
Abstract
The goal of this study was to determine the effects of chondrogenic predifferentiation on the ability of bone marrow-derived stromal cells (BMSCs) delivered to growth plate defects to restore growth function. Chondrogenesis was induced with transforming growth factor (TGF)-β1 treatment in high-density monolayer cultures of BMSCs in vitro. The predifferentiated or undifferentiated BMSCs were either seeded into agarose gels for continued in vitro culture, or injected into growth plate defects via an in situ gelling agarose. Predifferentiated BMSCs had higher Sox-9, type II collagen, and aggrecan mRNA levels compared to undifferentiated cells after high-density monolayer culture. After transfer to agarose gels, predifferentiated cells did not produce a cartilaginous matrix, even with continued TGF-β1 stimulation, whereas undifferentiated cells produced a cartilaginous matrix in this system. Three-dimensional images of the growth plate created from microcomputed tomography scans showed that delivery of either predifferentiated or undifferentiated cells to defects resulted in a decrease in mineralized tether formation (fusion) in the growth plate tissue surrounding the defect to normal levels. Limb length discrepancy between injured and control limbs was corrected after treatment with undifferentiated, but not predifferentiated, cells. These results indicate that cell therapy may be an effective treatment to reduce growth dysfunction after growth plate injury, perhaps by maintaining the health of the uninjured growth plate tissue, and that the cell differentiation state plays a role in restoring the growth potential of the injured limb.
Introduction
The challenge of treating growth plate injury has inspired several animal model studies to investigate the application of cartilage tissue-engineering principles to promote regeneration of the damaged region of the growth plate and restore growth function. Stem cells derived from various tissue sources, including bone marrow (BMSCs), adipose, and muscle, are an attractive choice for this application.9,10 Growth factor stimulation is required to induce chondrogenesis of stem cells, although the specific treatment protocol depends on the tissue source from which the cells are derived. 11 Our laboratory and others have shown that the degree of monolayer expansion, serial application of growth factors, and culture environment have profound effects on chondrogenic differentiation and the degree of matrix accumulation.12,13
Evidence is beginning to emerge that suggests that the degree of predifferentiation of BMSCs in vitro influences the behavior of the cells after implantation in vivo. The pathway of bone healing after cell implantation, for example, is dependent on whether undifferentiated BMSCs or osteoblasts are implanted into a defect. 14 Scotti et al. showed that the ability of chondrogenic stem cells to form bone at an ectopic site in vivo was dependent on how far down the chondrocytic pathway the cells were pushed in vitro before implantation. 15 In a study comparing the ability of articular chondrocytes to undifferentiated MSCs to repair intervertebral disc defects in vivo, Acosta et al. found that there was no repair response with MSC implantation. 16
The overall objective of this study was to determine the effect of stem cell differentiation state on healing of the growth plate in an established small-animal model of growth plate injury. We hypothesized that stem cells stimulated to undergo chondrogenesis in high-density monolayer would retain their chondrogenic phenotype after transfer into an injectable in situ gelling agarose and promote restoration of growth function after implantation. Using microcomputed tomography (micro-CT) techniques reported previously, 8 changes in growth plate and whole-bone morphology in response to delivery of agarose and BMSCs were quantified. We found that the differentiation state of the injected cells had a significant effect on the rate of growth plate fusion and limb length discrepancy in this model.
Materials and Methods
In vitro chondrogenesis of rat BMSCs in high-density monolayer
Cell isolation
Bone marrow stromal cells were retrieved as previously described. 17 Briefly, six male 45-day-old Sprague-Dawley (Charles Rivers Laboratory) rats were euthanized by CO2 asphyxiation, and their femurs and tibiae removed under an IACUC-approved protocol. Bone marrow was flushed from diaphyses with an α-minimum essential medium (α-MEM) supplemented with 1% antibiotic/antimycotic (100 U penicillin G, 100 μg streptomycin sulfate, and 0.025 μg amphotericin B/mL; Invitrogen) and a selected lot of fetal bovine serum (FBS; Hyclone) using a 20-gauge needle attached to a 10-mL syringe. The cell suspension was centrifuged at 1,200 RPM for 20 min, resuspended in the medium, and plated at one-leg/100-mm dish (Corning) for 30 min. Unattached cells were collected, replated in 150-cm2 T-flasks at a density of 150×106cells/flask, and covered with 35 mL of medium. Media were changed after 4 days to remove nonadherent cells. The cells grew in circular patches that became confluent after 8 days, at which time they were considered to be at primary confluence (P0). The cells were then lifted from the surface using 0.05% trypsin/0.53 mM EDTA (Invitrogen) and frozen until the experiment was performed.
