
Abstract
Multipotential stromal cells/mesenchymal stem cells (MSCs) are attractive candidates for regenerative therapy due to the ability of these cells to differentiate and positively influence neighboring cells. However, on implantation for wound reconstruction, these cells are lost as they are challenged by nonspecific inflammation signals generated in the wound environment and in response to any implanted foreign body. We have previously shown that sustained and surface-restricted epidermal growth factor receptor (EGFR) signaling by a tethered form of its prototypal ligand EGF enhances survival of MSC in the presence of death cytokines such as FasL, serum deprivation, and low oxygen in vitro. This was proposed to be due to the plasma membrane restriction of EGFR signaling. Interestingly, during wound repair, an extracellular matrix (ECM) component Tenascin-C (TNC) containing EGF-like repeats (EGFL) and fibronectin-like repeats (FNL) is upregulated. A few of the 14 EGFL on each of the 6 arms, especially the 14th, bind as low-affinity/high-avidity ligands to EGFR causing sustained surface-restricted EGFR signaling. We queried whether signaling by this physiologically relevant EGFR matrikine also protects MSCs from FasL-induced death. MSCs grown on TNC and Collagen I (as TNC by itself is antiadhesive) displayed a survival advantage in the presence of FasL. TNC neither sequestered nor neutralized FasL; rather, the effects of survival were via cell signaling. This survival was dependent on TNC activating EGFR and downstream pathways of Erk and Akt through EGFL; to a much lesser extent, the FNL of TNC also contributed to survival. Taken together, these results suggest that providing MSCs with a nonimmunogenic naturally occurring ECM moiety such as TNC enhances their survival in the presence of death factors, and this advantage occurs via signaling through EGFR primarily and integrins only to a minor extent. This matrix component is proposed to supplement MSC delivery on the scaffolds to provide a survival advantage against death upon in vivo implantation.
Introduction
The effects of nonspecific inflammation on the loss of MSC numbers have been studied extensively. While factors such as tumor necrosis factor-alpha, 12 interferon-gamma, 12 and TNF-related apoptosis-inducing ligand (TRAIL) 13 have been reported to kill MSCs both in vitro and in vivo, the most potent death initiator in MSCs is the Fas-FasL pathway,12–15 with FasL expressed by both natural killer cells 16 and T-lymphocytes.12,17 To counter this cell death and provide MSCs with a survival advantage, our group initially used a tethered form of EGF (tEGF), a growth factor found to increase the MSC cell numbers, but not interfere with MSC differentiation, 18 to improve MSC survival in the presence of FasL and TRAIL.13,19 tEGF was found to signal for survival in MSCs by causing sustained cell surface activation of its receptor epidermal growth factor receptor (EGFR) and its downstream pathways, mainly Erk.
In this study, we investigated whether a protein that presents the EGFR ligands in a nonengineered manner can offer a survival advantage to MSCs, as native proteins are expected to avoid immunogenic responses sometimes observed with engineered therapeutics. Tenascin-C (TNC) is tightly regulated in the adult body, being spatially and temporally restricted to actively remodeling tissues during wound healing 20 and inflammation, 21 in addition to being present during development and in invasive tumors. 22 TNC is a six-armed extracellular glycoprotein with the accessible rod-like arms of the protein containing 14.5 EGF-like repeats (EGFL). 22 Each EGFL is 2-nm long, approximately the same length as that between the N and C termini of EGF 23 with a subset of these capable of binding to and signaling via EGFR. 24 The EGFL of TNC however binds EGFR with a low affinity, three orders of magnitude lower than that of EGF, which is insufficient to cause EGFR internalization and degradation, leading to cell surface-restricted EGFR signaling. 25 The multimeric nature of the TNC causes EGFL to bind EGFR with a high avidity, causing sustained EGFR signaling. 25 Since TNC is a matrix protein abundant during wound healing and since its EGFL repeats can bind and signal EGFR from the cell surface similar to tethered ligands such as tEGF, we hypothesized that TNC should be able to promote survival in MSCs under challenges such as FasL.
Materials and Methods
Materials
Human TNC-purified protein (CC065) and fluorescent inhibitor of caspase activity (FLICA) reagent (APT503), as well as inhibitors PD98059, PD153035, and LY294002, were obtained from EMD-Millipore. Rat tail collagen I (Col I; 354236) was procured from BD Biosciences (San Jose, CA). Human recombinant Super FasL (ALX-522-020-3005) was from Enzo life sciences. Neutralizing antibodies to fibronectin-like repeat (FNL) (MAB2138) and EGFL (MAB3358) were obtained from R&D Systems; antibody to TNC for immunofluorescence (SC-13578) was from Santa Cruz Biotechnology and to EEA1 (C45B10) was from Cell Signaling Technology.
Cell culture
Primary human bone marrow-derived multipotential stromal cells (prhMSC) were obtained from Dr. Darwin Prockop's laboratory (Texas A&M) and maintained in an undifferentiated/proliferative state in alpha-minimum essential medium without ribonucleotides or deoxyribonucleotides supplemented with 16.5% fetal bovine serum (FBS; Atlanta Biologicals), 100 units/mL penicillin/streptomycin, and 2 mM
Coating culture plates with Col I and TNC
Cell culture dishes were coated for 16 h at 37°C with 1 μg/cm2 Col I, or 1 μg/cm2 Col I and 1 μg/cm2 TNC diluted in phosphate-buffered saline (PBS). PBS was aspirated, and the coated surfaces were placed under UV light for 10 min before cell seeding. Deposition of TNC on the surfaces was confirmed by immunofluorescence.
