
Abstract
Human adipose-derived stem cells (hASCs) are currently a point of focus for bone tissue engineering applications. However, the ex vivo expansion of stem cells before clinical application remains a challenge. Fetal bovine serum (FBS) is largely used as a medium supplement and exposes the recipient to infections and immunological reactions. In this study, we evaluated the osteogenic differentiation process of hASCs in poly-3-hydroxybutyrate-co-3-hydroxyvalerate (PHB-HV) scaffolds with the osteogenic medium supplemented with pooled allogeneic human serum (aHS). The hASCs grown in the presence of FBS or aHS did not show remarkable differences in morphology or immunophenotype. The PHB-HV scaffolds, which were developed by the freeze-drying technique, showed an adequate porous structure and mechanical performance as observed by micro-computed tomography, scanning electron microscopy (SEM), and compression test. The three-dimensional structure was suitable for allowing cell colonization, which was revealed by SEM micrographs. Moreover, these scaffolds were not toxic to cells as shown by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. The differentiation capacity of hASCs seeded on scaffolds was confirmed by the reduction of the proliferation, the alkaline phosphatase (AP) activity, expression of osteogenic gene markers (AP, collagen type I, Runx2, and osteocalcin), and the expression of bone markers, such as osteopontin, osteocalcin, and collagen type I. The osteogenic capacity of hASCs seeded on PHB-HV scaffolds indicates that this scaffold is adequate for cell growth and differentiation and that aHS is a promising supplement for the in vitro expansion of hASCs. In conclusion, this strategy seems to be useful and safe for application in bone tissue engineering.
Introduction
Tissue engineering, a multidisciplinary field that combines cells, scaffolds, and growth and differentiation factors to develop biological substitutes for the restoration, maintenance, or improvement of tissue function, 4 is an attractive science to bypass the limitations of autologous bone grafting. The scaffolds act as a temporary matrix for cell guidance and extracellular matrix deposition. Several materials, such as metals, ceramics, and polymers, have been used. Nevertheless, biodegradable polymers of natural or synthetic origin are believed to be ideal to develop scaffolds for bone regeneration. 5
Among the natural polymers, poly-3-hydroxybutyrate-co-3-hydroxyvalerate (PHB-HV) is a polyester that is produced by a variety of microorganisms. PHB-HV is an excellent biomaterial candidate due its properties, such as natural origin, biodegradability, biocompatibility, nontoxicity, stereospecificity, piezoelectricity, optical activity, and thermoplasticity.6,7 These attractive features enable the manipulation of PHB-HV to obtain the appropriated characteristics for bone substitutes. 8
Adipose tissue provides a suitable source of stem cells that can be combined with scaffolds in tissue engineering applications. Human adipose-derived stem cells (hASCs) can be harvested in large amounts from lipoaspirates, a disposable byproduct of cosmetic surgery, can differentiate into lineage-specific cells, including those of osteogenic lineage,9,10 in a controlled manner, 11 and can be safely and effectively transplanted into an autologous or allogeneic recipient.11,12 However, before hASCs can be used in clinical applications, it is necessary to expand these cells ex vivo to acquire the required amount of cells, and the media that are used for cell culture influence the growth and differentiation of these cells.
Proteins contained in fetal bovine serum (FBS), a common culture medium supplement, can start an immune response against the graft in patients who received a bone implant. The immune reaction can be triggered by the exposure to xenogeneic proteins and bacterial or viral infections. 12 Recently, some studies have been conducted to replace the FBS with human supplements.13,14
In this study, pooled allogeneic human serum (aHS), a culture medium supplement, was evaluated as a substitute to FBS during the isolation, growth, and osteogenic differentiation of hASCs in PHB-HV scaffolds.
Materials and Methods
aHS obtention
The aHS was obtained from the whole blood of healthy donors with blood group typed and described in Table 1. In an attempt to produce a pool of aHS, the blood from different blood types was mixed. From each donor, 50 mL of whole blood was collected with vacutainer tubes (Biosciences) without coagulants and was allowed to clot spontaneously overnight at 4°C. After the blood had clotted, the serum was separated by centrifugation at 252 g at 20°C for 15 min. The serum from at least five different donors was collected and mixed to produce the aHS. The aHS was inactivated at 56°C for 30 min. Subsequently, the aHS was aliquoted in sterile tubes and frozen at −20°C. This procedure was approved by the Ethics Committee in Research from the Universidade Federal de Minas Gerais (no. ETIC 0023.0.203.000-11).
Basal medium
Dulbecco's modified Eagle's medium–high glucose (Sigma-Aldrich) was supplemented with 5 mM sodium bicarbonate (Cinética Química Ltda), penicillin (100 U/mL), streptomycin (0.1 mg/mL), amphotericin B (0.25 μg/mL), (Sigma-Aldrich), gentamicin (60 mg/L; Schering-Plough), and 10% serum: aHS or FBS (Cripion Biotecnologia LTDA).
Osteogenic medium
Osteogenic medium (OM) consists of a basal medium supplemented with aHS or FBS, 50 μg/mL ascorbate-2-phosphate (Ecibra), 10 mM β-glycerophosphate (Sigma-Aldrich), and 0.1 μM dexamethasone (Aché). 10
Isolation and culture of hASCs
Human adipose tissue was harvested from healthy patients that had abdominal reduction surgery for aesthetic reasons at “Núcleo de Cirurgia Plástica,”. This study was approved by the Ethics Committee of the Universidade Federal de Minas Gerais (no. ETIC 0023.0.203.000-11). No diabetes, hepatitis, metabolic diseases, or other systemic complications were reported from these donors. The Núcleo de Cirurgia Plástica supplied the information about ABO blood and Rh group of lipoaspirate donors (Table 1).
The isolation and culture of hASCs was performed as described elsewhere. 10 Briefly, the raw lipoaspirates were washed with phosphate-buffered saline (PBS) and enzymatically digested with 0.075% collagenase type I (Life Technologies) in PBS at 37°C for 1 h. Subsequently, the stromal vascular fraction was isolated by centrifugation at 252 g for 10 min, and the pellet was resuspended in the basal medium and plated into cell culture flasks (Sarstedt). The flasks were incubated at 37°C in 5% CO2 and a humidified atmosphere. After 12–28 h of incubation, the flasks were extensively washed to remove the nonadherent cells, and the plastic adherent cells that remained were termed hASCs.
The hASCs were maintained at subconfluent levels with 3 weekly medium changes. When the cells reached approximately 80% confluence, they were treated with 0.05% Trypsin-ethylenediaminetetraacetic acid (Invitrogen) for 3–5 min to detach from the surface of the culture flask. The resulting suspension was replated in new cell culture flasks. The cells were expanded this way until passage 4 before they were used in the assays.
The cell population doubling time (DT) was determined by harvesting and counting cells with a Neubauer chamber (HBG). The DT was calculated by using the formula y=(T2|T1)×log10 2÷log10(N2÷N1), where N1 is the plated cell number, N2 is the cell number at harvest, T1 is the time when the cells were plated, T2 is the time when the cells were harvested, and y is the DT.