High-density monolayer
All experiments were performed using P1 cells. Cells were plated at a density of 6.67×103 cells/cm2 in six-well plates (35-mm dish) for a time-course analysis of chondrogenesis in monolayer culture. During expansion, the passaging medium was supplemented with 1 ng/mL fibroblast growth factor (FGF)-2 (R & D Systems) based on our previous study demonstrating that FGF-2 increases matrix production by these cells. 12 At confluence, cells were switched to a basal medium (high-glucose Dulbecco's minimum essential medium (DMEM) with 1% antibiotic/antimycotic, 0.1 mM nonessential amino acids (Invitrogen), and 50 μg/mL ascorbic acid-2-phosphate (AA-2P; Sigma) supplemented with 2% FBS. Control samples continued to receive 1 ng/mL FGF-2, and chondrogenic samples were switched to receiving 10 ng/mL transforming growth factor (TGF)-β1. Samples were given 1.5 mL medium every day. Media and cells samples were taken on days 0, 1, 2, 3, and 5 for sulfated glycosaminoglycans (sGAGs) and gene expression, respectively.
Hydrogel culture
From the results of the monolayer time-course study, we determined that the expression of chondrogenic genes (Sox9, collagen type II, and aggrecan) and matrix proteins (sGAGs) was highest after 2 days of exposure to TGF-β1. Thus, the cells for hydrogel experiments were prepared as follows. Cells were plated at a density of 6.67×103 cells/cm2 in 185-cm2 T-flasks. At confluence, the cells were switched to the chondrogenic medium as described above for high-density monolayer and cultured for 2 days more. Again, control samples continued to receive 1 ng/mL FGF-2, and chondrogenic samples were switched to receiving 10 ng/mL TGF-β1. To isolate cells from monolayer culture, 185-cm2 T-flasks were treated with a cocktail of 0.3% dispase and 0.2% collagenase (Invitrogen) made up in the α-MEM. Flasks were agitated at 37°C for 30 min to release the cells. Cells were then passed through a 70-μm filter, and the solution was diluted with three volumes of α-MEM containing 10% FBS. The cells were collected by spinning at 1,200 RPM for 7 min, resuspended in PBS, and counted on a hemocytometer. The cells were then resuspended in PBS at a density of 40×106 cells/mL.
The SeaKem® (Fisher Scientific) agarose properties were previously described. 8 A 1% solution of agarose was prepared by mixing the agarose powder with calcium- and magnesium-free phosphate-buffered saline (PBS). The mixture was autoclaved for 20 min to dissolve the powder and sterilize the solution. Three-dimensional constructs were produced with either P1 cells released from monolayer culture with trypsin or predifferentiated cells that were stimulated in high-density monolayer with TGF-β1 for 2 days. Cylindrical gels containing either undifferentiated P1 cells or TGF-β1-predifferentiated cells were cast as previously reported. 12 Briefly, cells in PBS were combined 1:1 with 1% SeaKem agarose for a final cell density of 20×106 cells/mL in 0.5% agarose. The gel plus cell solutions were pipetted into a well of a custom-designed mold and allowed to polymerize for 30 min at room temperature. The resulting gels measured 4 mm in diameter and were 2-mm thick. Gels were rinsed in a high-glucose DMEM (DMEM-HG) supplemented with 1% antibiotic/antimycotic after polymerization. The constructs were then placed into a basal medium with the following supplements: (A) 10% FBS, (B) 1% ITS+ (BD Biosciences), or (C) 1% ITS+ and 10 ng/mL TGF-β1. Gels were covered with 1 mL of medium and cultured in a humidified atmosphere of 5% CO2 at 37°C. Media were changed every 2–3 days for the duration of the experiments. TGF-β1 was added fresh at every medium change.
sGAG released into the medium
sGAG release into the culture medium was measured using a modification of the dimethylmethylene blue (DMMB) dye assay developed by Farndale et al. 18 Briefly, medium samples (1 mL) were lyophilized overnight and resuspended in 200 μL water. Whole agarose gels were first heated at 70°C in ammonium acetate until softened, and then cooled to 42°C for 1 h before adding 1.5 U/mL agarose. Samples were incubated at 42°C overnight to break up the agarose and then digested with proteinase k for 16 h at 60°C. Samples and standards created from shark chondroitin sulfate (Sigma) were mixed with 200 μL of DMMB dye, and the absorbance of each was measured at 525 nm.
Cell viability
For monolayer cultures, cells from 35-mm wells were trypsinized, resuspended in 3 mL media, and counted using trypan blue exclusion. Cell viability within agarose constructs was determined using the Live/Dead Assay (Molecular Probes). Briefly, samples were incubated in a solution containing 4 mM ethidium bromide and 4 mM calcein for 45 min. After rinsing in PBS, gels were imaged on a Zeiss LSM 510 confocal microscope.
