2-GG-RAD16-I). The peptide (at 10 mg/mL) reacted with the labile pentafluorophenyl group and, thereby, membranes with immobilized RAD16-I peptides were obtained. After autoclaving, 5 μL of PuraMatrix at 1% (w/v) was loaded on each membrane and was incubated for 1 h on the bench to allow any possible self-assembling between the soluble peptides and the ones immobilized on the membrane. Afterward, the membranes were dipped 10 times into deionized water to remove nonassembled peptide, which led to a thin hydrogel layer of self-assembling RAD16-I peptide. Modified membranes were placed in 24-well plates, and then incubated with a suspension of MC3T3-E1 cells in the control medium at a final density of 2.6×104 cells/cm2. After 8 h, nonattached cells were removed and the medium was changed to a fresh one.
ROCK inhibitor assays
Three-dimensional cultures in self-assembling peptide gels were also cultured in the presence of the Rho kinase (ROCK) inhibitor Y-27632 (Sigma) to evaluate the effect of cellular contractility on the 3D system. The inhibitor was diluted in deionized water at a stock concentration of 1 mM. Inhibitor-containing media were prepared by diluting the stock in the control medium at 1 and 10 μM final concentrations. Cultures in inhibitor-containing media were carried out in parallel with cultures in the control medium. The medium was changed daily in the way stated before in the section Cell encapsulation in self-assembling peptide hydrogel (3D cultures).
Osteogenic differentiation
To induce osteogenesis, the encapsulated cells (3D cultures) and cells cultured on 2D substrates (2D cultures) were cultured in the control medium for 4 days and, thereafter, the medium was changed to the osteogenic media (50 μg/mL ascorbic acid (AA) and 10 mM beta-glycerophosphate (βGP)) in the presence or absence of dexamethasone (D) (0.1 μM dexamethasone).
RNA isolation and quantitative reverse transcriptase–polymerase chain reaction
Following the manufacturer's instructions, mRNA was isolated and purified using a peqGOLD total RNA kit (PeqLab). Each 3D culture (cells+peptidic hydrogel) was lysed with 200 μL of the lysis buffer at 4, 8, 16, or 24 days of culture in control or osteogenic media (as indicated in the text and figures). Cells grown on flasks for 1 or 4 days in the control medium were used as controls and lysed with 500 μL of the lysis buffer. After degradation of genomic DNA and column purification, mRNA yields were measured by spectrophotometry (Bio-Rad SmartSpec Plus spectrophotometer) using trUView™ cuvettes for low volumes (Bio-Rad). Equal amounts of mRNA (150 ng) were reverse transcribed to cDNA using the QuantiTect Reverse Transcription kit (Qiagen). For quantitative PCR, 2 μL cDNA of each sample was mixed with SYBR Green master mix (QuantiTect SYBR Green RT-PCR kit; Qiagen) and the corresponding commercial primers from Qiagen: 18s (QT01036875), Col1a1 (QT00162204), Ibsp1 (QT00115304), and Bglap1 (QT00259406). Samples were run in a 7500 Real-time PCR system (Applied Biosystems). Expression of the genes of interest was normalized to the ribosomal unit 18s as a housekeeping gene and compared to the gene expression in 2D cultures in the control medium at 4 days, unless otherwise stated.
Phenotype assessment by staining
Alkaline phosphatase staining
The 3D and 2D cultures were rinsed with a buffer containing no phosphate (0.1 M Tris base [2-amino-2-hydroxymethyl-propane-1,3-diol], 100 mM NaCl, 5 mM MgCl2, pH 9.5). Thereafter, they were covered with the substrate solution containing 330 μg/mL nitrotetrazolium blue (NBT; Sigma) and 165 μg/mL 5-bromo-4-chloro-3-indolyl phosphate (BCIP; Sigma) in the buffer used for rinsing. The color of the culture was monitored for 10 min, and the reaction was stopped by washing the samples with deionized water. The formation of a blue precipitate indicates alkaline phosphatase (ALP) activity.
von Kossa staining for mineralization
The 3D and 2D cultures were fixed with 1% (w/v) p-formaldehyde, followed by extensive rinsing steps with deionized water to ensure phosphate ions removal. Then, samples were covered with 5% (w/v) silver nitrate solution in deionized water. After 1 h incubation in the dark, constructs were washed again with abundant deionized water and placed under a strong light source for approximately 10 min. Samples were inspected visually or under a stereoscopic microscope. Positive calcium mineralization would stain dark brown to black.
Osteopontin immunofluorescence
Two-dimensional cultures were fixed with 1% (w/v) p-formaldehyde. After fixation, samples were incubated in a blocking buffer (PBS containing 20% (v/v) FBS, 0.1% (v/v) Triton X-100, and 1% (v/v) dimethylsulfoxide) overnight at 4°C. Afterward, cultures were incubated with an anti-mouse osteopontin primary antibody (Millipore cat # AB10910, at 1:500 dilution) followed by the secondary antibody anti-rabbit IgG FITC conjugated (Sigma cat # F9887, at 1:160 dilution). Finally, the samples were visualized under a fluorescent microscope (Nikon Eclipse TS100).