Flow cytometry
Immunophenotypic analyses were performed at all passages with the samples of hASCs cultured in the basal medium supplemented with aHS or FBS. The protocol was adapted from a study that has been previously described. 15 Briefly, 1×106 cells were incubated with each monoclonal antibody for 30 min at 4°C. Then, the cells were washed twice with PBS. For some groups of markers (unconjugated antibodies), the cells were incubated with a secondary antibody for 30 min at 4°C and were protected from light. Finally, the cells were washed in PBS and fixed with 200 μL of 1% paraformaldehyde. The hASCs were incubated with only Alexa Fluor 488 goat anti-mouse immunoglobulin G (IgG) (H+L) (Cat. A11001; Invitrogen) to assess the background fluorescence of the secondary antibody as a control. The following unconjugated mouse monoclonal antibodies were used: integrin β1chain—CD29 (Cat. sc-9970; Santa Cruz Biotechnology), HCAM—CD44 (Cat. sc-7297; Santa Cruz Biotechnology), hematopoietic stem cell (HSC)-associated marker—CD34 (Cat. ab8147-500; Abcam), and pan-leukocyte marker CD45 (Cat. 616256; BD Biosciences), which were stained with Alexa Fluor 488 goat anti-mouse IgG (H+L) as a secondary antibody. The following conjugated antibodies were used: ecto-5′ nucleotidase—CD73-phycoerythrin (Cat. 550257; BD Biosciences), human leukocyte antigens (HLA)-ABC-fluorescein isothiocyanate (FITC) (Cat. ab20313-100; Abcam), and HLA-DR-FITC (Cat. ab36775-500; Abcam). The hASCs were analyzed by flow cytometry with a FACSCalibur (Becton Dickinson Immunocytometry System) using CELLQuest acquisition software (BD Biosciences). A minimum of 20,000 events were acquired for each sample of cells analyzed for each surface marker. Cell marker expression was determined by comparison with control and the data were analyzed with WinMDI 2.8 software.
Scaffold processing
The PHB-HV polymer that was used in this study was provided by PHB Industrial, has a molecular weight of approximately 425,692 kDa and was used with no further purification. The scaffolds were produced by the freeze-drying technique. Briefly, the polymer was dissolved in an accurately measured amount of chloroform (Sigma-Aldrich) at 60°C under constant magnetic agitation until the complete dissolution of the polymer; a polymer solution with a final concentration of 7.5% PHB-HV (m/v) was obtained. After generation of a homogeneous polymer solution, a water phase (Acetic Acid Panreac) was added to obtain an emulsion that was frozen at −80°C and subsequently freeze dried (Telstar) for 94 h. The produced scaffolds were cut into smaller pieces, approximately 6.0 mm in diameter and height, sterilized by 15 kGy gamma radiation for 30 min, and used for the cell culture studies.
Scaffold characterization
Scanning electron microscopy
The architecture of the scaffolds in terms of the surface characteristics, pore sizes, and pore distribution was preliminarily observed by scanning electron microscopy (SEM). For these analyses, the scaffolds were coated with gold before observation by SEM (DSM950; Zeiss) at 15 kV and a working distance of 20 mm in the Microscopy Center of Biological Sciences Institute (CEMEL), Universidade Federal de Minas Gerais.
Micro-computed tomography
Micro-computed tomography (micro-CT; SkyScan 1072) was used as a nondestructive technique to evaluate the internal morphology of the scaffolds and to quantify the porosity and pore size. The scaffolds were analyzed with a high-resolution mode of 6.9 μm x/y/z and an exposure time of 1792 ms. The energy parameters defined in the scanner were 50 kV and 185 μA. After the scaffolds were scanned, the obtained data were analyzed by using NRecon and CT-An to produce binary (white and black) images. A threshold (to distinguish the polymeric material from pore voids) of 60–255 was chosen and maintained constantly for all of the scanned specimens. CT-Vol image software (Sky Scan) was also used for the three-dimensional reconstruction of the scaffolds.
Compression test
Compression tests were performed with a universal tensile testing machine (Instron 4505 Universal Machine). The tests were performed under compression loading using a cross-head speed of 2 mm min−1. The scaffold's mechanical properties were analyzed in both the transverse and radial (perpendicular to transverse) directions to confirm if the observed morphological anisotropy would affect the mechanical behavior. Each specimen measured 6 mm in diameter and 4 mm in thickness. For each tested direction, the presented results are the average of at least five specimens.
Energy dispersive spectroscopy
The scaffolds were coated with gold and analyzed in the Microscopy Center of Universidade Federal de Minas Gerais with energy dispersive spectroscopy equipment (Quanta 200-FEG-FEI 2006). The elements that constitute the scaffolds after the polymer processing were confirmed.
Seeding hASCs on PHB-HV scaffolds
The hASCs that were cultured in aHS or FBS were seeded at a density of 5×103 cells/scaffold under static conditions in a 48-well plate. After 2 h of cell attachment at 37°C, 5% CO2 and humidified atmosphere, 2 mL of the basal medium with aHS or FBS was added to each well, and the constructs were incubated. For some assays, to induce osteogenic differentiation, the basal medium was changed to OM supplemented with aHS or FBS and the constructs were cultured for periods of 7–28 days. The medium was replaced three times weekly.
SEM sample preparation for cell adhesion on scaffolds and morphological analysis
The adhesion, distribution, and morphology of hASCs seeded on PHB-HV scaffolds were observed by SEM. The constructs were washed in PBS and fixed with 2.5% glutaraldehyde in PBS for at least 2 h. Afterward, the samples were washed three times with PBS for 10 min and incubated with 1% osmium tetroxide (Electron Microscopy Sciences) in PBS at 4°C in the dark for 1 h. The constructs were again washed with PBS and incubated in 1% tannic acid in water for 20 min. The constructs were subsequently washed with PBS and incubated in 1% osmium tetroxide in water at 4°C in the dark for 1 h. Then, the constructs were washed with PBS and dehydrated through stepwise incubation in a series of graded ethyl alcohols: 30%, 50%, 70%, 90%, and 100% ethanol; each construct was incubated twice in each alcohol solution for 10 min. Finally, the constructs were reduced in size with liquid nitrogen. The resulting fragments were dried with a Critical Point Dryer (Balzers CPD-020). The samples were coated with gold and analyzed in the Microscopy CEMEL, Universidade Federal de Minas Gerais by SEM (DSM950; Zeiss) at 10 kV and a working distance of 14 mm.
In vitro cytotoxicity and cell proliferation during differentiation
Cell viability and proliferation were assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Cat. M-6494; Invitrogen) as previously described. 16 The constructs were cultured in the basal medium and OM supplemented with aHS or FBS at 37°C, 5% CO2, and humidified atmosphere. At the end of each time point, the medium was removed and 170 μL of the MTT solution (5 mg/mL) and 210 μL of the new basal medium were added to each well. Two hours later, the formazan crystals were dissolved with 210 μL of sodium dodecyl sulfate (SDS)-10% HCl. After 18 h, 100 μL of solution was transferred to a 96-well plate, and the optical density (OD) was measured at 595 nm. Scaffolds without cells were subjected to the same procedure, as a control.