Histology
Agarose constructs were taken down on days 7, 14, and 21 for histological analysis. Samples were fixed in 10% neutral buffered formalin at 4°C for 1 week and stored in 70% ethanol at 4°C until histologic processing. Four-micron sections were taken from paraffin-embedded samples through the cross-section and stained with safranin-O.
RNA isolation and quantitative reverse transcription-polymerase chain reaction
Cells in monolayer culture were rinsed with PBS and then covered with 350 μL RLT buffer (Qiagen) containing 1% β-mercaptoethanol. The cells were then scraped from 35-mm dishes and disrupted by passing through a Qiashredder according to the manufacturer's protocol (Qiagen). RNA was isolated using RNeasy spin columns according to the manufacturer's protocol. One microgram of DNase-treated RNA was converted to cDNA using the Promega Reverse Transcriptase kit (Promega), and the samples were stored at −20°C until polymerase chain reaction amplification. The yield of the purified isolate was read at 260 and 280 nm on an UV-1601 spectrophotometer (Shimadzu). Samples were probed with custom primers for aggrecan, collagen type II, Sox9, and collagen type I using SYBR Green dye on an ABI PRISM 7700 system (Applied Biosystems) using the sequences listed in Table 1. cDNA was mixed with random hexamers and the specific primer at an optimal concentrations (between 400 nM and 900 nM). The mRNA concentration was determined from standard curves created with known amounts of product.
Growth plate defect animal model
Surgical procedure
All procedures were performed under the Georgia Institute of Technology IACUC-approved guidelines. Sprague-Dawley male rats (body weight 100–125 grams, ∼5-weeks old; Charles Rivers Laboratory) were used in all studies. The defects were made as previously described. 8 Briefly, animals were anesthetized; the skin over both knees was sterilely prepared and draped, and the trochlear groove of the distal femur was exposed. Using a hand drill and a 2-mm drill bit, a central defect was created across the physis of the distal femur by drilling through the articular cartilage between the condyles, normal to the cross-sectional plane of the bone shaft, and then further up the intramedullary canal to a depth of 6 mm. The depth of drilling into the metaphyses was controlled with a mechanical stop. After irrigation with sterile saline, Gelfoam® (Pfizer) was inserted into the bottom of each cavity and then packed with gauze. After 20 min, the gauze was removed, and the site was inspected to confirm the cessation of bleeding. Previous studies in our laboratory have demonstrated that Gelfoam insertion into the bottom of the defect had no effect on subsequent defect healing (unpublished data). The defects were then either left empty, filled with 0.5% agarose (Control), filled with 0.5% agarose containing BMSCs predifferentiated with TGF-β1 (Prediff), or filled with 0.5% agarose containing undifferentiated P1 BMSCs (Undiff) (see Figure 1). All cells were suspended in agarose at a density of 20×106 cells/mL. After gel polymerization, the patella was relocated to its original position, and the medial joint capsule was closed with a 4-0 vicryl suture. The skin was closed using wound clips, and the surgical area was washed with hydrogen peroxide. The wound clips were removed 10–14 days postsurgery. The left leg was left unoperated and used as the control for all analyses. Animals were euthanized by CO2 asphyxiation on day 56 (n=4–5 per group; 18 animals total) postsurgery, and both femurs removed.

Experimental design. This illustration outlines the in vitro treatment of undifferentiated and predifferentiated bone marrow-derived stromal cells (BMSCs) before injection into the defect. FGF, fibroblast growth factor; TGF, transforming growth factor.
Microcomputed tomography
Total length and growth plate morphological parameters were assessed with micro-CT imaging as previously described.8,19 The epiphyseal region of the defect legs were scanned at a 20-μm voxel size on a VivaCT 40 system (Scanco Medical). The volume of bone infiltration into the defect (bone bridge formation) was measured by isolating a 2 mm×15 slice (0.32 mm) cylindrical region within the original defect contained entirely within the growth plate as shown in Figure 2A. This parameter was termed the defect bone volume fraction (Defect BVF). The bone length was determined from 3D images rendered from whole-bone scans at a voxel size of 36 μm (Fig. 2B). Finally, the growth plate was isolated from the surrounding bone tissue in the micro-CT images by manual contouring of 2D slices of sagittal images. The contoured regions were compiled to render 3D images (Fig. 2C). From these images, the total growth plate volume was calculated, and thickness maps were generated. To determine the effect of the defect on the natural course of growth plate closure in Sprague-Dawley rats, the volume of mineralized tethers within the growth plate tissue excluding the bone bridge within the defect was quantified (Tether BVF).