Characterization of construct stiffness by atomic force microscopy
The stiffness of both hydrogels and 3D constructs was measured with a custom-built atomic force microscope attached to an inverted optical microscope (TE2000; Nikon) by using a previously described method.38,39 Gels were probed with a Si3N4 V-shape Au-coated cantilever with a pyramidal tip on its apex and with a nominal spring constant (k) of 0.01 N/m (MLCT; Bruker). The cantilever was displaced in 3D with a nanometric resolution by means of piezo actuators coupled to strain gauge sensors (Physik Instrumente) to measure the displacement of the cantilever (z). The deflection of the cantilever (d) was measured using the optical lever method. The force (F) on the cantilever was computed as F=kd. The slope of a force–displacement (F-z) curve obtained at a bare region of the coverslip was used to calibrate the relationship between the photodiode signal and cantilever deflection. Gels were measured at 3–5 different points (separated by ∼50 μm) located at the central region of the sample. At each measurement point, five F-z curves were obtained by ramping the cantilever in the vertical direction at constant speed (amplitude=5 μm, frequency=1 Hz, and indentation up to ∼1 μm). The indentation of the sample (δ) was computed as δ=(z–zc)–(d–doff), where zc is the position of the contact point and doff the offset of the photodiode. Force–displacement curves were analyzed with the pyramidal Hertz model
where θ is the semi-included angle of the tip, E is Young's modulus of the sample, and ν is its Poisson's ratio, assumed to be 0.5. The nonlinear least-squares fit (Matlab; TheMathWorks, Inc.) was used to estimate E from the loading branch of each F-z curve. The average E obtained from the five F-z curves recorded at each measurement point was computed. The shear modulus of the sample is related to E as G=E/[2(1+ν)].
Statistical analysis
Data are expressed as mean values±standard deviation. Quantitative reverse transcriptase–polymerase chain reaction (RT-PCR) data were collected from three independent samples for each condition. The degree of contraction was evaluated from six independent samples. For normally distributed data, statistical analysis was carried out by 1-way or 2-way ANOVA, followed by the Tukey or Games-Howell comparisons, as appropriate in each case. In case of non-normal distribution, the nonparametric Kruskal–Wallis test was used and data were compared with the post hoc Mann–Whitney U test. Analyses were performed using SPSS Statistics v17.0 software.
Results
Cell morphology and cell–cell interactions depending on hydrogel stiffness
Initially, MC3T3-E1 cells were cultured in hydrogels with different RAD16-I self-assembling peptide concentrations, since small changes in the peptide concentration implied a variation in the material stiffness, as determined by rheometry by Sieminski et al. 29 and by atomic force microscopy (AFM) (see values in Table 1). Just after encapsulation, cells showed a round morphology at any peptide concentration (Fig. 1a). After 24 h, half of the cell population extended cellular processes and invaded the matrix in 0.15% gels, corresponding to a shear modulus of 120 Pa—obtained by rheometry (Fig. 1b)—while every cell remained round in 0.30% gels (510 Pa) (Fig. 1c). This distinct behavior was even more noticeable at 2 days of culture (Fig. 1d, e). After 4 days, cells in 0.15% hydrogels were able to elongate and interact, creating a visible and rich cellular network (Fig. 1f). As expected, in stiffer hydrogels, a higher ratio of cells remained round and the cellular network was less dense or even did not happen, as depicted for 0.30% gels (Fig. 1g). At longer periods of time, mostly the cellular cluster formation, instead of the cellular network, was favored in those stiffer matrices, while the cellular network was maintained and became even denser in compliant gels.
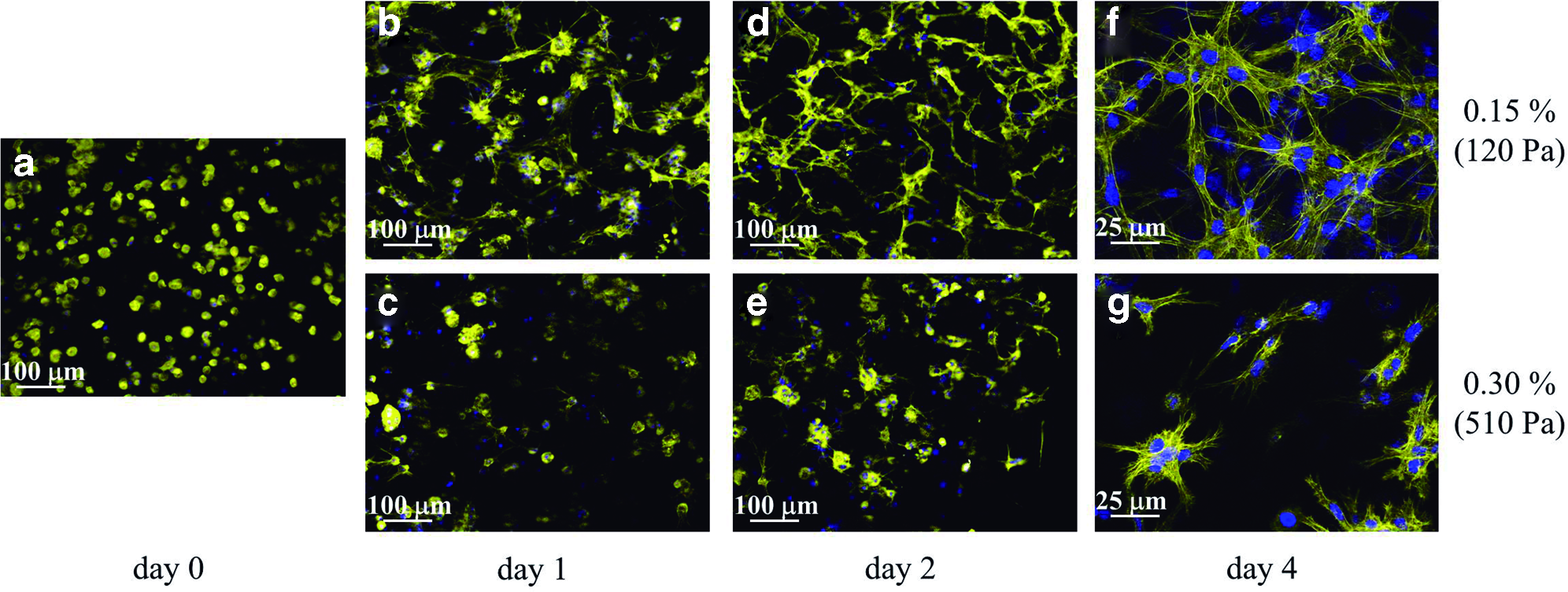
Cell morphology and network formation is influenced by matrix stiffness. MC3T3-E1 cells cultured in three-dimensional (3D) self-assembling peptide hydrogels were visualized under fluorescence microscopy just after encapsulation
nd, not determined; AFM, atomic force microscopy.