Cell adhesion assay
To quantify cell adhesion on the extracellular matrix (ECM)-coated tissue culture plastic, six-well plates were coated with 1 μg/cm2 Col I, or 1 μg/cm2 Col I and 1 μg/cm2 TNC diluted in PBS for 24 h, after which 500,000 cells were plated per six-well dish, allowed to attach, and spread for 2 h. One set of wells was enumerated after washing, staining for DAPI, and counting DAPI-positive cells in the representative micrographs. The other set of wells underwent inverted centrifugation before enumeration. In brief, cells in the six-well plate were washed, and plates completely filled with a medium eliminating air bubbles by inverting plates into a bucket containing the medium. The plates were sealed in a box designed to fit in the rotor of a centrifuge and centrifuged in an inverted position at 1750 rpm for 5 min. Postcentrifugation, the media were aspirated, and the cells remaining on the surface were stained with DAPI and counted. The number of cells remaining on surfaces in centrifuged plates was plotted relative to the cell numbers in plates that were not centrifuged.
Proliferation assay
Cell proliferation was measured by counting of cells using the Millipore Scepter cell counter. imhMSC and prhMSC were seeded on 12-well plates with or without coated ECM as described, at a density of 80,000 cells/cm2. Twenty-four hours postseeding of cells, the media were changed from the proliferation medium to a medium containing 2% FBS. Seventy-six hours postseeding, the medium was replaced again with a proliferation medium containing only 2% FBS. Ninety-six hours postseeding, the cells were trypsinized and counted using the Millipore Scepter. Cell numbers at 96 h were normalized to the cell numbers initially plated.
Apoptosis measurement
Fluorochrome inhibitor of apoptosis (FLICA) is a cell-permeable and noncytotoxic inhibitor of caspase-3 bound to sulforhodamine, which emits red fluorescence when bound to active caspase-3. Lab-tek 8 chamber slides were coated for 24 h with 1 μg/cm2 Col I, or 1 μg/cm2 Col I and 1 μg/cm2 TNC diluted in PBS, or left uncoated. MSCs were grown on these surfaces for 24 h, followed by treatment with 100 ng/mL FasL or 100 ng/mL FasL with 20 μM cycloheximide (CHX) for 8 h. At 7 h post–treatment, FLICA reagent and Hoechst 33342 dye were added to cells in the medium and incubated for 1 h at 37°C. The media were aspirated, cells washed, and live cells imaged for caspase-3 fluorescence.
In situ cell death detection kit (fluorescein) (11 684 795 001) from Roche was used for the TUNEL assay. After 8 h of treatment with 100 ng/mL FasL or 100 ng/mL FasL with 20 μM CHX for 8 h, the cells were fixed in 4% paraformaldehyde and treated with TUNEL reagent for 1 h, followed with staining of the nuclei with DAPI. Cells were imaged for fluorescein fluorescence. Fluorescein-positive and fluorescein-negative cells in five representative photomicrographs of treatments were enumerated for quantification.
Immunofluorescence
MSCs grown on Col I alone left untreated, or Col I treated with EGF for 15 min, or MSCs grown on Col I and TNC, were fixed in 4% formaldehyde for 30 min, rinsed in PBS, permeabilized with 0.2% Triton in PBS on ice for 5 min, blocked in 1% BSA in PBS for 30 min at room temperature, and incubated with 1:100 phospho-EGFR antibody (sc-12351R from Santa Cruz Biotechnology) or 1:100 phospho-Erk antibody (#4370 Cell Signaling Technology) at 4°C overnight. Cells were washed in PBS, incubated with 1:500 Alexa Fluor 594 for 1 h, washed again, and counterstained with DAPI for 1 min, before imaging of cells.
Animals
Control, age-matched C57Bl6/J mice were obtained from Jackson Laboratories. Additional mice were bred in house and genotyped in house. Serological analyses did not detect blood-borne pathogens or evidence of infection. Mice were housed in individual cages after wounding and maintained under a 12-h light/dark cycle and temperature. All studies on these animals were performed after approval by and in compliance with the Institutional Animal Care and Use Committees of the Veteran's Administration and University of Pittsburgh; these animals were housed in a facility of the Veteran's Affair Medical Center (Pittsburgh, PA) accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Cell labeling and gel suspension
Culture dishes were coated for 24 h with 1 μg/cm2 Col I and 1 μg/cm2 TNC. Approximately 1×106 human MSCs were tracked with 1 μM long-term carbocyanine dye (CellTracker CM-DiI; Molecular Probes, Inc.) according to the manufacturer's standard protocol and plated on the coated culture dishes. Twenty-four hours later, the carbocyanine-tracked human MSCs were scraped gently in the presence of the medium to allow for MSCs to remain attached to the coated matrix. This MSC-ECM mix was resuspended in 50 μL of hyaluronic acid (HA) non-cross-linked polymer (Hylan A gel; Genzyme Biosurgery). Each treatment therefore consisted of 50 μL HA with 1×106 carbocyanine-labeled human MSCs on Col I and TNC.
Cell transplantation
A minimum of eight mice in each group were used in the experiments based on statistical considerations. Full-thickness 8-mm punch wounds were created in the dorsal skin of the animals. A 1-mL syringe with a 24-gauge needle was then used to xenotransplant ∼50 μL of the HA-Col-TNC-MSC mixture to each wound. Control wounds received HA only, or HA-MSC only, with no Col I-TNC. The wounds were left untouched for 15–20 min while mice remained anesthetized using the ketamine–xylazine mixture. Mice were kept in a light-sensitive room for 24 h postwounding in consideration of bleaching of the carbocyanine dye.
Histological analysis
Wound bed biopsies surrounded by a margin of nonwounded skin were collected after euthanasia at 3, 7, 14, and 28 days postwounding. Wound biopsies were fixed in 10% buffered formalin, processed, and embedded in paraffin blocks using standard protocols or OCT embedded with liquid nitrogen for frozen sections. Tissue sections (5 μm) were stained with hematoxylin and eosin and analyzed for general tissue and cellular morphology. Frozen-section wound sections were analyzed using an imaging microscope. Fluorescence data were collected using a rhodamine filter (excitation 553; emission 570 nm). All images were collected at original magnification 10×and 20×.