Microcomputed tomography (micro-CT) analysis. Images demonstrating
Statistical analysis
The effects of culture or growth plate defect treatment conditions on group means were assessed using the analysis-of-variance general linear model. Statistical significance (p<0.05) between individual group samples was determined using the Tukey post hoc test for multiple comparisons. All graphs represent the mean±SEM unless otherwise noted.
Results
TGF-β1 stimulates chondrogenesis of BMSCs in monolayer culture
Cells treated with TGF-β1 detached from the plates after 2 days and remained a floating monolayer until day 5. Due to this phenomenon, cells could not be counted on day 5. BMSCs in high-density monolayer showed very little proliferation after switching to the chondrogenic medium in both FGF-2- and TGF-β1-treated samples over the culture period (Fig. 3). The viability of the isolated cells remained above 90% in both FGF-2- and TGF-β1-treated samples, whether cells were released with trypsin or dispase and collagenase (Fig. 3). sGAG released into the medium was higher in TGF-β1 cultures until after day 2 (Fig. 4). BMSCs treated with TGF-β1 also had significantly higher expression of aggrecan mRNA from day 1 and collagen type II from day 2. There was no significant expression of Sox9 or collagen type I until day 5 (Fig. 5).
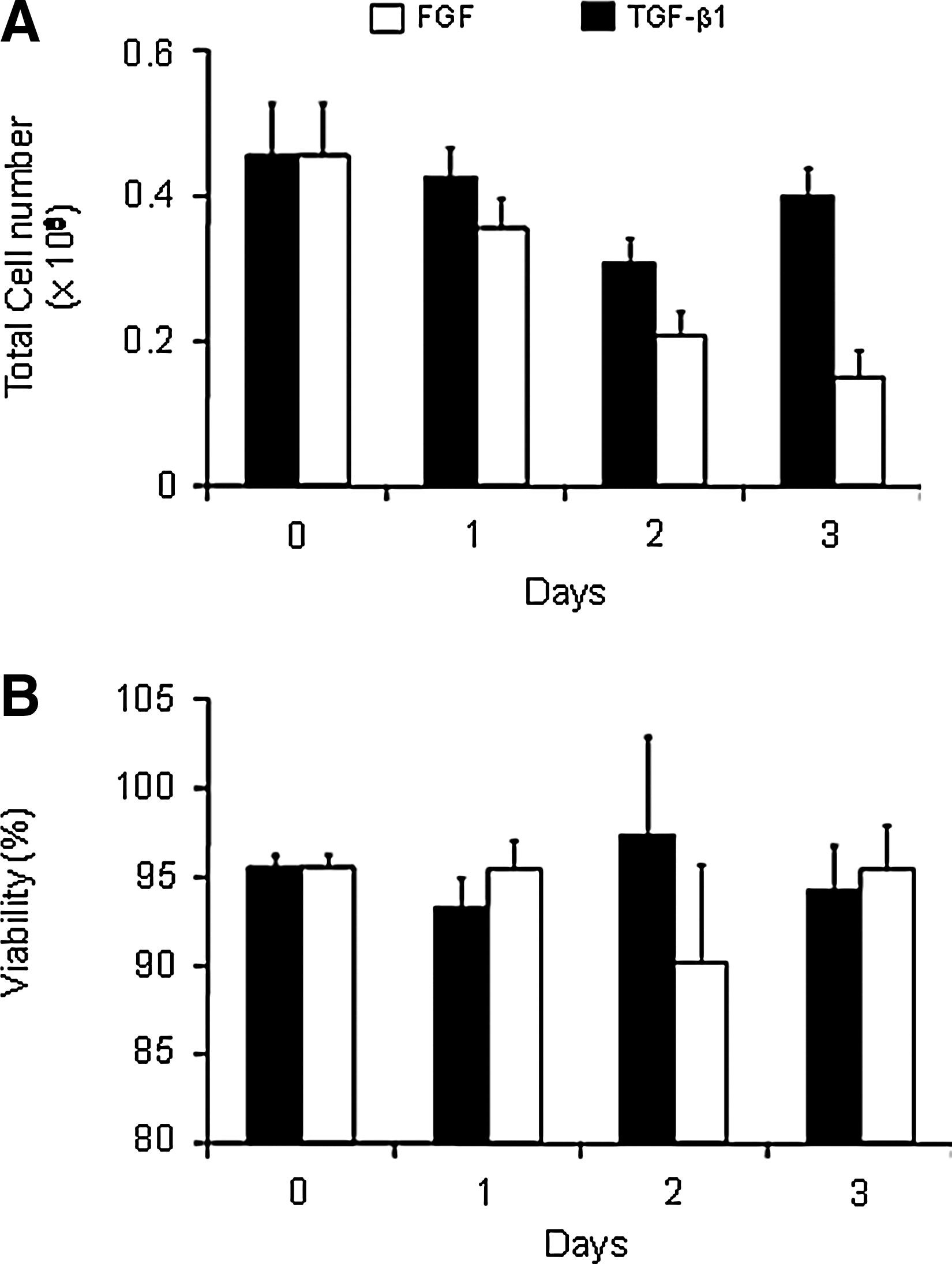
Total number and viability of cells in monolayer culture. Cells were trypsinized from 35-mm plates. Cell number and viability were determined by trypan blue exclusion on a hemocytometer.