Morphological changes of the construct and cellular viability
In the biomechanical conditions where cells were able to spread and interconnect (0.15% peptide concentration), the construct underwent morphological changes over time (Fig. 2a). Cells were observed to pull all together—especially in the disk edge—dragging the hydrogel with them and forming a smaller disk (Fig. 2a, a1) with a higher cellular density at the edges (Fig. 2b). Further contraction generated a dense cellular ring along the construct perimeter (Fig. 2a, a2), and, finally, the construct became smaller until a size plateau was reached (Fig. 2a, a3).

Cellular constructs undergo morphological changes over time.
Analysis with field emission gun scanning electron microscopy (FEG-SEM) showed a cohesive layer of elongated cells on the surface of the contracted sample (Fig. 2c, c1). A close-up of a cell revealed structures, such as microvilli or microspikes, on the membrane and a nanofibrous matrix underneath the cell (Fig. 2c, c2, c3). When the cell layer was carefully ripped off, the underlying material (Fig. 2d, d1) looked radically different from the original self-assembling peptide hydrogel (Fig. 2d, d2). Fibers were mostly arranged in parallel and they were less branched and thicker (around 60–70 nm in diameter) than the RAD16-I nanofibers, which are highly branched and present a diameter around 10 nm. Therefore, this would suggest that cells had remodeled the initial matrix and secreted a new one. Interestingly, proliferating cells were also found on the surface of the construct (Fig. 2c, c4 and c5). Furthermore, cells—both on the surface and within the matrix—remained viable after 3D encapsulation and culture, even after 42 days (Fig. 3a).

Encapsulated MC3T3-E1 cells remain viable at different conditions. Constructs were stained with LIVE/DEAD kit and visualized under fluorescent microscopy. Viable cells stain green and dead cells stain red.
Effect of initial hydrogel stiffness on the contraction process and final mechanical properties
The contraction degree of the 3D cultures over time depended on the initial concentration of the self-assembling peptide (Fig. 4a). After 4 days, the most compliant hydrogels (0.15%, 120 Pa) had contracted by a 30% of its initial diameter, while gels at 0.20% (220 Pa) and 0.25% (350 Pa) peptide concentration contracted at 15% and 10%, respectively. Moreover, stiffer hydrogels hardly contracted (1%–4%). Figure 4b depicts the dramatic difference in final morphology of two constructs at 0.15% and 0.25% gels after 25 days of culture. Regarding cellular viability, it was qualitatively assessed that higher matrix stiffness did not induce cellular death, since only few cells stained red (dead cells) both in 0.15% and 0.30% gels (Fig. 3b, pictures in the first row).

Cell morphology and construct contraction is influenced by initial matrix stiffness and cytoskeletal contractility.
Interestingly, the contraction of the 3D cultures had a remarkable effect in the mechanical properties of the constructs over time (Fig. 5). The exploration of the specimens by AFM revealed that cells in the softest hydrogel (a 0.15% peptide concentration) eventually produced much stiffer tissue-like constructs than the ones cultured in initially stiffer hydrogels (0.25%). The mean shear modulus (G) obtained for the 0.15% hydrogel was 63 Pa. Once cells were encapsulated in this material for 4 days, the G value was observed to slightly rise to 84 Pa. However, more importantly, the G value was found to be greatly increased up to 8000 Pa after the whole process of contraction (at 30 days of culture). In contrast, when cells were encapsulated in 0.25% gels (with a modulus around 70 Pa), the final constructs at 30 days could only stiffen up to 1200 Pa (Fig. 5). The variability of the values is associated to the heterogeneity of the samples.