Flow cytometry
Twelve-millimeter punch biopsies of wounds and 12-mm punch biopsies of unwounded regions were isolated for quantification of MSC engraftment in wounded skin at days 3, 7, 14, and 28. Tissue was minced and incubated in 0.5 mg/mL Liberase TL (Roche Applied Science) for 1 h at 37°C. Tissue was filtered; cells were washed, and labeled for analysis. PE channel flow analysis was performed for carbocyanine dye-positive MSCs in wound. Appropriate isotype controls, unstained cells, and untreated wounds were used as controls. Flow cytometry was performed on an LSR II Cytometer (BD Biosciences) and subsequently analyzed using FlowJo digital FACS software (Tree Star, Inc.).
TNC dose–response
Lab-tek 8-chamber slide were coated for 24 h with increasing concentrations of TNC (0.1, 0.5, 1, and 2 μg/cm2) diluted in PBS in a 1:1 ratio with Collagen. MSCs were grown on these surfaces for 24 h, followed by treatment with 100 ng/mL FasL (CHX) for 8 h. In situ cell death detection kit (fluorescein) (11 684 795 001) from Roche was used for TUNEL assay to percent cell death by DNA breakage. Five representative photomicrographs of treatments were enumerated for quantification.
Statistical analysis
Treatments were compared using paired t-test. p<0.05 and p<0.01 were considered statistically significant.
Results
TNC in combination with Col I provides an adhesive surface for MSCs while not causing MSC differentiation
TNC reduces the number of focal adhesions and decreases cell adhesiveness in several cell types. 26 To test for MSC adhesiveness on TNC, an inverted centrifugation assay was performed 2 h postseeding MSCs on tissue culture plastic, and the number of cells attached after centrifugation was compared to the number of cells attached on a similarly coated surface that had not undergone centrifugation (Fig. 1A). As expected, MSCs grown on TNC detached from the surfaces after inverted centrifugation. To increase adhesiveness of cells on TNC, we coated mixed Col I, a matrix naturally occurring in the in vivo bone environment and one that promotes MSC attachment and spreading, in a 1:1 ratio with TNC for coating substrates. Cells on TNC were only half as adhesive as the cells on Col I (Fig. 1A, B). Growing cells on a mixed matrix comprised of Col I and TNC was able to significantly increase MSC adhesiveness. For all further studies, a combination of Col I and TNC was used.
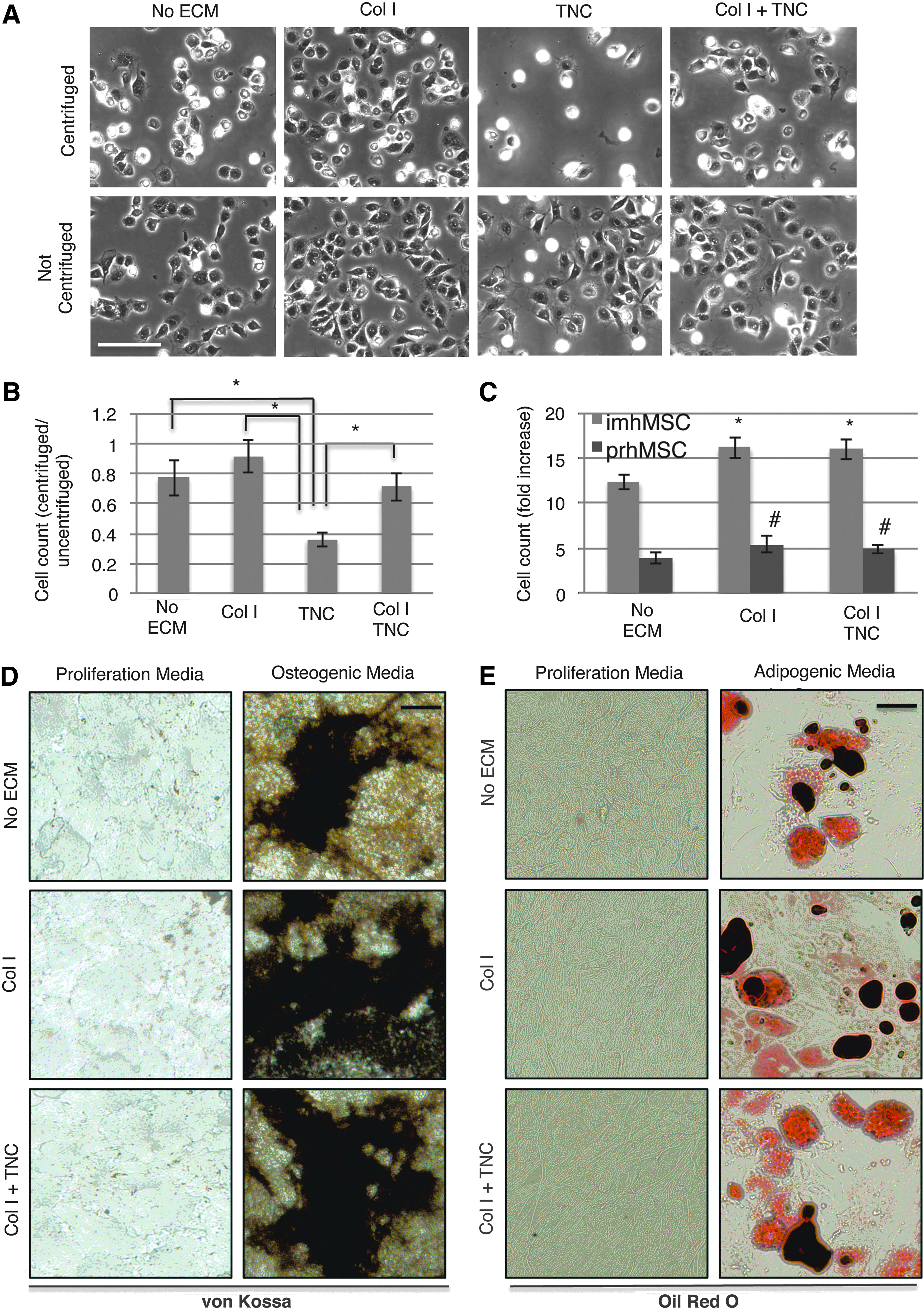
Tenascin-C (TNC) and Collagen I (Col I) aid in multipotential stromal cell/mesenchymal stem cell (MSC) attachment and do not cause MSC differentiation. Phase-contrast images of MSCs after inverted centrifugation or without inverted centrifugation 2 h postseeding on the indicated matrices (scale bar 50 μm)
During wound healing, MSC proliferation and differentiation are temporally distinct events. The use of a survival factor, which promotes (or at the least allows for) proliferation, but does not induce differentiation, would cause MSC expansion postincorporation into tissue, promoting sufficient numbers of cells to form to regenerate the wounded tissue. 27 We first tested if our ECM factors had any influence on MSC proliferation. TNC with Col I, like Col I by itself, was seen to promote both imhMSC and prhMSC proliferation 96 h postseeding, relative to cell numbers initially plated (Fig. 1C).