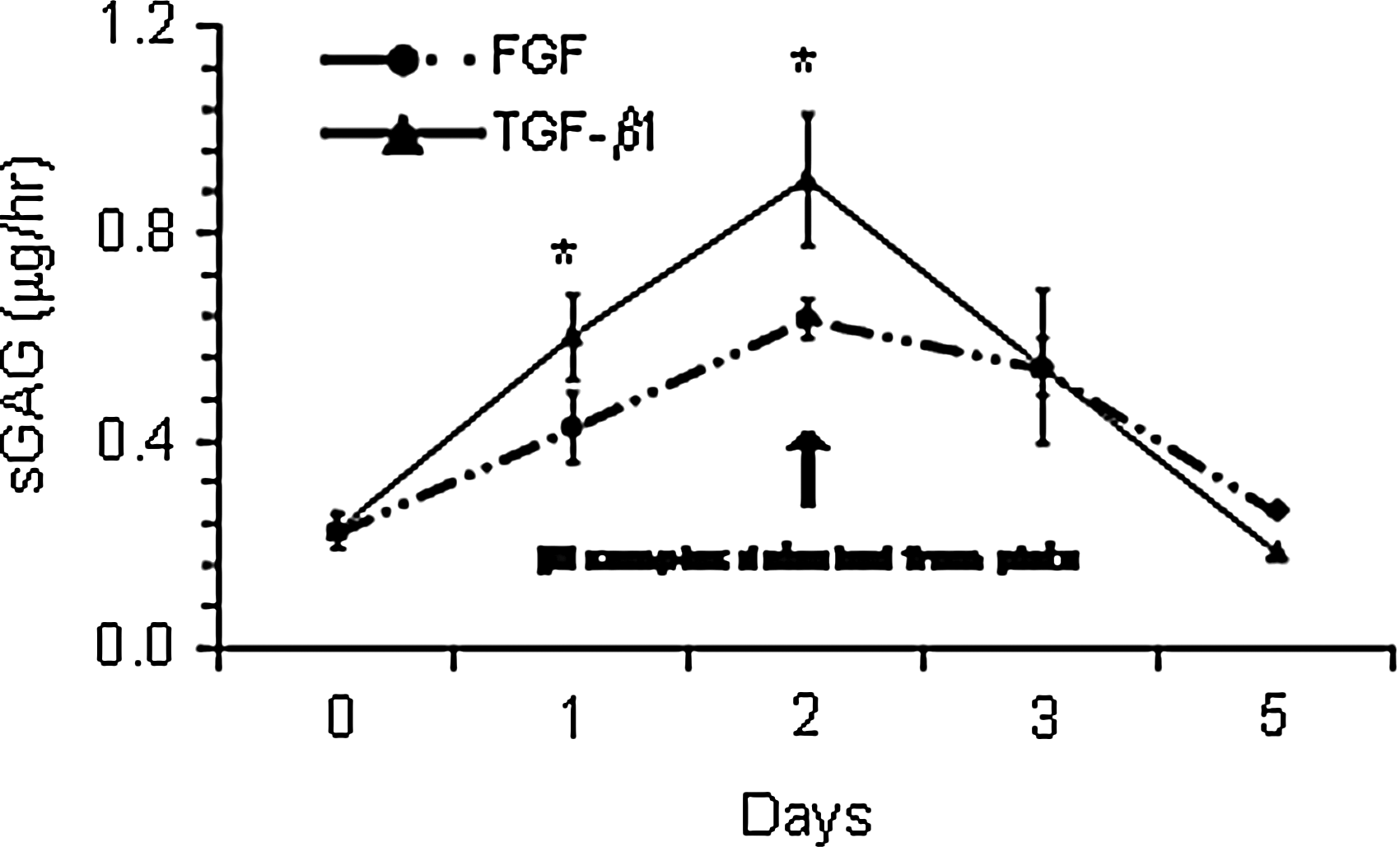
Sulfated glycosaminoglycans (sGAGs) released into the medium during monolayer chondrogenesis. *indicates difference from fibroblast growth factor (FGF) treatment.