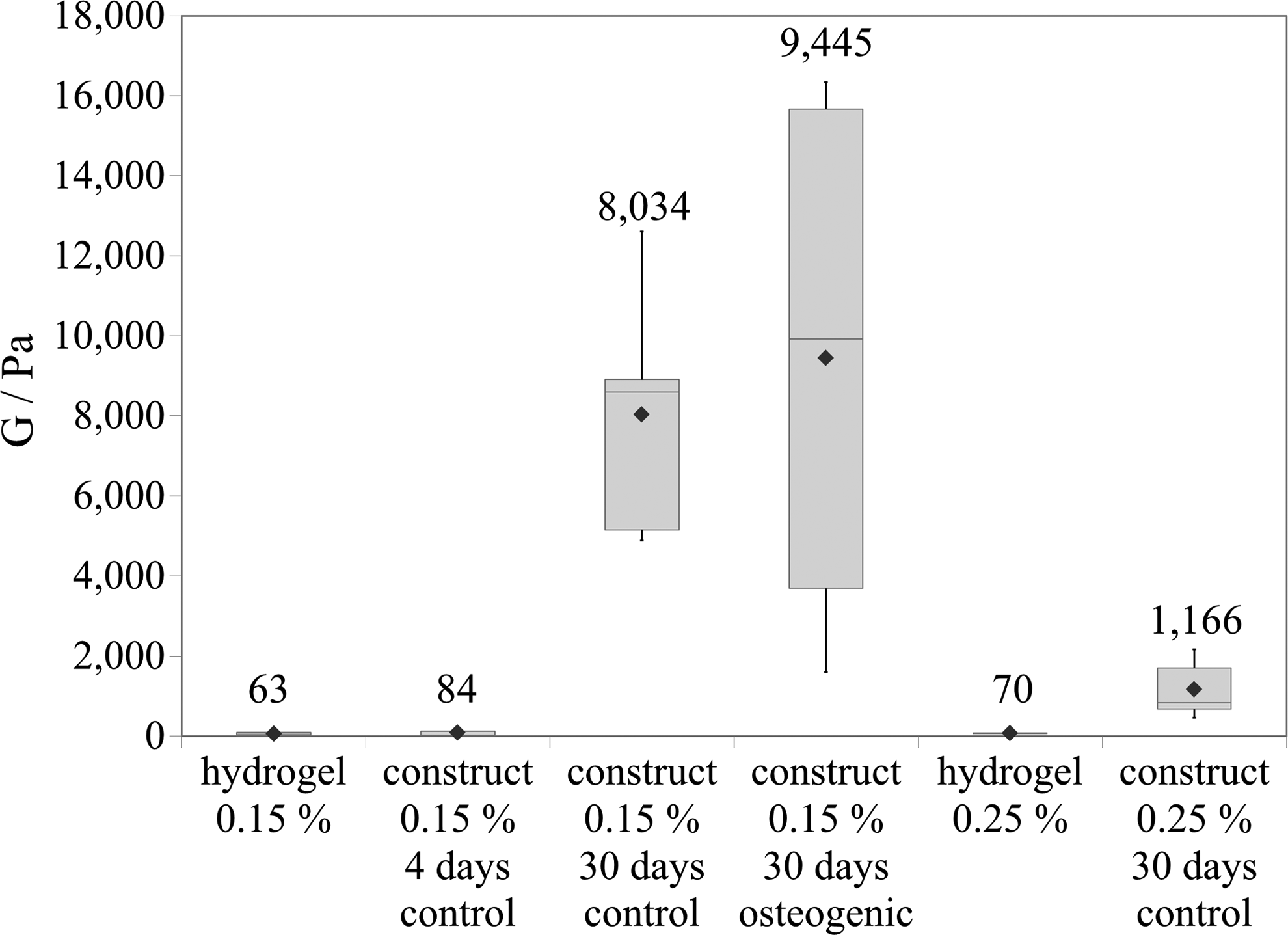
Shear modulus (G) was determined by atomic force microscopy (AFM) for hydrogels and cellular constructs. Data are depicted in a box plot and the rhombus represents the mean (value drawn over the boxes). Different samples were analyzed: the bare hydrogel at 0.15% peptide concentration, two constructs in 0.15% gel at 4 and 30 days of culture in control medium, a mineralized construct in 0.15% gel—cultured in osteogenic medium containing dexamethasone for 30 days—the bare hydrogel at 0.25% peptide concentration, and a construct in 0.25% cultured for 30 days in control medium.
Disruption of cellular elongation and 3D culture contraction by ROCK inhibitor
To evaluate the role that cytoskeleton contractility plays in a 3D culture contraction, constructs at a 0.15% peptide concentration were cultured in the presence of the ROCK inhibitor Y-27632 at 1 and 10 μM. ROCK is known to act downstream Rho and to promote myosin II contractility by phosphorylation of myosin light chain. After 1 day of culture, it was observed that the number of cells presenting protrusions substantially dropped in cultures treated with Y-27632 in a dose-depending manner (Fig. 4c). Examination of cell viability indicated that the inability to elongate was not due to an increased cellular death, since it was equivalent in control and inhibitor-containing media (Fig. 4c). Moreover, contraction of the cultures was significantly restricted in the presence of the ROCK inhibitor, especially at 10 μM concentration, suggesting that the morphological change of the construct depends on cell contractility (Fig. 4d).
Expression of bone-related markers over time
To further characterize this system, the expressions of certain bone-related proteins, such as collagen type I, bone sialoprotein, and osteocalcin, were determined at the mRNA level compared to its basal level in 2D cultures in the control medium at 4 days (Fig. 6a). Surprisingly, in the absence of any osteogenic inducer, bone-related genes were found to be upregulated over time. The expression of collagen type I was significantly increased by 3.8-fold after 24 days of culture. Bone sialoprotein showed a more pronounced upregulation, with a 29-fold and 54-fold increase at 16 and 24 days, respectively. Furthermore, osteocalcin was strongly upregulated over time and reached up to a 164-fold increase after 24 days of culture in the control medium. At that point, osteocalcin expression was studied in 2D and 3D cultures at 4 and 8 days to compare the upregulation levels between both systems. In that case, gene expression values were compared to osteocalcin expression in 1-day 2D cultures. Interestingly, osteocalcin expression was significantly higher in 3D cultures compared to 2D (Fig. 6b).