Next, we needed to determine whether TNC by itself, in the absence of specific differentiation cues, promotes MSC differentiation. We examined osteoblast or adipose cell formation from MSCs grown on TNC and Col I, in the presence of either a proliferation or differentiation medium. von Kossa staining of MSCs grown for 30 days in a proliferation medium (Fig. 1D) showed that neither Col I by itself nor TNC and Col I caused hydroxyapatite deposition. However, in the presence of the differentiation medium, TNC did not interfere with osteogenic differentiation (Fig. 1D). Similarly, TNC did not promote adipocyte formation on its own, neither did it prevent adipocyte formation in the presence of adipogenic inducers (Fig. 1E). The matrix mix of TNC and Col I thus supported MSC attachment and growth and did not cause MSCs to differentiate nor did interfere with differentiation.
TNC protects MSCs from FasL-induced cell death
Several groups, including ours, have previously shown that MSCs are most susceptible to cell death via the Fas death pathway.12–16 We induced cell death in imhMSC (data not shown) and prhMSC by treating with FasL and found intense capsase3 activation as highlighted by FLICA after 8 h (for imhMSC) or 12 h (for prhMSC) of treatment, as seen by us previously. 14 Cells were also treated with a low concentration of the protein synthesis inhibitor CHX in addition to FasL. CHX, being a protein synthesis inhibitor, increases cell stress, imitating the starvation stress seen in vivo in a wound setting. Cells grown on tissue culture plastic with no matrix displayed caspase-3 activation after treatment with FasL or CHX and FasL. Cells grown on Col I and TNC displayed a survival advantage and substantially reduced cell death in the presence of FasL or CHX and FasL. TNC (1 μg/cm2) was found to be optimal to promote survival in MSCs in the presence of FasL based on a dose response, as determined by TUNEL and FLICA staining (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea). We attributed the protective effect to TNC in the matrix, since MSCs grown on Col I surfaces did not show protection to MSCs in the presence of FasL or CHX and FasL (Fig. 2A, B). Similarly, we saw that TNC and Col I, but not Col I by itself, protected MSCs from nuclear DNA damage induced by FasL, indicated by TUNEL assay (Fig. 2C, D). These results support our hypothesis that TNC plays a role in increasing cell survival in MSCs in the presence of threats such as FasL.

TNC is protective to MSCs in the face of FasL-induced death. Fluorescent inhibitor of caspase activity (FLICA)-stained primary human bone marrow-derived multipotential stromal cells (prhMSC) 12 h post-treatment with FasL/cycloheximide (CHX) and FasL on indicated surfaces (scale bar 50 μm)
TNC supports MSC survival by signaling via EGFL and not by sequestering or neutralizing FasL
Since TNC contains both EGFL repeats, which can bind and activate EGFR as well as FNL repeats, which can bind and signal integrins, we needed to determine which moieties provided the survival effects. To test this, postpreparation of coated surfaces and before seeding of MSCs, neutralizing antibodies to the EGFL and FNL domains were added for 24 h. Immortalized hMSCs were then seeded for 24 h, kept treated (Fig. 3A, top rows), or untreated (Fig. 3A, bottom rows) with FasL for 8 h as done in previous experiments, followed by an FLICA assay to test for caspase-3 activation. Presence of neutralizing antibodies for FNL and EGFL was confirmed before seeding of cells. Blocking of FNL allowed for a small amount of caspase-3 activation, but much less than that without antibody blockade. On the other hand, the neutralization of EGFL permitted cell death to the extent of no blockade (Fig. 3A). MSCs seeded on Col I alone with no neutralizing antibody and treated with FasL showed caspase-3 activation as shown earlier. Addition of neutralizing antibodies did not have a toxic effect on MSCs. This was confirmed by growing MSCs on Col I and TNC-coated surfaces with a neutralizing antibody, untreated, and testing for caspase-3 activation. These results indicate that although both the EGFL and FNL repeats of TNC are involved in survival signaling in MSCs, the dominant survival effect is via EGFL.