Alkaline phosphatase activity
The alkaline phosphatase (AP) activity was evaluated with the 5-Bromo-4-chloro-3-Indolyl phosphate (BCIP)/Nitroblue tetrazolium salt (NBT) Kit assay as described by the manufacturer (Cat. 00-2209; Invitrogen). The constructs were cultured with the basal medium and the OM supplemented with aHS or FBS at 37°C, 5% CO2, and humidified atmosphere. On days 7, 14, 21, and 28 of induction, the supernatant of each well was removed and the constructs were washed twice with PBS. Then, 200 μL of the BCIP-NBT solution was added to each well and incubated. Two hours later, the insoluble purple precipitants were dissolved with SDS-10% HCl. After 18 h, 100 μL of solution was transferred to a 96-well plate and the OD was measured at 595 nm. Scaffolds without cells were subjected to the same procedure, as a control.
Indirect immunofluorescence
The constructs from each time of induction were processed for cryosection for indirect immunofluorescence and to confirm osteogenic differentiation.
Briefly, the constructs were washed twice in PBS and fixed in 4% paraformaldehyde for 24 h. Afterward, the constructs were washed in PBS, incubated for 6 h in 10% sucrose solution at 4°C, and then transferred to another solution of 20% of sucrose for 24 h at 4°C. The constructs were embedded in a frozen gel medium (Easy Path) at −30°C. Subsequently, 10-μm cryosections were obtained with a cryostat (Leica CM1510S-Leica; Mycrosystems).
The cryosections were thawed for 10 min and hydrated through incubation in a series of graded ethyl alcohols (100%, 95%, and 80%), water, and PBS; each incubation was performed twice for 5 min. For permeabilization, the cryosections were incubated for 1 h in PBS containing 0.25% Triton X-100 and 2% bovine serum albumin (BSA). Afterward, the cryosections were incubated for 1 h in PBS containing 2% BSA 0.1% Tween 20. The cryosections were blocked for 1 h at room temperature with a blocking buffer containing 1% BSA and 5% goat serum in PBS. Subsequently, the cryosections were incubated for 2 h in the blocking buffer containing the following antibodies: the rabbit polyclonal antibody against osteopontin (Cat. ab8448; Abcam), the mouse monoclonal antibody against collagen type I (Cat. ab6308-100; Abcam), and the mouse monoclonal antibody against osteocalcin (Cat. ab13418; Abcam). Afterward, the cryosections were washed with PBS and incubated with the respective Alexa Fluor 488 goat anti-rabbit IgG (H+L) (Cat. A11008; Invitrogen) and Alexa Fluor 488 goat anti-mouse IgG (H+L) (Cat. A11001; Invitrogen) antibodies for 1 h. The cryosections were washed with PBS, and the nuclei were stained with 1 μg/mL Hoeschst 33258 pentahydrate (Cat. H3569; Invitrogen) for 20 min.
The slides were mounted with Hydramount Aqueous media (Cat. HS-106; National diagnostics) and analyzed with a Zeiss LSM 510—Meta confocal microscope. The images were taken at CEMEL, Universidade Federal de Minas Gerais.
Reverse transcriptase–polymerase chain reaction
Reverse transcriptase–polymerase chain reaction (RT-PCR) was performed to evaluate the relative mRNA levels expressed by the AP, collagen type I, Runx2, and osteocalcin genes at constructs cultured with OM-aHS from each time of induction and hASCs in monolayer cultured in the basal medium supplemented with aHS (non induced) as a control of expression.
Total cellular RNA was extracted from constructs up to 7, 14, 21, and 28 days and hASCs cultured in monolayer with Trizol (Invitrogen), as described by the manufacturer. Total RNA samples were treated with the DNase (Promega; Cat. #M6101), according usage information. A NanoDrop ND-1000 Microspectrophotometer was used to measure the total RNA amount (ng/μL). Total RNA of each sample were treated with the RevertAid™ H Minus M-MuLV RT (Fermentas; Cat. #K1631) to generate cDNA using an oligo(dT) adapter primer. Next, PCR amplification was performed for AP, collagen type I, Runx2, and osteocalcin, and the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA. The specific primers (AP, Runx2, osteocalcin, collagen type I, and GAPDH) used were described at Table 2. The RT-PCR products were analyzed through 1% agarose gel electrophoresis and visualized with ethidium bromide.
Statistical analyses
All experiments were repeated three or more times with triplicate samples. Statistical analyses were performed by using Graph Pad Prism 5.0. The values were represented as the mean±standard error of the mean. Statistical differences were calculated by using analysis of variance and Bonferroni's post-tests. Differences were considered significant at p<0.05.
Results
Isolation and culture of hASCs
The isolation of hASCs in the basal medium supplemented with aHS was successful. The adherent cells formed several colonies and spread easily with aHS. Little differences in morphology could be observed; hASCs cultured in aHS and in FBS had a fibroblastoid morphology, but the hASCs were smaller when they were cultured in aHS (Fig. 1). The cells cultured in aHS were more dense and were less adherent to the tissue culture plastic. The time required for the cells to detach from the tissue culture plastic during trypsinization was approximately 3 min for the hASCs cultured with aHS and approximately 5 min for the hASCs cultured with FBS. Moreover, when the hASCs reached confluency, the cells cultured in aHS had a tendency to cluster.
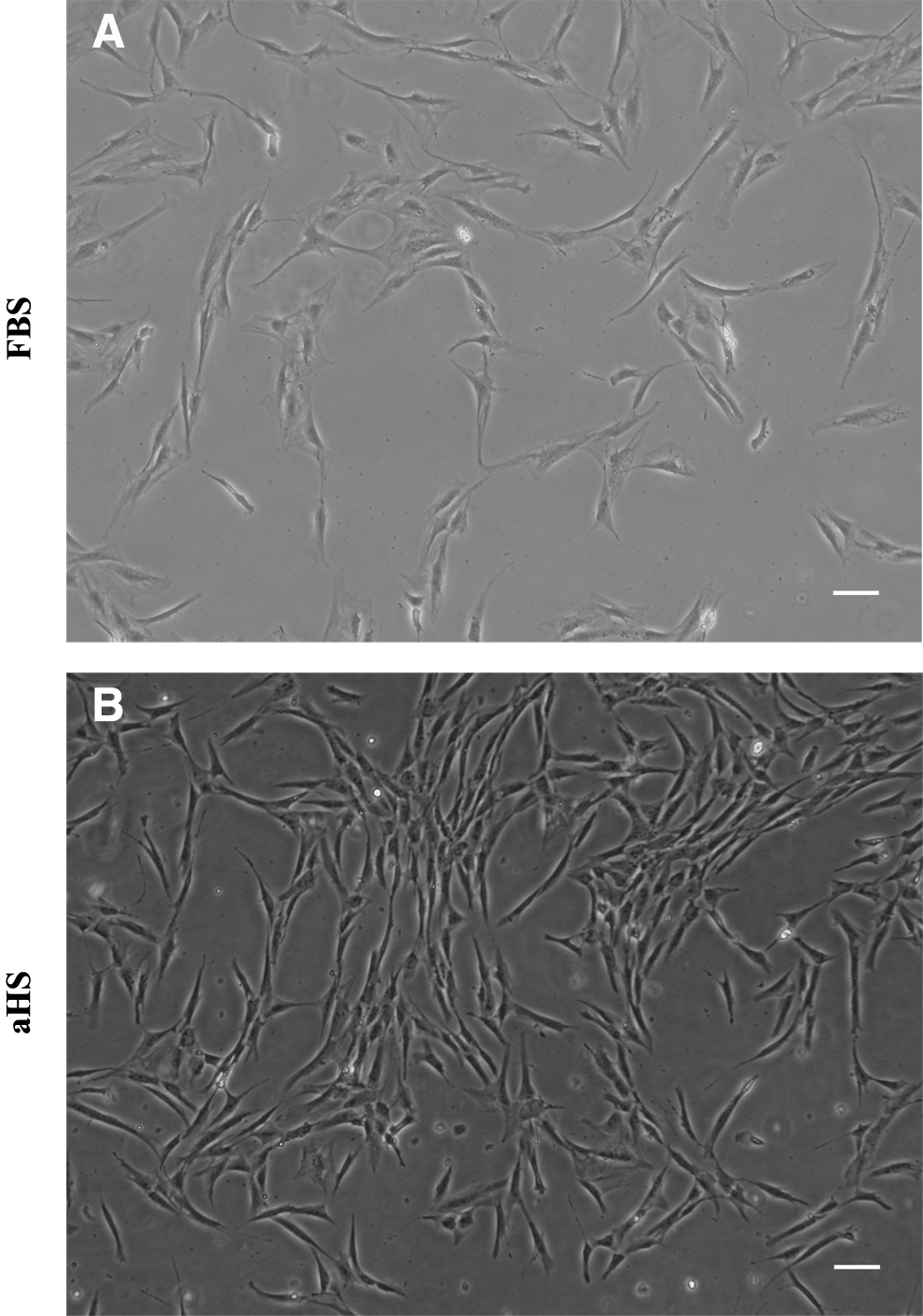
The morphology of human adipose-derived stem cell (hASC) cultures, at passage 4, from a representative donor. The hASCs were cultured in a medium supplemented with
The DT was 120±17 h for hASCs in FBS and was 28±5 h for hASCs in aHS.