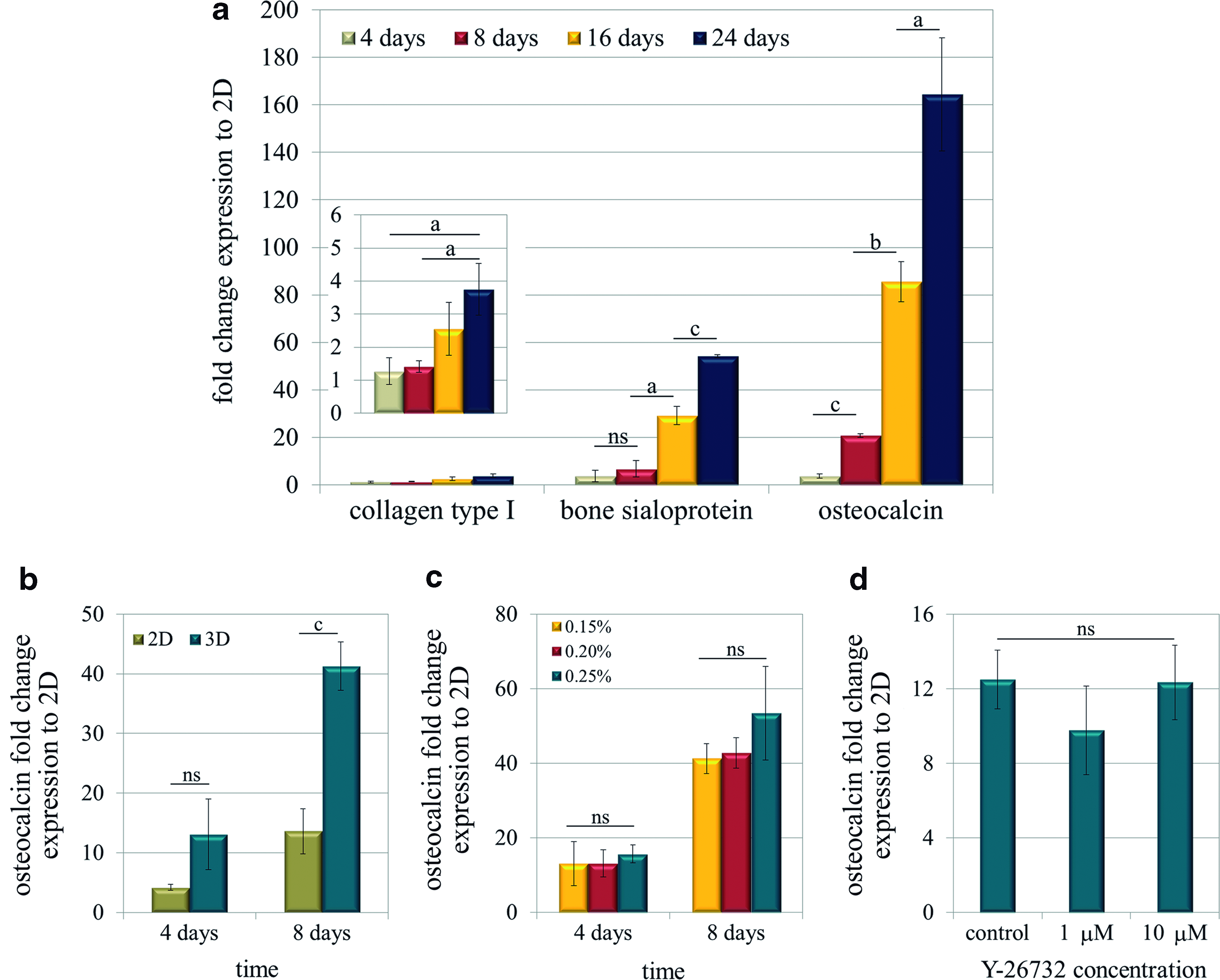
MC3T3-E1 cells cultured in self-assembling peptide scaffolds spontaneously upregulate bone-related genes over time, independently of construct biomechanics. Quantitative reverse transcriptase–polymerase chain reaction (RT-PCR) was used to determine the gene expressions shown, relative to the expression in 2D cultures at 4 days, unless otherwise stated.
Effect of disrupting 3D culture contraction on osteocalcin expression
To assess if osteogenesis was associated to construct shrinkage, the contraction process was disrupted mechanically and chemically, either by increasing gel stiffness or by the addition of the ROCK inhibitor. Under those conditions, osteocalcin mRNA expression was assessed as a marker for osteogenesis. Cells encapsulated at three concentrations of self-assembling peptide (0.15%, 0.20%, and 0.25% (w/v)) did not show any significant change in osteocalcin expression at 4 and 8 days (Fig. 6c). On the other hand, chemical disruption of contraction by the ROCK inhibitor Y-27632 did not involve a variation of the osteocalcin expression level (Fig. 6d).