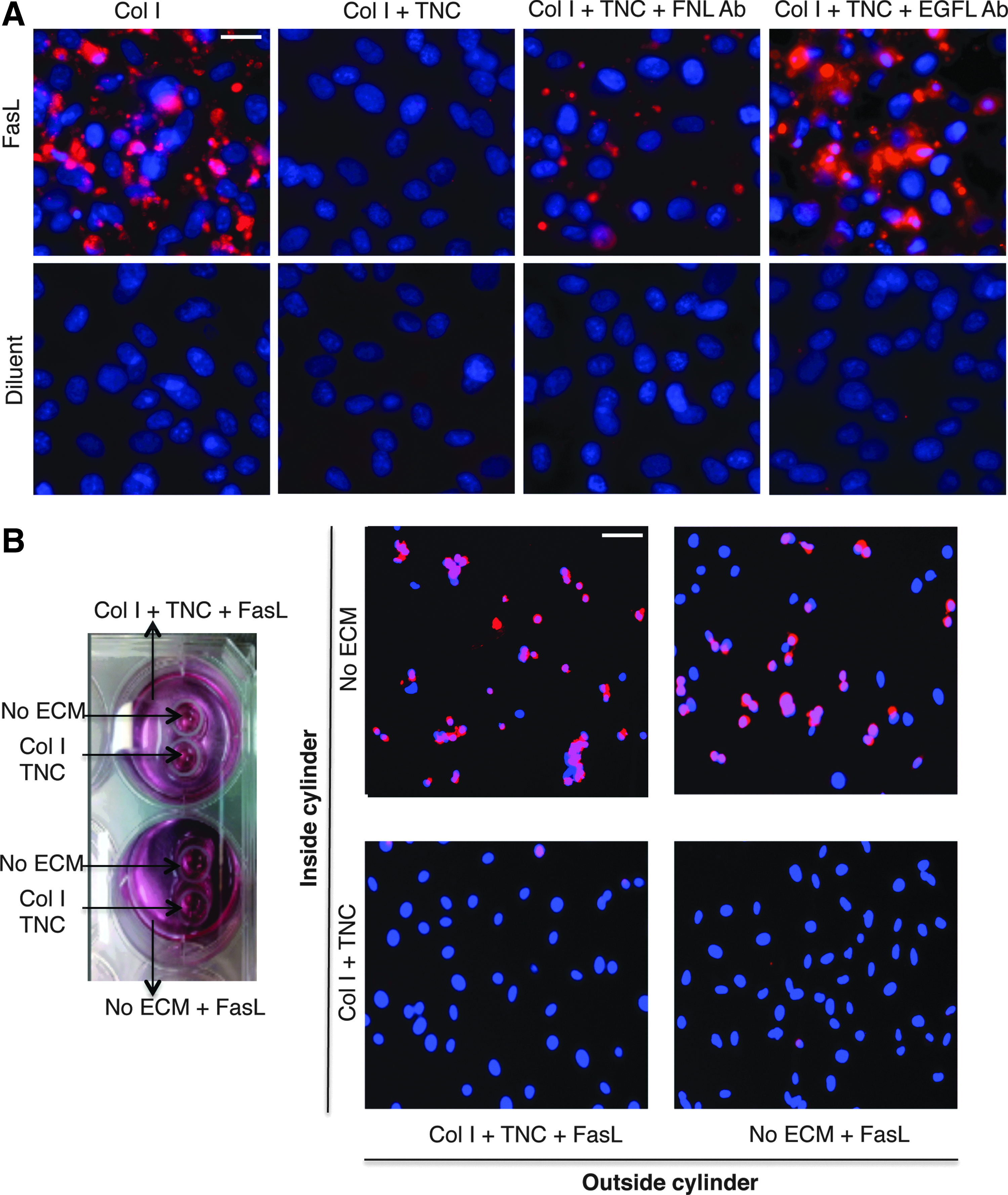
TNC protects MSCs from FasL-mediated cell death via signaling through its EGF-like repeats (EGFL) and not by sequestering of FasL. FLICA-stained immortalized human MSCs 12 h post-treatment with FasL grown on the indicated surfaces with or without neutralizing antibodies to the fibronectin-like (FNL) repeats of TNC or EGFL of TNC (scale bar 10 μm)
We next questioned whether TNC might contribute to survival of MSCs by sequestering FasL and prevention of FasL from binding its receptor Fas on MSC. To test this, we grew MSCs inside two cloning cylinders placed in wells of a 6-well dish, enclosing areas without any ECM, or with Col I and TNC, on which imhMSC were grown. One well had Col I and TNC coating surrounding the cloning cylinders, covered with a medium and FasL, and the second well had only a medium with FasL surrounding the cloning cylinders, with no TNC (Fig. 3B). When the cloning cylinders were taken out and the medium came in contact with cells within the cloning cylinders, there was comparable cell death seen in cells grown on uncoated surfaces in both wells (Fig. 3B). Cells coated on TNC did not show caspase-3 activation as seen earlier and expected. This indicated that TNC was neither sequestering nor neutralizing FasL.
TNC activates EGFR on MSCs and protects MSCs via EGFR signaling
To determine that EGFR is the operative receptor for the survival advantage, prhMSC were grown on Col I- and TNC-coated surfaces, and treated with the inhibitors of EGFR activation PD153035, Erk activation PD98059, or Akt activation LY294002, 30 min before addition of FasL. The inhibitors were present during the entire 8 h of FasL treatment, after which an FLICA assay was performed. Blocking of either EGFR or its downstream pathways of Akt and Erk blocked the survival advantage that TNC and Col I provide MSCs in the presence of FasL (Fig. 4A). The controls of only-inhibitor treatment PD153035, or PD98059, or LY294002 on MSCs grown on Col I and TNC did not have a toxic effect on cells. As seen earlier, MSCs grown on Col I and TNC and treated with FasL or CHX and FasL survived. These results indicate that activation of EGFR and its downstream pathways of Erk and Akt by TNC are necessary for promoting survival in MSCs.