Flow cytometry
Cell size and granularity were measured by using the forward- and side-scatter settings shown in Figure 2A and C. Representative histograms are shown in Figure 2B and D. The levels of marker expression did not change significantly over the passages and between cells cultured in medium supplemented with aHS or FBS. The most of hASCs cultivated in aHS as in FBS were positive for mesenchymal stem cell (MSC) markers. Approximately 94.35%±5.00% of hASCs cultured in aHS expressed CD29, 98.04%±1.34% expressed CD44 and 90.18%±9.96% expressed CD73. There was low expression of HSC-associated markers: 1.48%±0.42% of hASCs expressed CD34 and 1.02%±0.71% expressed CD45. Moreover 95.98%±2.45% stained positively for HLA-ABC and lacked expression of HLA-DR, with only 0.59%±0.96% of cells stained positively. There was no significant difference in the percentage of expression of markers in cells cultivated in FBS, approximately 92.33%±2.00% of hASCs expressed CD29, 97.40%±1.22% expressed CD44, 96.55%±0.76% expressed CD73 and 91.76%±1.45% expressed HLA-ABC, 3.8%±0.45% expressed CD34, 0.11%±0.03% expressed CD45 and 0.08%±0.04% expressed HLA-DR.

The expression of mesenchymal stem cell markers, as assessed by flow cytometry, by different donor-derived human adipose stem cells at passage 4 and cultured in
Scaffold characterization
The conducted morphological analyzes have shown that PHB-HV scaffolds possess highly interconnected porosity with an anisotropic pore shape: more round pores in the transverse direction and laminar pores in the radial direction. Moreover, micro-CT analysis revealed that the scaffolds exhibit a porosity of 88.1%±0.3% and an average pore size of 163.5±0.1 μm (Fig. 3).

Representative
The mechanical tests revealed that the processed scaffolds have better mechanical properties in the transverse direction than in the radial direction. The measured compressive moduli for both directions were 32.09±0.273 MPa and 1.24±0.307 MPa in the transverse direction and in the radial direction, respectively.
The energy-dispersive X-ray spectroscopy (EDS) analysis of the scaffolds detected the presence of C, O, and Au (from the processing technique) elements on the surface, which confirmed the organic composition of the specimens.
Cell adhesion on scaffolds and morphological analysis
The hASCs seeded on PHB-HV scaffolds were able to adhere to the surface of the scaffolds from 7 days of culture onward, as observed by SEM (Fig. 4). The SEM micrographs show high cell density and viability in the inner regions, without occlusion of the pores. The cells were flattened and adhered to the scaffold surface by cytoplasmic extensions (Fig. 4B). There was cell communication between neighboring cytoplasmic fibers (Fig. 4C), and some cells presented vesicles on the surface of the cell membrane (Fig. 4D).
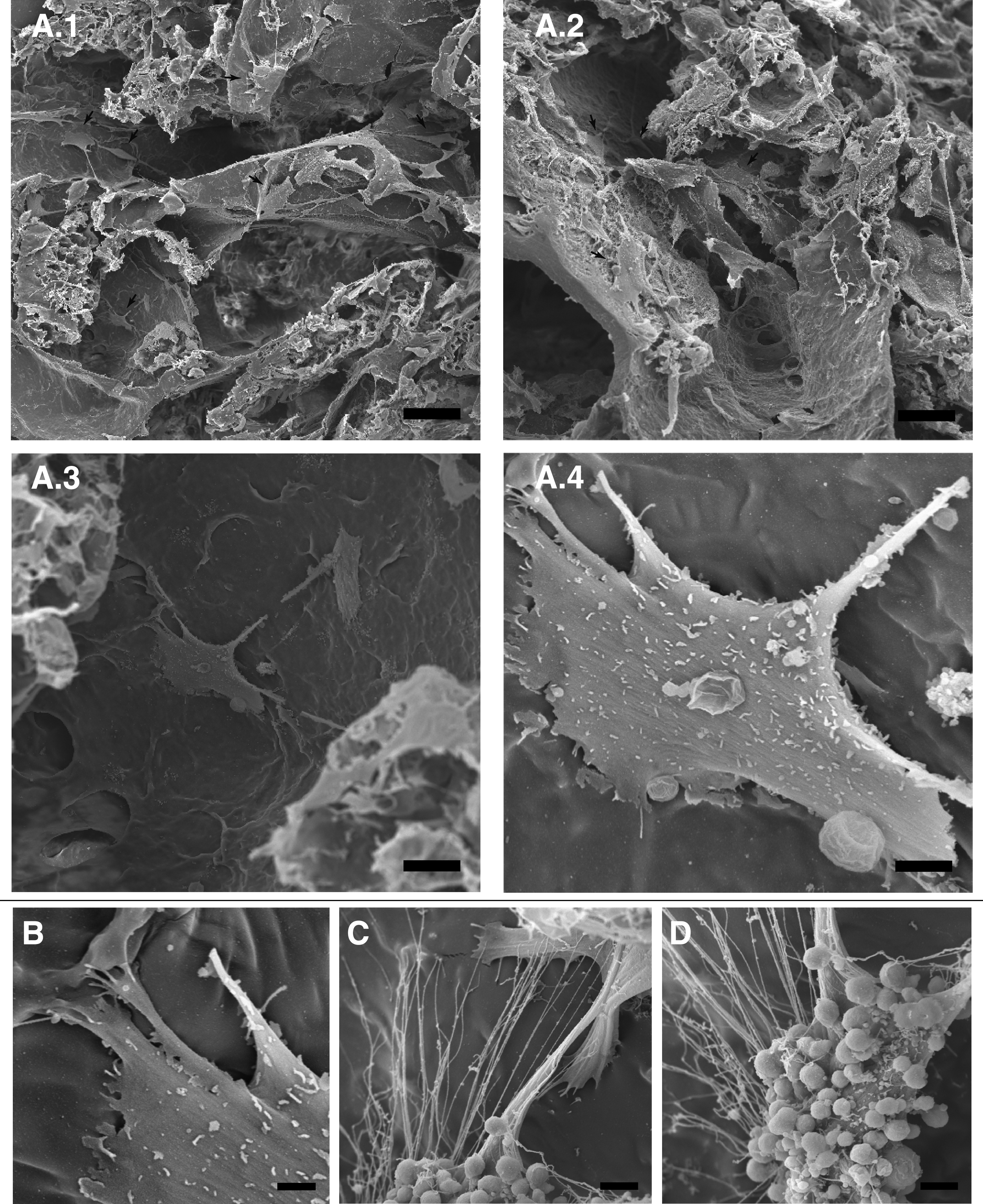
SEM micrographs of hASCs
In vitro cytotoxicity and cell proliferation during differentiation
The hASCs seeded on PHB-HV scaffolds were able to metabolize the MTT into formazan crystals, which demonstrated high viability and proliferation as a function of time (Fig. 5). During the osteogenic induction of hASCs, the results of the OD showed an initial high proliferation rate until day 14, when they exhibited superior cell proliferation compared to hASCs cultured in the basal medium in both FBS and aHS. The results showed that after day 14, the cells stopped proliferating, while the hASCs cultured in the basal medium continued to proliferate (Fig. 5).