Effect of chemical inducers on osteogenic differentiation in 3D
Finally, long-term differentiation was studied not only in the control medium, but also in two induction media, containing AA and βGP, with or without dexamethasone (D). All the experiments were performed on cells encapsulated in 0.15% (w/v) self-assembling peptide, a condition that allowed the formation of a homogeneous cellular network, and osteogenic media were added always at day 4 of culture. First, a slight increase in cellular death was detected only in cultures treated with dexamethasone (Fig. 3b). Expression of collagen type I, bone sialoprotein, and osteocalcin in the osteogenic medium without dexamethasone was upregulated (Fig. 7) as expected, since AA and βGP are commonly used to differentiate MC3T3-E1 cells in traditional 2D cultures. 40 However, in most of the cases, the increase was not significantly different from the results obtained in the control medium (Fig. 7). On the other hand, when dexamethasone was added into the osteogenic media, expression of collagen and osteocalcin was strongly downregulated, especially after 24 days. Also, ALP stained positive in all three media, but again the presence of dexamethasone was found to decrease ALP activity (Fig. 8a). Finally, as a definitive marker for osteogenesis, the presence of calcium deposits was assessed by von Kossa staining. Surprisingly, only in the presence of dexamethasone, a fully mineralized matrix was obtained (Fig. 8a). Neither in the control nor in osteogenic medium (without dexamethasone) was the presence of black stained nodules ever observed, even after 60 days of culture. Regarding the mechanical properties of the mineralized construct, the AFM analysis elicited an average shear modulus of 9500 Pa, while it was 8000 Pa in the control 3D construct (both at 30 days). Interestingly, the variability of the results was very high, particularly in the case of the mineralized matrix. Since mineral deposition might be a nonhomogeneous process, results indicate that it occurred in patches or nucleated clusters with a higher shear modulus (around 16,000 Pa) separated by zones of much lower values (around 4000 Pa) (see Fig. 5).