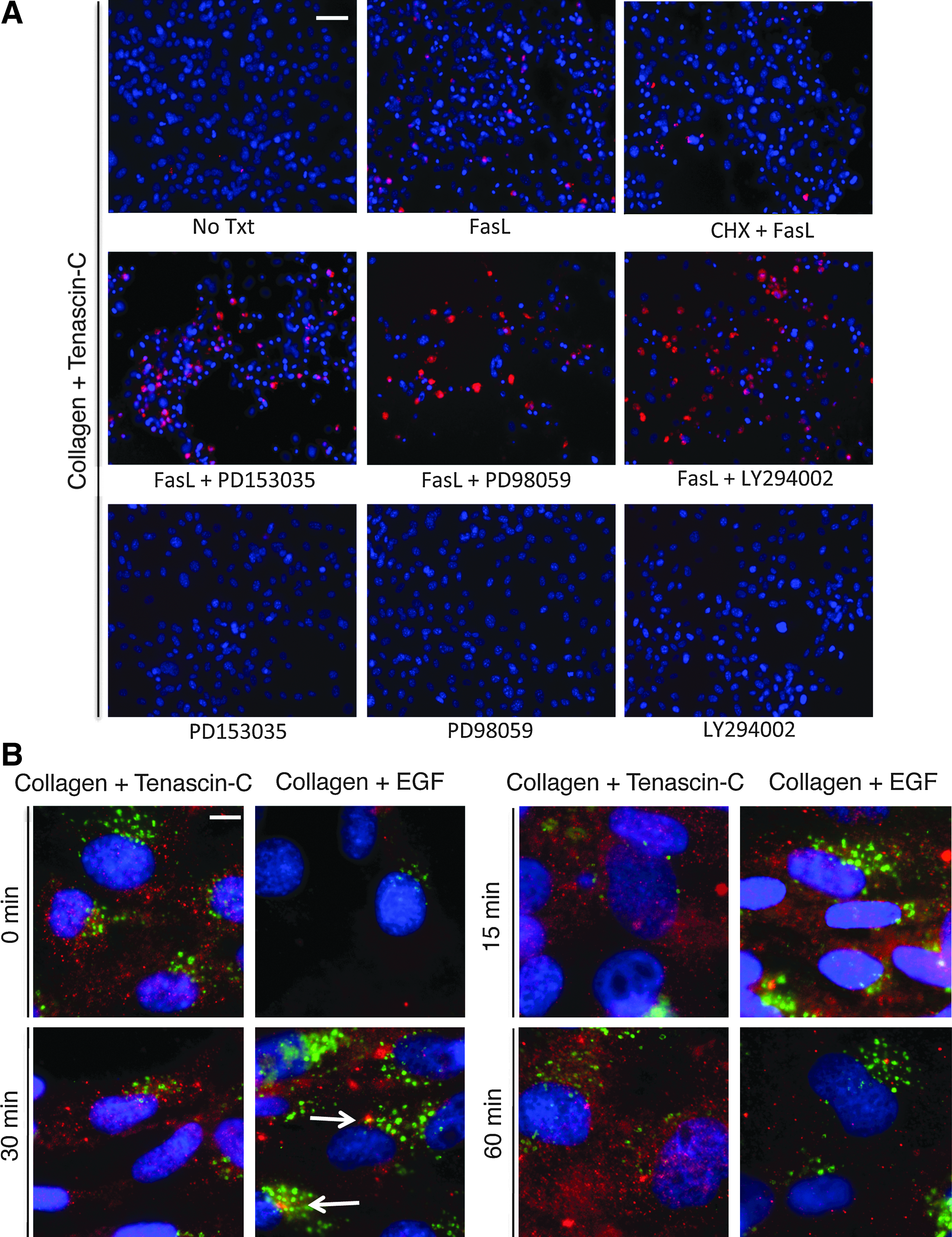
TNC activates epidermal growth factor receptor (EGFR) on MSCs, and activation of EGFR is essential for the survival effects of TNC. FLICA-stained prhMSC 12 h post-treatment with FasL/CHX and FasL on Col I- and TNC-coated surfaces in the presence or absence of the indicated inhibitors (scale bar: 50 μm)
Upon finding that TNC did provide a survival effect on MSCs in the presence of FasL, and that this mapped to the EGFL, we tested if TNC was able to activate EGFR on the surface of MSCs, and if this activation remained sustained. MSCs have low levels of EGFR, which may limit activation by the ultra-low-affinity EGFL. 18 Immortalized hMSCs were plated on the surfaces with Col I alone or Col I and TNC for 24 h. After 24 h, (considered 0 min), immunofluorescence for phospho-EGFR (red fluorescence) and the early endosome marker EEA1 (green fluorescence) was done on these samples. Immortalized hMSCs on Col I and TNC showed phospho-EGFR, while cells on Col I alone did not show phospho-EGFR staining showing specific activation of EGFR by TNC (Fig. 4B). Immortalized hMSCs seeded on Col I were treated with EGF, the prototypal EGFR ligand, known to cause EGFR internalization for 15, 30, and 60 min before fixation, to induce phosphorylation of EGFR. At the same timepoints, immunofluorescence was done on the cells grown on Col I and TNC. As expected, the cells treated with EGF stained positive for phospho-EGFR after 15 min of treatment, and phospho-EGFR colocalized with the endosome marker EEA1 at 15 and 30 min of treatment, indicating activation and internalization of EGFR by EGF. At 60 min post-treatment with EGF, there was very little phospho-EGFR signal indicating a near-complete internalization of receptors. Phospho-EGFR on TNC and Col I surfaces however continued to be expressed at 15, 30, and 60 min, indicating sustained activation of EGFR on the cell surface by TNC compared to EGF.
TNC causes sustained activation of phospho-Erk in MSC via EGFR signaling
Since TNC was found to cause sustained EGFR signaling in MSCs, we wanted to test if this signal translated to sustained downstream signals in MSCs related to survival. We specifically examined that of Erk. Immortalized hMSCs were plated on the surfaces with Col I alone or Col I and TNC for 24 h. After 24 h (considered 0 min), immunofluorescence for phospho-Erk (green fluorescence) was tested either with or without the EGFR inhibitor PD153035. TNC was found to activate phospho-Erk in MSC at 0, 30, and 60 min (Fig. 5A). In the presence of the EGFR inhibitor, this activation was reduced, but not completely inhibited, indicating the presence of other modes of Erk activation in MSCs. There was Erk activation seen in the presence of Col I alone, however, significantly lesser than that with TNC (Fig. 5B). These results suggest that TNC causes sustained EGFR and Erk signaling, promoting survival in MSCs.