Timecourse of mRNA expression of rat BMSCs in monolayer. *indicates difference from FGF treatment.
Cells released from day 2 TGF-β1 monolayer cultures and P1 FGF-treated cells were seeded in agarose to determine if predifferentiation of cells in monolayer culture influenced matrix accumulation in 3D. Very little matrix was produced by predifferentiated BMSCs in serum, ITS+, or with continued TGF-β1 stimulation. Light safranin-O was seen only in undifferentiated cells that were treated with TGF-β1 for 21 days in agarose culture (Fig. 6). After 21 days of culture in agarose, cell viability remained high (Fig. 6 inset).
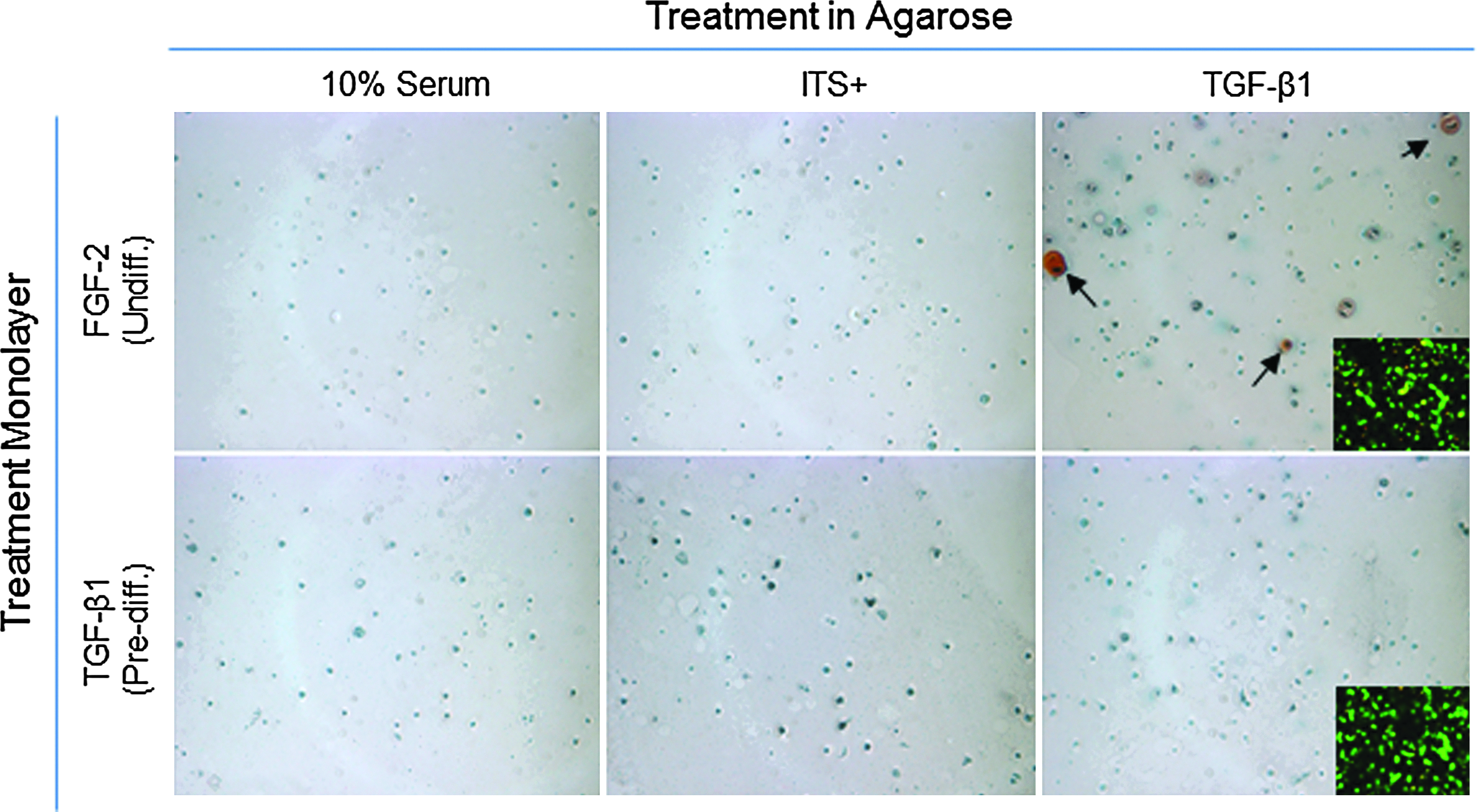
Safrinin-O staining and viability. Safrinin-O staining of GAG production after continued culture in agarose for 21 days. Proteoglycans were expressed only in agarose gel-containing cells that were expanded in the presence of FGF and cultured in the presence of transforming growth factor-β1 (black arrows). Inset: Viability of the BMSCs after transfer to agarose and continued culture for 7 days. Original magnification on both set of images was 10×. Color images available online at www.liebertpub.com/tea
BMSCs affect growth plate healing in a differentiation-dependent manner
To determine the effect of the BMSC differentiation state on the healing of growth plate defects, undifferentiated or predifferentiated cells were suspended in 0.5% SeaKem agarose and delivered into the defect. These were compared to defects left empty or filled with agarose alone. In agreement with our previous results, there were no differences in the volume fraction of the bone bridge formed within the defect in any of the treatment groups 56 days postsurgery (Fig. 7A).