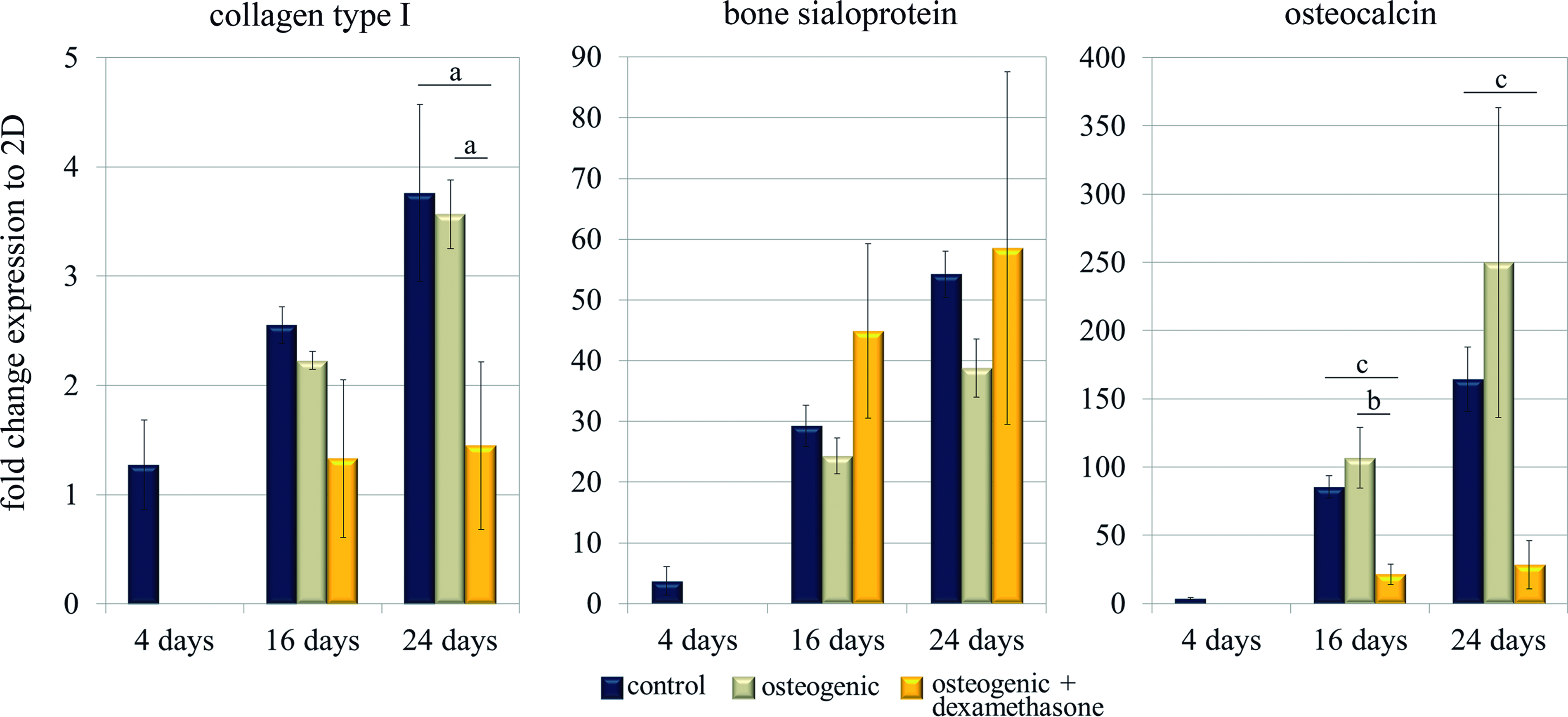
Osteogenic gene expression in 3D is influenced by osteogenic chemical inducers, in particular, dexamethasone. Quantitative RT-PCR was performed to determine the gene expressions of collagen type I, bone sialoprotein, and osteocalcin over time, relative to the corresponding expression in 2D cultures at 4 days. All 3D constructs were initially cultured in control medium and media was changed to control, osteogenic (with AA and βGP), and osteogenic with dexamethasone (AA, βGP, and D) media at day 4. Statistical differences are indicated as: “a” for p<0.05, “b” for p<0.01, and “c” for p<0.001. Color images available online at www.liebertpub.com/tea
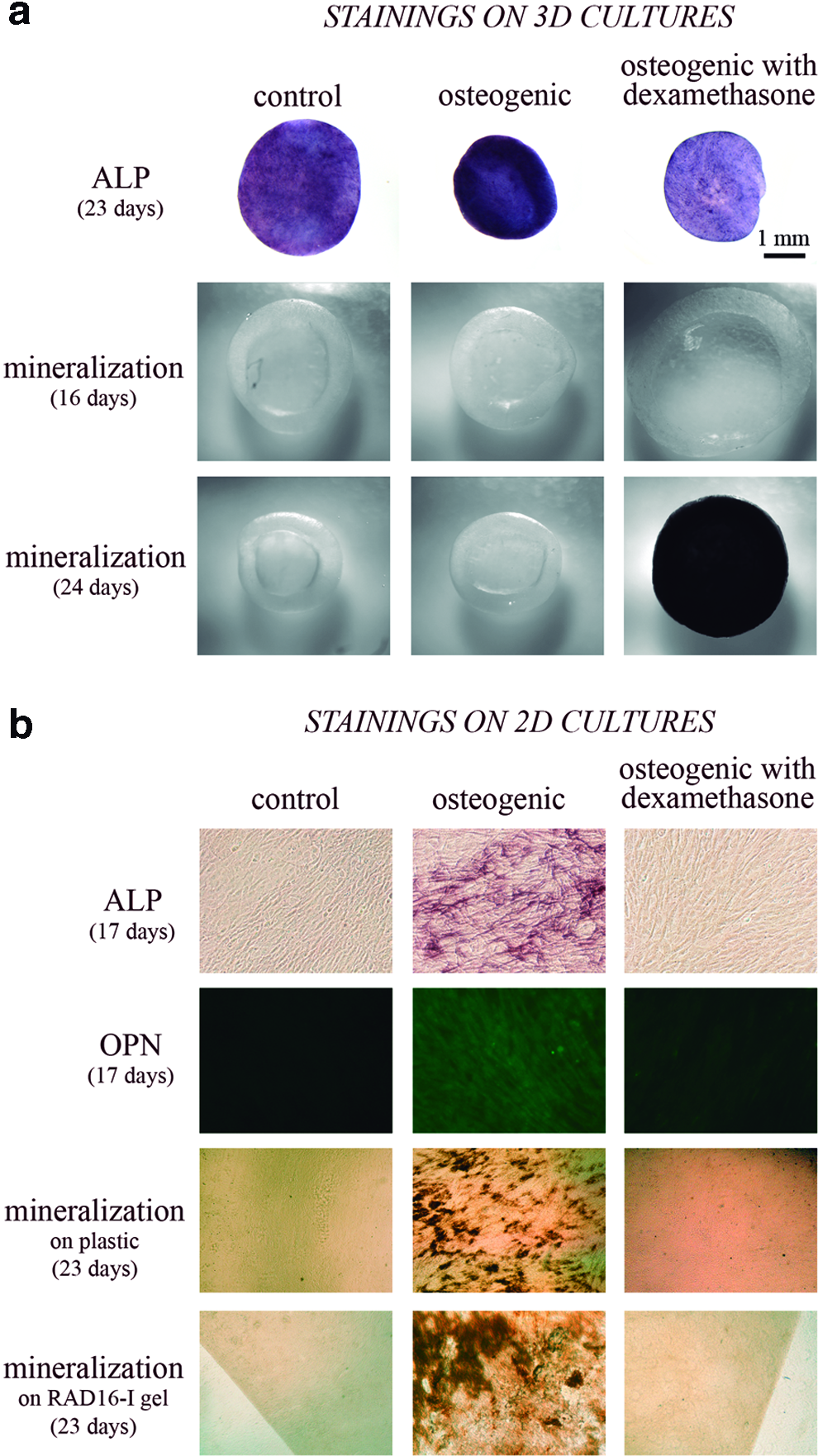
MC3T3-E1 cells show differential osteogenic behavior in self-assembling peptide hydrogels (3D cultures) compared to 2D cultures. Bone-like phenotype was further assessed by different stainings in cells cultured in three media conditions: control, osteogenic (with AA and βGP), and osteogenic with dexamethasone (with AA, βGP, and D).
These results contrast with the ones obtained in 2D cultures, where MC3T3-E1 cells clearly show a final osteoblastic phenotype only when cultured in the osteogenic medium without dexamethasone, as observed by ALP, osteopontin immunofluorescence, and von Kossa staining (Fig. 8b). To corroborate that this difference was not only caused by the peptide itself, preosteoblastic cells were also cultured on membranes that were previously treated to attach a thin self-assembling peptide hydrogel layer. Again in this case, cells only showed mineralized matrix without dexamethasone, indicating that the different requirements for a final mineralization might be due to the 3D arrangement of the cells.