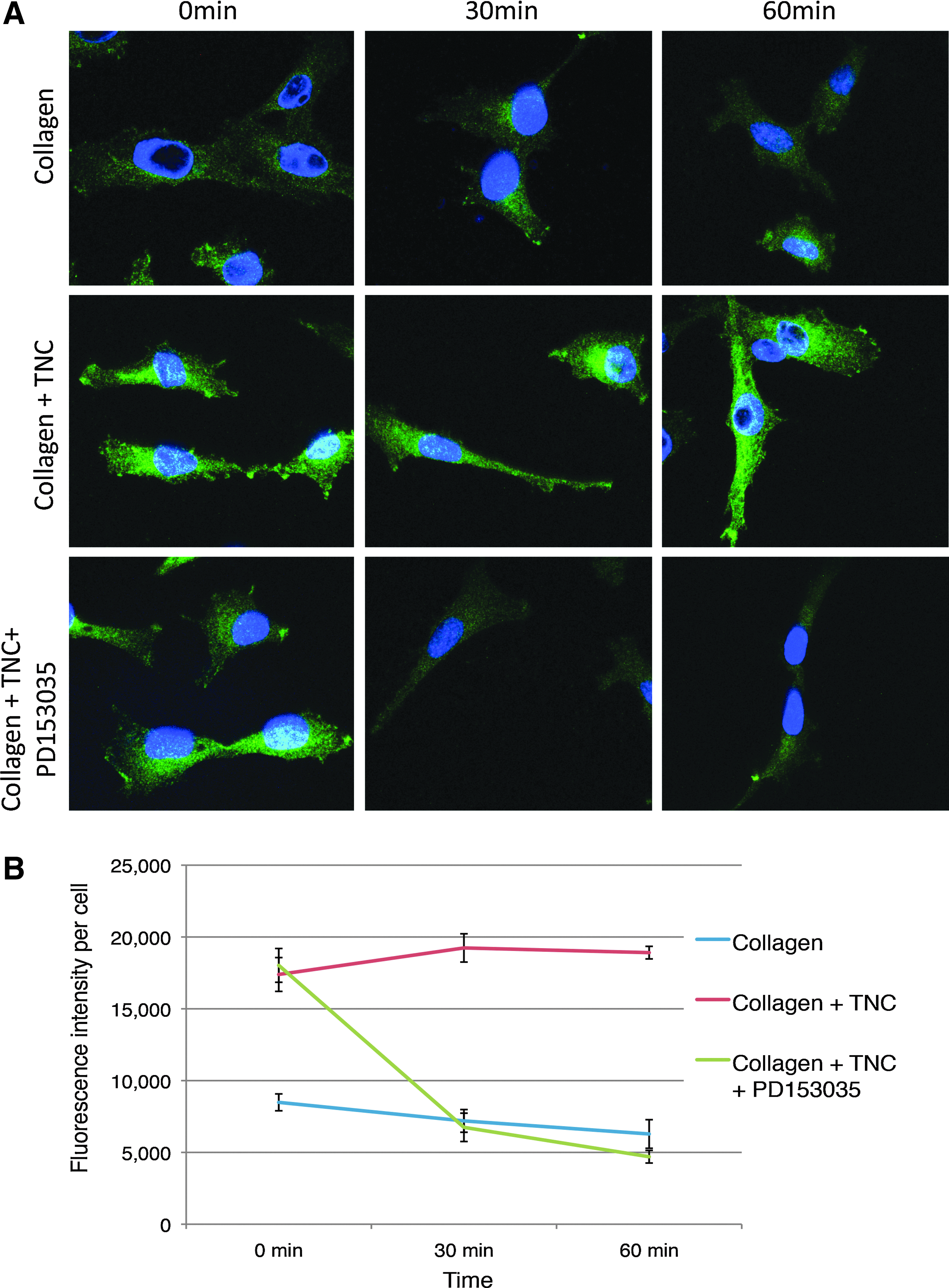
TNC causes sustained activation of Erk through activation of EGFR. Immunofluorescence for phosphorylated Erk (green fluorescence) in MSCs grown on either Col I or Col I and TNC, at times 0, 30, and 60 min, in the presence or absence of the EGFR inhibitor PD153035, shows sustained Erk activity by TNC dependent on EGFR activation (scale bar: 10 μm)
TNC allows for MSC survival in vivo
To assess the survival advantage TNC provides to MSCs during cell transplantation, we used a hydrogel delivery system to deliver carboxycyanine-tracked human MSCs in a mouse wound model. Full-thickness 8-mm punch biopsy wounds were created in wild-type mice. These wounds were then treated with HA alone, human MSCs in HA, human MSCs in HA-Col, or human MSCs in HA-Col-TNC. At days 3, 7, 14, and 28, wound biopsies were assessed for the survival and maintenance of carboxycyanine-tracked human MSCs. Interestingly, donor cells delivered in the HA-Col-TNC-MSC gel exhibited strong survival within the wound bed at day 7 postwounding (Fig. 6A). The cryosections did not display carboxycyanine-positive MSCs in HA without TNC. This trend was seen on day 14 as well as day 28 (data not shown). These data further support the in vitro finding that suggests that TNC promotes survival of MSC and can provide a novel strategy for maintaining MSC survival in wounds.

TNC is protective of MSCs in vivo. Cryosections of wounded skin containing carbocyanine-stained MSCs implanted with hyaluronic acid (HA) alone or HA-containing Col I and TNC on days 7, 14, and 28 show either the presence (red fluorescence) or absence (no fluorescence) of MSCs
Immunofluorescent sections of the wound tissue demonstrate increased MSC survival by TNC, but are not quantitative. Therefore, the survival advantage of TNC on MSCs in the wound was further investigated by flow cytometry analysis of single cells isolated from wound tissue. Wound tissue was treated with liberase to separate out single cells, and the carbocyanine-tracked MSCs were analyzed by Flow Cytometry using the PE channel. Flow cytometry of dissociated wound beds at days 3, 7, 14, and 28 postwounding validated the histological analyses, demonstrating higher survival of HA-Col-TNC-MSC gel delivery versus that of HA-MSCs or HA-Col-MSCs by a significantly higher percentage (Fig. 6B). At day 7, there were 21% PE-positive cells in the HA-Col-TNC-MSC population vs 10% positive cells in HA-Col-MSCs and 8% positive cells in the HA-MSC population. Taken together, TNC and Col I extended a survival effect to MSCs not just in vitro, but also in vivo in a wound site.