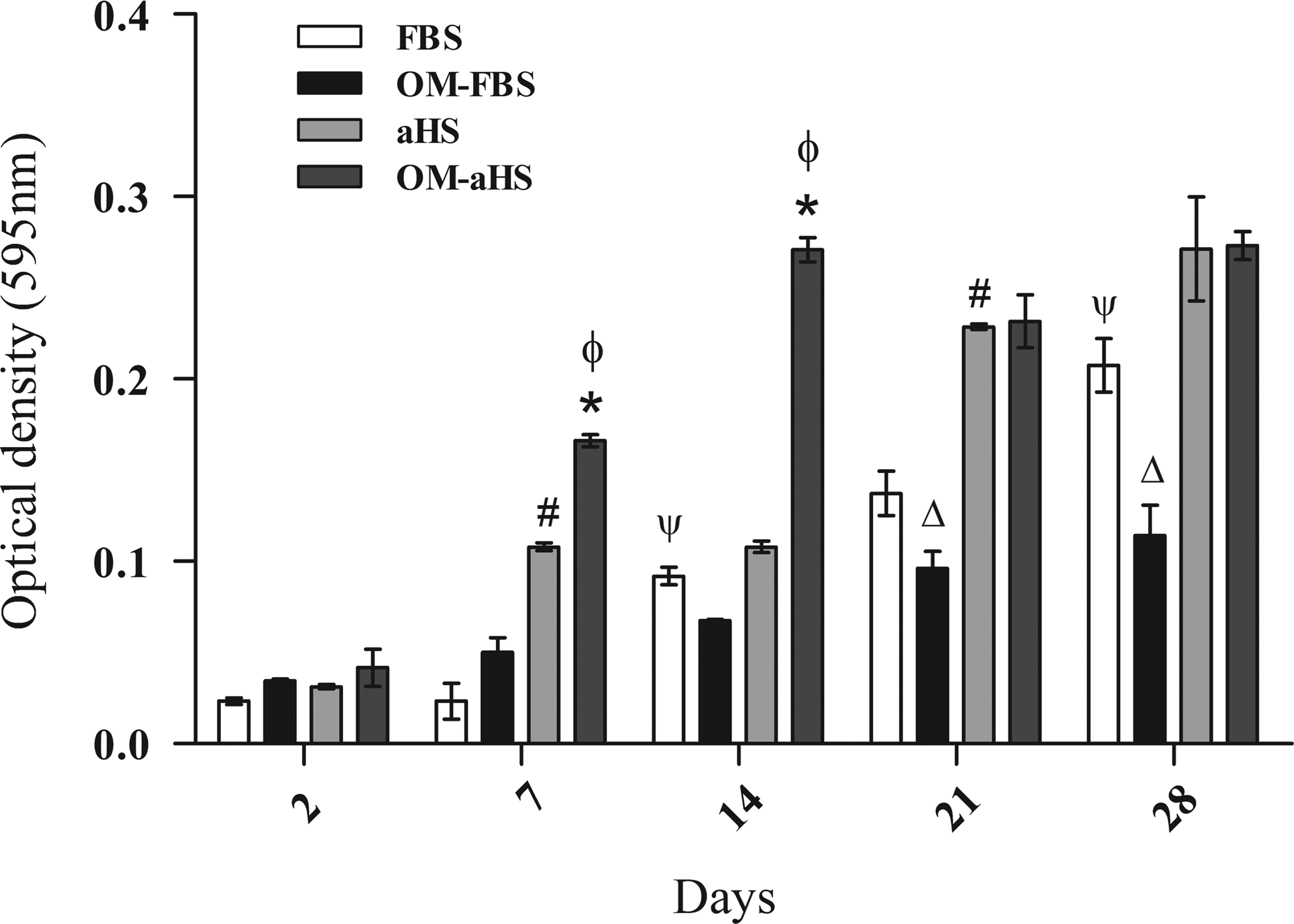
The cytotoxicity and proliferation of hASCs seeded on PHB-HV scaffolds as assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay during osteogenic differentiation. The results are based on the optical density at 595 nm and represent the viability and proliferation of hASCs seeded on PHB-HV scaffolds as a function of time in basal medium supplemented with FBS or aHS and OM-FBS or OM-aHS. n=3, from different cultures. The results represent the mean±standard error of the means. *p<0.05 comparing the condition of OM as a function of time for aHS; #p<0.05 comparing the condition of basal medium with aHS or Ψp<0.05 with FBS as a function of time; and øp<0.05 comparing the two groups-basal medium×OM for aHS and Δp<0.05 for FBS. OM, osteogenic medium.
AP activity
The results obtained from the NBT-BCIP assay showed a typical pattern of AP activity from hASCs cultured in OM that was similar or less than the AP activity of the control group at all the evaluated times, except at day 21 for OM-aHS, when the cultures in OM-aHS presented higher levels of activity. After day 21, the AP activity again decreased in these cultures (Fig. 6). The high level of AP activity at OM-FBS cultures was observed at day 14, but the difference was not statistically significant regarding the basal medium.