Bone length and growth plate morphological parameters.
The tether BVF (an indication of growth plate fusion) of the growth plate surrounding the defect region in the empty and agarose-alone groups was higher than their respective contralateral controls. Defects that received cells in addition to agarose showed no increase in tether formation over their contralateral controls (Fig. 7B). All groups had a thinner defect growth plate than their contralateral control leg, and there was a trend suggesting that injection of agarose or agarose and cells improved the outcome of this parameter (p=0.062; Fig. 7C).
Limb length discrepancy (expressed as the percent reduction from the control leg) was improved (i.e., decreased) in all animals that received agarose injection into the defect (4.5%—hydrogel alone, 5.2%—predifferentiated, and 2.9%—undifferentiated) compared to the empty defect (7.8%). The limb length discrepancy was corrected in only the group treated with undifferentiated cells, and the defect legs of this group were significantly longer then the empty defect limbs (Fig. 7D). Representative micro-CT images are shown in Figure 7E, demonstrating the differences in growth plate morphology with treatment.
Discussion
Previous studies have found that stem cells isolated from various tissues can contribute to growth plate healing in vivo.20,21 These include the insertion of articular chondrocytes, growth plate chondrocytes, and stem cells.9–12,17,19–24 Hui et al. found that BMSCs decreased limb length discrepancy, but did not completely restore function. 21 Chen et al. found that periosteal-derived MSCs delivered in agarose generated a new growth plate and completely corrected angular deformity. 20 Based on these studies, further work is needed not only to characterize the chondrogenic potential of MSCs from various sources but also to determine the effect of the MSC differentiation state on the morphology of the remaining growth plate tissue and restoration of limb growth.
Interest has developed in applying a multistep approach to chondrogenic predifferentiation of BMSCs.25,26 Worster et al. found that equine BMSCs first stimulated with TGF-β1 in high-density monolayer and then seeded in fibrin discs continued to accumulate sGAGs once placed in hydrogel culture, and that additional IGF-I stimulation in 3D culture enhanced matrix production. 27 Using human BMSCs, Masuda et al. found that a cocktail of TGF-β3, IGF-I, and dexamethasone stimulates higher levels of aggrecan mRNA expression than cells seeded in 3D gel/polymer amalgams, and that chondrogenic predifferentiation of cells in 2D before seeding in 3D enhanced overall collagen II and aggrecan mRNA expression. 13 These studies demonstrate not only that BMSCs undergo chondrogenesis in high-density monolayer, but also that the chondrocytic phenotype is maintained after transfer to 3D culture, and that predifferentiation in monolayer increased expression of chondrogenic markers and matrix accumulation over 3D culture alone. In this study, we have shown that male rat BMSCs expressed the chondrocytic genotype in high-density monolayer culture with TGF-β1 stimulation. After transfer to agarose, however, they failed to produce a cartilaginous matrix even with continued TGF-β1 stimulation for 21 days. In contrast, P1 cells treated with FGF-2, but not TGF-β1, during 2D expansion did undergo chondrogenesis when placed in agarose and stimulated with TGF-β1 as previously reported. 12
A number of aspects may have contributed to the lack of in vitro matrix production of chondrogenic BMSCs seeded into agarose gels, including the passage point and culture conditions. Previously, we showed that female cells exposed to dexamethasone in agarose culture had high loss of cell viability, but dexamethasone also stimulated the greatest amount of matrix accumulation in these cells. 12 Passage 1, male BMSCs expanded in the presence of FGF-2 were chosen for this study due to their ability to maintain high viability in agarose culture. However, expansion with FGF-2 resulted in minimal matrix production in vitro, in agreement with our previous study. 12 The effects of dexamethasone were not evaluated in this study due to the high loss of cell viability we observed in female cells.