Discussion
During embryonic bone development, the concentration of progenitor cells that occurs in mesenchymal condensation is considered a fundamental step, both in intramembranous and endochondral mechanisms, because it promotes increased cell-to-cell communication, which is critical in tissue formation. 15 At the moment, many osteogenic models in hydrogels, such as the commonly used polyethylene glycol (PEG) and alginates, usually constrain cells within the matrix, just impeding the aforementioned cellular connections.11–13,17 Hence, as an alternative, we devised a 3D osteogenic model, which could somehow recapitulate the first steps of bone formation by favoring the migration and interaction of preosteoblasts. For this purpose, a soft nanofiber hydrogel made of self-assembling peptides was used as scaffold material. Under the appropriate biomechanical conditions—that is at initial matrix stiffness around 100 Pa—MC3T3-E1 cells were able to elongate, migrate, and interact to each other creating a rich interconnected cell–cell network within the first 3–4 days of culture (Fig. 1). Recently, Ehrbar et al. also described similar cellular network formation with MC3T3-E1 cells in very soft PEG hydrogels (around 60–100 Pa) especially when modified with MMP-sensitive sites, although in that case, dense cell–cell contacts were not reported until after 3 weeks of culture. 23
In the system reported here, the establishment of a cellular network allowed the system to undergo morphological changes over time (Fig. 2a). Interestingly, this pattern was already observed using mouse embryonic fibroblasts in the RAD-16 self-assembling peptide gel, suggesting that some cells undergo a similar organizational process in this 3D environment. 41 However, it was demonstrated that cell elongation and construct contraction were clearly hindered at slightly higher stiffness values (220 or 350 Pa), where cells could hardly overcome the matrix resistance (Figs. 1 and 4a, b). These findings are in agreement with other works that use compliant PEG gels modified with proteolytic sites to study cellular mechanics.22,23 As a consequence, those stiffer matrices led cells to the formation of undesired isolated cellular aggregates at longer periods of culture. Moreover, the cellular mechanical machinery involved in the contraction process was abrogated by inhibiting the Rho kinase (ROCK), confirming that construct contraction operates through myosin II contractility, as expected. More importantly, the contraction process produces an increase in the cell density and cell–cell interaction, as well as matrix components concentration, which leads to a stiffer construct, as proved by the measurements of the shear modulus by AFM. For this reason, very soft hydrogels ultimately produce mechanically improved tissue-like constructs (around 8000 Pa) over stiffer gels that yield mechanically poorer cultures (around 1200 Pa), due to the hindered cellular connections and 3D culture contractility (see Table 1).
Interestingly, along with the described contraction process, preosteoblastic MC3T3-E1 cells were found to spontaneously upregulate the expression of bone-related genes (collagen type I, bone sialoprotein, and osteocalcin) in the absence of any osteogenic inducer (Fig. 6a). Moreover, those expressions reached the levels in constructs cultured under chemical induction with AA and βGP. Therefore, the 3D environment promotes gene expression by itself, regardless of the presence of chemical induction. Nevertheless, a more potent inducer, such as dexamethasone, was required to acquire a final osteogenic phenotype, typically characterized by matrix mineralization. Along with mineral formation, collagen type I and osteocalcin were downregulated under those conditions. As reviewed in the literature, this inhibition typically occurs in glucocorticoid-induced osteogenesis, 42 when cells are in a mature osteoblastic stage, 43 which correlates with the results shown here. On the other hand, dexamethasone was found to be counterproductive for osteogenesis in MC3T3-E1 cultures in 2D (Fig. 8b), which agrees with other authors' findings.44,45 More importantly, this difference between 2D and 3D supports that even committed cells may change their behavior in these two conformations and, thus, results should not be directly translated from 2D to 3D.
The inhibition of the contraction process, either mechanically—by increasing hydrogel stiffness—or biochemically—by using the ROCK inhibitor—did not affect the expression of osteocalcin at short periods of culture time (up to 8 days). Therefore, once in soft self-assembling peptide hydrogel, MC3T3-E1 cells seemed to engage into a default pathway of osteogenesis, regardless of cell contractility and network formation. Moreover, the stiffness values used in this work (120–350 Pa) are too low to expect a mechanical induction of osteogenesis, as described in other reports that observed enhanced bone formation at much higher matrix stiffness values (around 30 and 60 kPa).11,13 However, it is not dismissed that the contraction process could be mechanically stimulating bone formation at long term, since it has been proved that stiffness importantly increases with contraction. In agreement with this system, others have shown that preosteogenic cells in compliant matrices with contractile behavior displayed greater osteogenic maturation than in stiffer noncontractile scaffolds. 46
Ensuring cellular viability is a key issue when dealing with new tissue-engineered products. This system was proven to support cellular viability for prolonged culture periods, regardless on the final contraction degree or the initial matrix stiffness (Fig. 3). Only in constructs reaching terminal differentiation (cultured in the osteogenic medium containing dexamethasone), cell death was significantly detected. This finding is actually in agreement with the natural cell death occurring after cellular specialization and maturation in vivo. 47
Conclusions
In this work, we developed a simple 3D model for osteogenesis using a soft nanofiber matrix and the well-defined preosteoblastic MC3T3-E1 cell line. This synthetic matrix favored cellular interaction in a 3D conformation, which might be enough to engage cells into a default osteogenic pathway. We showed that extremely compliant gels can support bone formation, while allowing the cells to communicate and self-organize, creating an intricate network and producing a mechanically superior final 3D construct. Although network formation does not seem to be essential for the system to express certain osteogenic markers, it provides cellular interactions and tissue-like cohesion.
Footnotes
Acknowledgments
We greatly thank the Nanotechnology Platform at Parc Científic de Barcelona and Ana Sancho and Elena de Juan-Pardo at CEIT and TECNUN (Universidad de Navarra) for sample preparation and FEG-SEM analysis. This work was supported by the Translational Centre for Regenerative Medicine, Leipzig University (Germany), Award 1098 NF to CES and by the Ministry of Science and Innovation grant PI11-00089 to DN. NMB also acknowledges financial support from DURSI (Generalitat de Catalunya) and the European Social Fund.
Disclosure Statement
No competing financial interests exist.