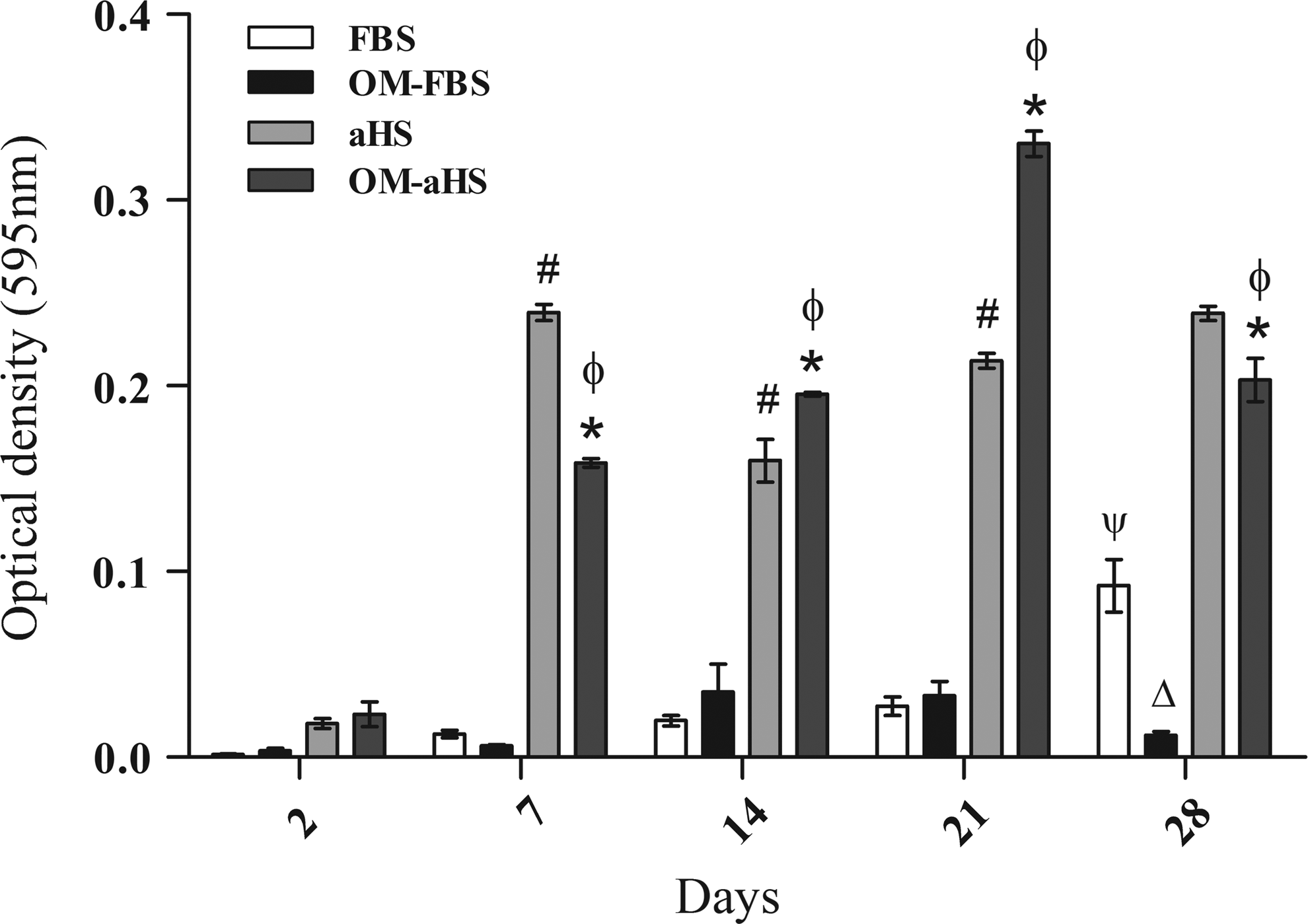
Alkaline phosphatase (AP) assay of the hASCs seeded on PHB-HV scaffolds during osteogenic differentiation. The highest levels of AP were reached at 21 days of induction with OM-aHS and at 14 days of induction with OM-FBS. n=3, from different cultures. The results represent the mean±standard error of the means *p<0.05 comparing the condition of OM-aHS as a function of time; #p<0.05 comparing the condition of basal medium with aHS as a function of time and Ψp<0.05 with FBS; and øp<0.05 comparing the two groups-basal medium×OM for aHS and Δp<0.05 for FBS.
Indirect immunofluorescence
To further analyze the osteogenic differentiation of hASCs seeded on PHB-HV scaffolds and cultured in OM, the expression of specific bone cell proteins was analyzed by immunofluorescent staining of the cryosections obtained at each time point. The constructs at day 14 expressed osteopontin, collagen type I, and osteocalcin (Fig. 7). The architecture of the scaffolds and the patterns of protein expression, which were not restricted to around the nuclei, were observed by confocal microscopy.
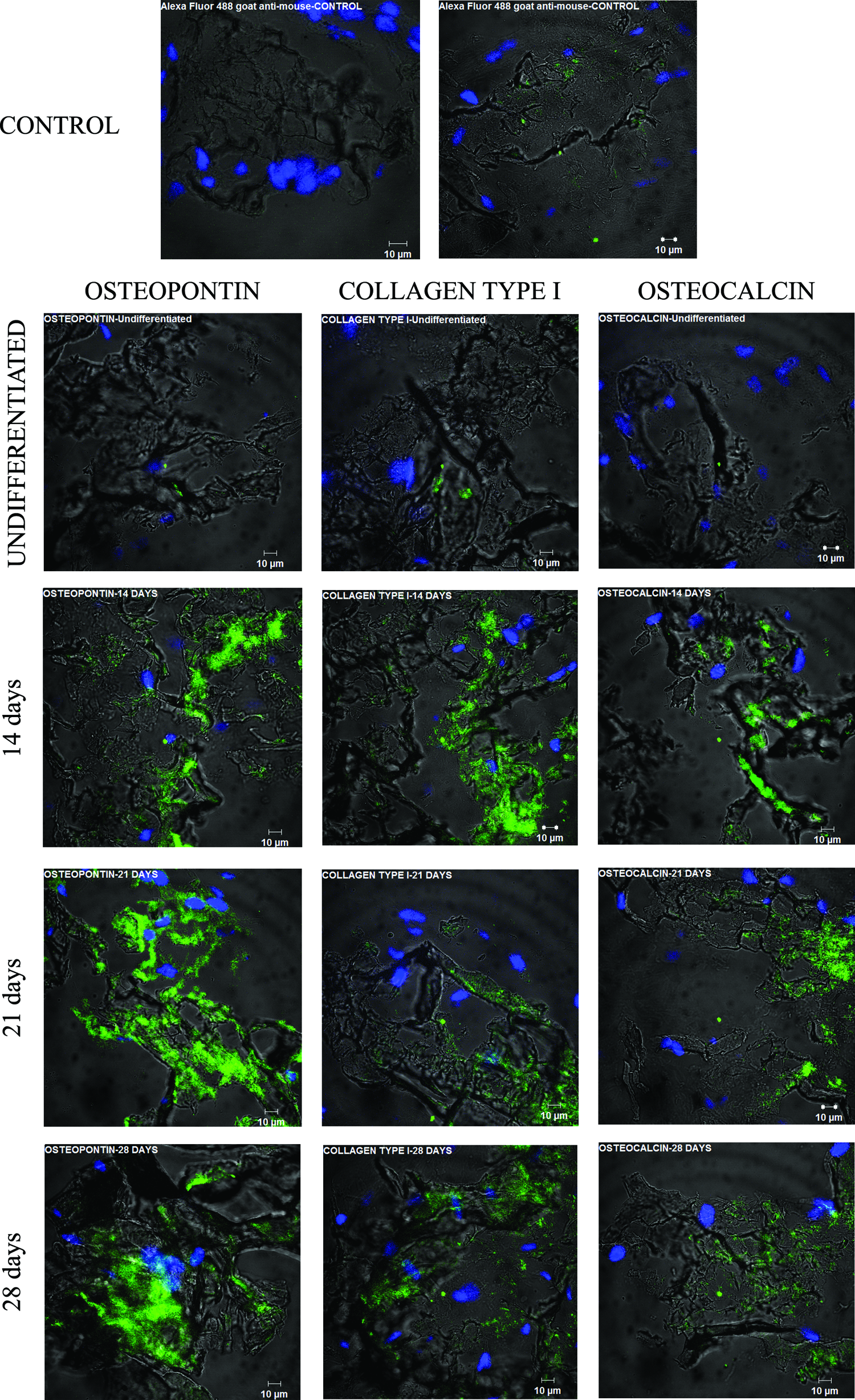
Protein expression of hASCs seeded on PHB-HV scaffolds during osteogenic differentiation from 14 to 28 days of induction and undifferentiated. The expression of bone proteins osteopontin, collagen type I, and osteocalcin were examined. Green fluorescence represents protein expression. Blue color stains the nuclei. Color images available online at www.liebertpub.com/tea
Reverse transcriptase–polymerase chain reaction
For more evidence of osteogenic differentiation, the mRNA expression over time of selected osteogenic markers by constructs induced and hASCs in monolayer noninduced were analyzed by RT-PCR. The analysis shows the expression of specific genes related to osteogenic lineage (Fig. 8). The transcription factor Runx2, AP and collagen type I were detected at all time points, including at noninduced cells sample, being more pronounced at induced cells cultured on scaffolds. The osteocalcin gene expression pattern differ from others, it was not detected at hASCs cultured in monolayer without osteogenic induction, only at all time points of induction. These results indicate that the cells cultured on PHB-HV scaffolds successfully differentiated into the osteogenic phenotype.