Discussion
Tissue replacement or regeneration by stem cells requires the implanted cells to survive in a harsh microenvironment. The challenges include hypoxia, nutrient deprivation, and, especially, death cytokines. 28 The latter occur even in the absence of a wound, as the introduction of foreign bodies elicits nonspecific inflammation. It is this harsh environment that likely underlies the dramatic loss of implanted MSCs.
Our group has pursued strategies to promote survival of implanted MSC/cells to repair bone 29 and skin. 30 We previously reported that EGF in its soluble form (sEGF) does not promote MSC survival due to the short duration of signaling.13,15 However, sustained, surface-restricted EGFR signaling by a tEGF construct protects MSCs from FasL induced cell death. This difference in signaling outcome between surface-restricted (tEGF) and internalized (sEGF) EGFR is not unexpected. Duration of signaling, subcellular localization, and intensity of activation are all factors that determine the outcome of growth factor receptor signaling.31–33 EGFR binds to various ligands: soluble ligands such as EGF and TGFα, bound ligands sequestered in the ECM such as heparin binding EGF, or ECM-based ligands such as TNC. Differential localization of activated EGFR can alter the strength of downstream signals to cause varied effects.34–37 EGFR localized to the cell membrane and activating PLCγ strongly and a subset of Erk trigger motility preferential to endosomally trafficked EGFR that drives p21ras signaling and strong Erk in the cytoplasm, leading to proliferation. 38 The low affinity binding of the TNC EGFL repeats allows for a staccato signaling in which the EGFR is not internalizaed, 38 thus limiting the signaling to the membrane with a lower level of sustained Erk and Akt activation. Such a sustained EGFR and Erk activation has been shown to be critical in bringing about cell survival in several cell types,39–41 including MSCs. 13
Thus, in this study, we investigated the matrikine TNC, naturally but transiently upregulated during wound healing, which contains EGFL repeats presented in multiple valency, and which binds and triggers EGFR at the cell membrane with a low affinity.42,43 This was done to find a natural molecule that would confer MSC survival, but not be immunogenic when delivered in vivo. We found that TNC promoted MSC detachment as noted in several other cell types, suggesting that MSCs were signaled by these repeats despite their very low levels of EGFR. 18 Combining TNC with Col I, an ECM, which unlike fibronectin does not interfere with TNC signaling, increased MSC attachment. 44 TNC has been shown previously to be associated with chondrogenic and osteogenic differentiation in vivo in chick embryos and has been shown to promote chondrogenesis in wing bud cultures of chick embryo in vitro.45–47 However, in the presence of a proliferation medium, the mixed matrix of Col I and TNC neither induced osteogenic or adipogenic differentiation nor interfered with subsequent induction of these states. Most importantly, we found that the mixture of TNC and Col I protected MSCs from FasL-induced cell death, and that the effects of survival were due to TNC and not Col I.
There are limited reports in the literature on the effects of TNC on survival, with a major emphasis on TNC-based motility signaling. Addition of TNC externally to smooth muscle cells brings about the interactions with α5B3-integrins, causing rearrangement of the actin cytoskeleton, clustering of activated EGFR on the cell surface near focal adhesion complexes, and survival signaling. 47 Chondrosarcoma cells grown on TNC display survival under serum deprivation due to Akt activation. 48 TNC has also been shown to have a survival effect on oligodendrocyte precursor cells. 49 It has been hypothesized that TNC by its nature of being a de-adhesive brings about survival by creating an intermediate state of cellular adhesiveness and causing motility. 50 On the other hand, there are also reports that suggest that TNC promotes inflammation by activating Toll-like receptors on macrophages, stimulating release of the proinflammatory cytokines. 51 In the skin, however, under several inflammatory conditions, TNC is upregulated and has been shown to limit the severity of the disease. 52 Thus, the main effect is on the survival of the MSCs.
We found that TNC is able to activate EGFR in MSCs, although to a lesser extent than EGF, and blocking EGFR activation or downstream Akt and Erk activation no longer supports survival of MSCs in the presence of FasL. Blocking of different regions on TNC using neutralizing antibodies to either EGFL or FNL shows that the survival effects of TNC on MSCs are primarily via its EGFL. The FNL is required, but not sufficient, to bring about MSC survival in the presence of FasL. Finally, since the ECM is known to sequester large numbers of growth factors and cytokines, 53 we tested and ruled out the possibility that the TNC and Col I matrix sequesters and neutralizes the effect of TNC. Rather, TNC directly signals the survival advantage.
Importantly, this protective effect extended to an in vivo situation. Inoculation of human MSCs in a polymer gel matrix into the perifascial region in mice resulted in rapid clearing of the xenotransplanted MSCs. However, fortifying this gel matrix with TNC significantly extended the persistence of these cells (the immunogenic nature of xenotransplanted human cells eventually overcame the survival signals). Endogenous TNC was not beneficial to transplanted MSCs, since TNC is transiently upregulated in the wound, with expression starting only after 2 days of wounding 54 and increasing thereafter with greater expression at the regenerating wound edges rather than the center. 55 Treatment of stalled and unhealing wounds would include MSCs being implanted at the center of the wound, with MSC death noted to occur within the first 48 h of implantation when endogenous TNC is low to nonexistent. External supply of TNC would therefore be a solution to aid in the survival of these implanted stem cells. While we could not directly dissect the signaling pathways in this avascular and nutrient-deficient matrix, the twofold increase in the MSC numbers out to 4 weeks argues strongly for this physiological component as an adjunct to MSC transplantation for tissue regeneration.
In conclusion, TNC enhanced the survival of MSCs in the presence of apoptotic factors such as FasL to which MSCs are most susceptible to, and that this survival was via binding and activation of EGFL to, EGFR. Further, since TNC does not by itself cause differentiation, but in the presence of differentiation cues does not hinder differentiation, three-dimensional scaffolds of TNC and Col I can be engineered for use with MSCs to improve the survival of MSCs on implantation.
Footnotes
Acknowledgments
This work was supported by the grants from the National Institutes of Health (USA) R01DE19523 and GM63569. We thank the members of the Wells and Griffith labs for comments and suggestions.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.