We have observed that the chondrogenic differentiation potential of male and female rat BMSCs is dissimilar (unpublished results), and sex-based disparities in the osteochondral differentiation of muscle-derived progenitor cells have also been noted. 28 Like female cells, there is likely an optimal point in male BMSC expansion for favorable chondrogenic matrix production. The cells remained viable throughout the in vitro culture period, so it is unlikely that cell death is the cause of the lack of matrix secretion. Further optimization of the regimen of growth factor and/or steroid stimulation for monolayer differentiation and 3D culture may improve matrix synthesis.
Despite the lack of matrix produced by these cells under 3D conditions in vitro, we were able to test our initial hypothesis and have shown that the differentiation state of the cells implanted into a centralized defect had an effect on growth plate morphological parameters and limb length discrepancy. Contrary to our hypothesis, predifferentiation did not have a positive effect on limb length discrepancy. When agarose with or without a cellular component was implanted into growth plate defects, there was no reduction of bone bridge formation within the defect or restoration of growth plate thickness in agreement with our previous study. 8 We also saw that while inclusion of cells in the agarose resulted in restoration of normal tether formation, there was no dependence of this parameter on the differentiation state of the implanted cells. However, a differentiation-dependent effect on growth of the injured limb was observed. Injection of undifferentiated BMSCs into the defects resulted in a recovery of growth in the defect limb resulting in a correction of limb length discrepancy.
This study and our previous work8,19 have raised some interesting questions about the role of tether formation in growth plate fusion. Tether formation has been noted in the normal closure dynamics of dog, porcine, and human histological sections,29,30 as well as other rat models.31,32 Despite the evidence that this process has a role in normal growth plate morphology and fusion, there are few investigations into the biomolecular mechanisms through which tethers form or the change in these pathways due to injury. Recently, we showed that tethers are present in the femoral growth plates of mice, and that their formation and three-dimensional distribution are regulated in part by vitamin D receptor signaling, 19 and that tether formation was highly correlated with limb length discrepancy. 8 In a growth plate defect model in rat tibiae, tethers were only observed in injured limbs, and associated increases in chondrocyte apoptosis and osteocalcin mRNA expression were found, 32 which suggests that tether formation in this system is induced by initiating the process of endochondral ossification.
In this study, the transfer of BMSCs into growth plate defects did inhibit excess tether formation in this model, but unlike undifferentiated cells, predifferentiated cells did not correct limb length discrepancies. The disconnection between these parameters in this study may be due to the differential response of chondrogenic cells at specific levels of chondrocyte maturation. 31 In the growth plate, the chondrocyte differentiation state is important to their response to autocrine and paracrine regulators of cell phenotype—progression from a resting state to a mineralizing one. For instance, it is well known that the predominant effect of Indian Hedgehog is on proliferating zone cells to stimulate parathyroid-related protein production, which in turn exerts its effect on cells in the hypertrophic zones. 33 The in vitro response of both cell types used in this study demonstrates that these cell types were phenotypically different. The outcome of this study suggests that both undifferentiated and predifferentiated BMSCs secrete factors that reduce tether formation, possibly through decreasing apoptosis in the adjacent healthy growth plate. In future studies, immunohistochemical analysis will be performed to characterize the change in the phenotype of the implanted cells with time. Further studies examining the process of tether formation and cross-talk between the implant and native tissue are required to elucidate the relationship between the implant cell phenotype, tether formation, and limb growth.
Conclusions
In this study, we have demonstrated that the potential for BMSCs to reduce limb length discrepancy due to a centralized defect through the growth plate of the distal femur is dependent on the differentiation state of the cells. Specifically, delivery of predifferentiated BMSCs did not restore growth function compared to empty defects, whereas undifferentiated BMSCs corrected the limb length discrepancy. Inclusion of cells in the implant also reduced tether formation to control levels. It is apparent from this and our previous study 8 that bone bridge formation within a growth plate defect is not a good indicator of growth plate healing potential, since the volume fraction of mineralized tissue within the bridge was consistently the same for every treatment group, whereas the health of the remaining growth plate tissue, as indicated by tether formation, did correspond with growth disturbance. The process of growth plate healing needs to be better understood on a tissue and cellular level to develop appropriate treatments. To our knowledge, this is the first study examining the role of cell differentiation state in the healing of growth plate defects.
Footnotes
Acknowledgments
The authors would like to thank Mela Johnson, Ken Dupont, and Yash Kolambkar for their help during surgeries, Angela Lin for aid in developing the micro-CT protocols, and Chris Dosier for his aid in collecting the micro-CT data. This work was funded by the Georgia Tech/Emory Center for the Engineering of Living Tissue (NSF EEC-9731643) and a Whitaker Foundation Graduate Fellowship.
Disclosure Statement
No competing financial interests exist.