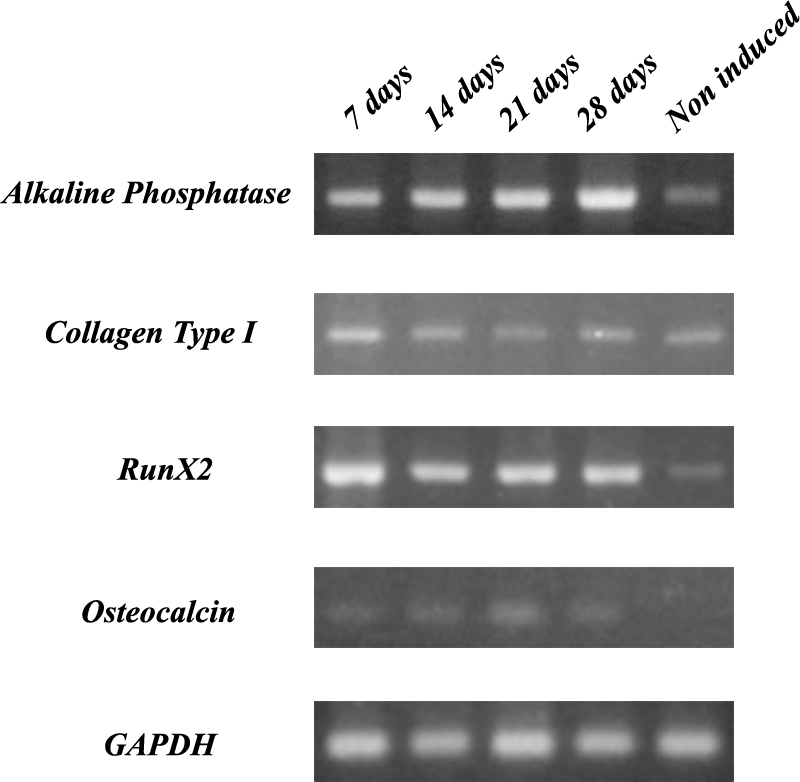
Reverse transcriptase–polymerase chain reaction analysis of AP, collagen type I, transcription factor Runx2, and osteocalcin mRNA expression by hASCs grown on PHB-HV scaffolds and OM-aHS over 28 days and hASCs in monolayer cultured under basal medium. Glyceraldehyde-3-phosphate dehydrogenase was the house-keeping gene.
Discussion
Currently, tissue engineering is a promising strategy in the area of bone regenerative medicine. 1 Stem cells, bioactive factors, and biomaterials are the functional elements of bone tissue engineering that are used to overcome the limitations of traditional orthopedic treatment. However, this strategy presents some challenges, including choosing adequate cell sources,17–20 methods for stem cell culture for transplantation,21,22 and three-dimensional porous scaffolds. 1 As cellular therapies are becoming more common, the clinical use of stem cells needs to follow good manufacturing practices (GMPs). 12
From a therapeutic perspective, the use of FBS is disadvantageous because its use exposes cells to xenogeneic proteins that can be internalized or can adhere to the surface of the cells,23–25 and provoke immunological reactions in the recipient. 26 Moreover, FBS contains endotoxins and can be a source of viruses, bacteria, and prions. 22 Therefore, it is necessary to replace FBS as a supplement in the culture medium.
Several studies have been conducted to use different FBS substitutes. Similarly, our research has been published recently by Aldahmash et al. 27 Their study showed that human serum supported proliferation and differentiation of a model of MSC immortalized by expression of the human telomerase reverse transcriptase gene (hMSC-TERT). 27 Human platelet-rich plasma (PRP) is an alternative serum source that also resulted in a higher expansion of bone marrow-derived stem cells (BMSCs), without influence on the phenotype and capacity of differentiation, which result in a shorter culture time required to get the amount of cells for clinical use. 28 Human platelet lysate (HPL) is considered another reliable FBS substitute, showing more pronounced osteogenic differentiation of BMSCs in HPL than in FBS. The components present in platelet, such as the platelet-derived growth factor (PDGF), the basic fibroblast growth factor (bFGF), and the transforming growth factor (TGF-β) seem to be needed for expansion of stem cells, being PDGF associated with MSC proliferation. 29
Animal serum is a complex mixture of components, such as cytokines, chemokines, hormones, attachment and binding proteins, vitamins, trace elements, and growth factors, essential for cell survival and growth. 30 However, the immunological reaction and batch-to-batch variability are another disadvantage of using animal sera at tissue engineering. 31
So, exists a need to develop chemically defined media, which can propagate the cultivation of stem cells without adversely affecting the cell function and the phenotype. 30 The advantage of using a serum-free chemically defined medium is to avoid cell contact with xenogenic components, ideal for clinical application; moreover, it enables the study of cell biology mechanisms induced by defined molecules without confounding effects of the possible presence of these components in the serum. 32
Recently, chemically defined media with inclusion of different compounds as growth factors (PDGF, epidermal growth factor, b-FGF, TGF-β),33,34 human recombinant insulin, human transferring, 32 retinoic acid 35 and others, have been developed for culturing MSC. The addition of physiological molecules, such as BMP-2, in the serum-free medium 36 and dexamethasone even at low serum concentration 31 enhance differentiation into osteogenic lineage.
However, for success of stem cell culture for clinical application, all compounds of maintenance and differentiation media should be extensively studied and disclosed for evaluation by other researches. 32 The information about formulation by the companies is often limited. And problems related to costs and ability of the media to support growth of cells in long-term cultures remained undetermined. So, the development of serum-free chemically defined cell culture media is difficult. Human serum and plasma are good alternatives to substitute FBS due to their availability 27 and regarding transmission of pathogens during cell transplantation is necessary an extensive testing of blood donate before use in cell culture to avoid this.
In this study, we reported the biological performance and osteogenic differentiation of hASCs seeded on PHB-HV scaffolds in the aHS medium.
In an attempt to substitute the FBS as a supplement of culture medium as a way to achieve safety and efficacy of hASCs culture 22 and attend the regulations of GMPs, 12 we proposed to use a pool of aHS produced from all types of blood group. This choice is justified because the expression of ABO antigens and other antigens based in proteins and carbohydrates are not detectable on the MSC surface. 37 The replacement of FBS for other human derivatives have already been demonstrated in other studies of BMSCs and hASCs, such as allogeneic AB serum,13,14,38,39 PRP,13,38 and HPL.38,40
The hASCs isolated and cultured in both FBS and aHS demonstrated satisfactory results. The cells presented a fibroblastoid morphology and were adherents to the tissue culture plastic surface. Some differences could be observed in the primary cultures of hASCs cultured in aHS, such as a smaller size, less granularity, decreased adherence to the tissue culture plastic surface, a much denser growth, and a tendency to grow in clusters. Similar characteristics were described for hASCs cultured in AB human serum and thrombin-activated PRP, 13 which corroborates our results. Bieback et al. 41 demonstrated that pooled blood group AB human serum and thrombin-activated platelet releasate plasma support the expansion of hASCs. And as showed by other studies, a whole-genome analyses detected a number of adhesion and extracellular matrix-associated molecules expressed at lower levels in hASCs cultivated in human supplements justifying the reduced adhesion of cells cultivated under these supplements. 41
Moreover, the primary cultures grown in aHS were more dense and had a DT that was approximately four times shorter than the primary cultures grown in FBS. These results makes aHS an attractive candidate for clinical use to optimize the ex vivo expansion of cells before transplants.
Besides the morphologic characterization, the phenotypic characterization with flow cytometry confirmed the appearance of the smaller size and lower granularity of hASCs cultured in the medium supplemented with aHS compared to cells cultured in FBS shown by the size versus granularity graph. Moreover, the flow cytometry revealed that hASCs cultured in aHS or FBS comprised a homogeneous population that expressed surface markers CD29, CD44, CD73, and HLA-ABC and almost did not express CD34, CD45, and HLA-DR. Thus, the aHS did not interfere with the characteristics of hASCs that are in accordance with the proposed profile 10 and accepted by the International Society of Cell Therapy.
PHB-HV arises as a new generation of natural polymers. Its properties, such as biocompatibility, biodegradability, nontoxicity, thermoplasticity, and piezoelectricity, make it suitable for several applications in medicine, including bone tissue engineering. 7
The developed PHB-HV scaffolds possess an anisotropic and highly interconnected porous structure, which is essential for successful colonization by cells, and better mechanical properties in the transverse direction than in the radial direction. Anisotropy is a behavior that is also observed in bone. 42 The EDS confirmed the typical composition of poly-esters. Therefore, the scaffold properties are in the expected range for bone substitutes. 5
hASCs adhered to the surface of the scaffolds and inside the pores, and proliferated and penetrated into the inner part of the scaffolds without occluding the pores. The cells were able to make connections to each other through cell processes, as observed by SEM. Therefore, the scaffold's structure was suitable to allow cell infiltration and ingrowth. Moreover, it was possible to observe some cells with several vesicles on their membranes, which could represent active protein synthesis and secretion.
The hASCs were viable and still proliferating in both culture supplements, FBS or aHS, during the time evaluated. Therefore, the substances that were released can be considered to be nontoxic. When the cells were induced for osteogenic differentiation, they continued to be viable with a high degree of proliferation in the beginning, but the proliferation was reduced after 14 days, which suggests the beginning of differentiation. This observation is in agreement with the intrinsic capacity of stem cells to auto renew with a proliferation potential that gradually decreases with differentiation. 43
AP is a marker of embryonic stem cells that was reported for hASCs. 44 Increased AP activity is considered to be an early marker of osteogenic differentiation.45,46 Our results showed that the AP activity of hASCs seeded on PHB-HV scaffolds and cultured in OM-aHS reached the maximum at day 21, which corroborates the proliferation assay and suggests an early osteogenic phenotype. The increase of AP activity was slighter at OM-FBS cultures, but occurred at day 14.
The expression of osteopontin, osteocalcin, and collagen type I could be evidenced by immunofluorescence of the sections. The photomicrographs revealed that the scaffolds provided the framework for adhesion, proliferation, and penetration into the inner scaffold and induced the expression of proteins usually associated with mineralization during the later stage of osteogenesis. 47 Collagen type I is essential for mineralization of the bone matrix. 5 Osteopontin and osteocalcin are involved in bone remodeling. Osteopontin, which is produced by osteoblasts during different stages of tissue maturation, can be expressed in several tissues, including bone. During in vitro bone growth, there is an initial production of osteopontin that continues at a higher level during the mineralization stage. The intracellular expression during proliferation seems to be related to cell migration. The extracellular osteopontin seems to regulate the mineralization process. 48 In contrast, osteocalcin is expressed more after the proliferation stage along with extracellular matrix mineralization. The expression of osteocalcin occurs later during the osteoblast development and contributes to the regulation of the mineral portion of bone. 47
The differentiation of hASCs cultured on PHB-HV scaffolds under OM supplemented with aHS toward osteogenic lineage was ultimately demonstrated by expression of genes that are usually associated with osteoblastogenesis, such as the transcription factor Runx2, which is associated with matrix mineralization, collagen type I, AP, and osteocalcin, which are matrix proteins. Runx2 is essential for osteoblastic differentiation and bone formation as a master gene for regulating osteoblast differentiation and growth. 49
The results indicate that hASCs seeded on PHB-HV scaffolds cultured in OM with aHS underwent the three different phenotypic stages of osteogenic differentiation as described by Lian and Stein 47 : cell proliferation, extracellular matrix formation and high AP activity and matrix mineralization, when the tissue has a higher expression of osteopontin and osteocalcin.
Conclusions
The present study shows that the aHS enhances hASCs proliferation without compromising the morphology and immunophenotype. The results provide an optimal alternative to producing constructs that could be widely available and produced in accordance with GMP. Moreover, the scaffolds developed by the freeze-drying technique presented an adequate structure, are noncytotoxic, and favor hACS colonization. The association of inductor factors in the presence of aHS, PHB-HV scaffolds, and hASCs was successful for the cells to acquire the osteogenic phenotype. Therefore, it can be concluded that this strategy represents a viable and safe alternative for bone tissue engineering application.
Footnotes
Acknowledgments
The authors thank “Núcleo de Cirurgia Plástica” especially Dr. Luíz Alberto Lamana dos Santos and PHB Industrial S.A, for providing the polymer. The authors also thank the Origen Clinic, Rogéria Serakides, and Jankerle Neves Boeloni. This work was financially supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq/Brazil), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES/Brazil), and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG/Brazil).
Disclosure Statement
No competing financial interests exist.