
Abstract
Chronic skin ulcers such as diabetic ulcers and venous leg ulcers are increasing and are a costly problem in healthcare. We have developed a novel artificial dermis, collagen/gelatin sponge (CGS), which is capable of sustained release of basic fibroblast growth factor (bFGF) for more than 10 days. The objective of this study was to investigate the safety and efficacy of CGS impregnated with bFGF in the treatment of chronic skin ulcers. Patients with chronic skin ulcers that had not healed in at least 4 weeks were treated with CGS impregnated with bFGF at 7 or 14 μg/cm2 after debridement, and the wound bed improvement was assessed 14 days after application. Wound bed improvement was defined as a granulated and epithelialized area on day 14 with a proportion to the baseline wound area after debridement of 50% or higher. The wound area, the wound area on day 14, and the granulation area on day 14 were independently measured by blinded reviewers in a central review using digital images of wounds taken with a calibrator. Patients were followed up until 28 days after application to observe the adverse reactions related to the application of CGS. From May 2010 to June 2011, 17 patients were enrolled and, in 16 patients, the wound bed improved. Among the randomized patients in step 2, no significant difference was seen between the low-dose group and the high-dose group. No serious adverse reactions were observed. Adverse reactions with a clear causal relationship to the study treatment were mild and patients quickly recovered from them. This study is the first-in-man clinical trial of CGS and showed the safety and efficacy of CGS impregnated with bFGF in the treatment of chronic skin ulcers. This combination therapy could be a promising therapy for chronic skin ulcers.
Introduction
In addition to these cell therapies, genetically derived growth factors have been developed in association with advances in molecular biology. Some growth factors such as platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), and epidermal growth factor have already been clinically used for the treatment of chronic skin ulcers.11–13 bFGF, which was identified in 1974, promotes the proliferation of fibroblasts and capillary formation and accelerates tissue regeneration.14,15 In Japan, human recombinant bFGF (FIBRAST SPRAY; Kaken Pharmaceutical, Tokyo, Japan) has been clinically used for chronic skin ulcers since 2001, and its clinical effectiveness has been demonstrated.14,16 Recently, combination therapy involving bFGF and artificial dermis has been reported to accelerate dermis-like tissue formation, not in the treatment of acute and chronic skin ulcers alone.17–19 A novel artificial dermis, collagen/gelatin sponge (CGS) containing a 10 wt% concentration of acidic gelatin, was developed in a previous study. 20 CGS can sustain the release of positively charged growth factors such as bFGF for more than 10 days. 20 In preclinical studies using mice and beagle dogs, CGS itself could be used as a scaffold for dermal regeneration, the same as conventional artificial dermis, and CGSs impregnated with 7 or 14 μg/cm2 bFGF accelerated neovascularization and the formation of dermis-like tissue two or three times earlier than with conventional artificial dermis.21–23
In view of the above, combination therapy with CGS and bFGF is expected to have an effect comparable to tissue engineering products containing cells on promoting wound healing, even in patients with chronic skin ulcers. Thus, an exploratory clinical study was designed to investigate the safety and efficacy of this combination therapy in the treatment of chronic skin ulcers. In chronic ulcers caused by diabetes mellitus, venous insufficiency, pressure sores, or collagen disease, granulation tissue is not easily formed. In this study, the null hypothesis that the proportion of patients with wound bed improvement was 10% or less within 14 days was tested.
Materials and Methods
Ethical considerations
This study was conducted in compliance with Good Clinical Practice and in agreement with the latest revision of the Declaration of Helsinki, Pharmaceutical Affairs Law, and all applicable Japanese laws and regulations, as well as any local laws and regulations and all applicable guidelines. This protocol and any amendments have Institutional Review Board approval from Kyoto University Hospital.
Study design and patients
This study was an open-label, randomized, multiple dose, controlled clinical trial to evaluate the safety and efficacy of CGS impregnated with bFGF in the treatment of chronic skin ulcers that were not expected to heal with conventional treatments. It was conducted at Kyoto University Hospital. 24
The design was previously reported in detail. 24 Two groups, low-dose (7 μg/cm2 bFGF impregnation) and high-dose (14 μg/cm2 bFGF impregnation) groups, were arranged. In the initial step (step 1), three patients were enrolled in the low-dose group and, after confirming the safety of this step, fourteen patients were randomized to the low-dose or high-dose bFGF group in step 2 at a ratio of 1:1 without stratification using a computer-generated random sequence (Fig. 1). Data management, randomization, and statistical analysis were managed by the independent data center in the Department of Clinical Trial Design and Management, Translational Research Center, Kyoto University Hospital.
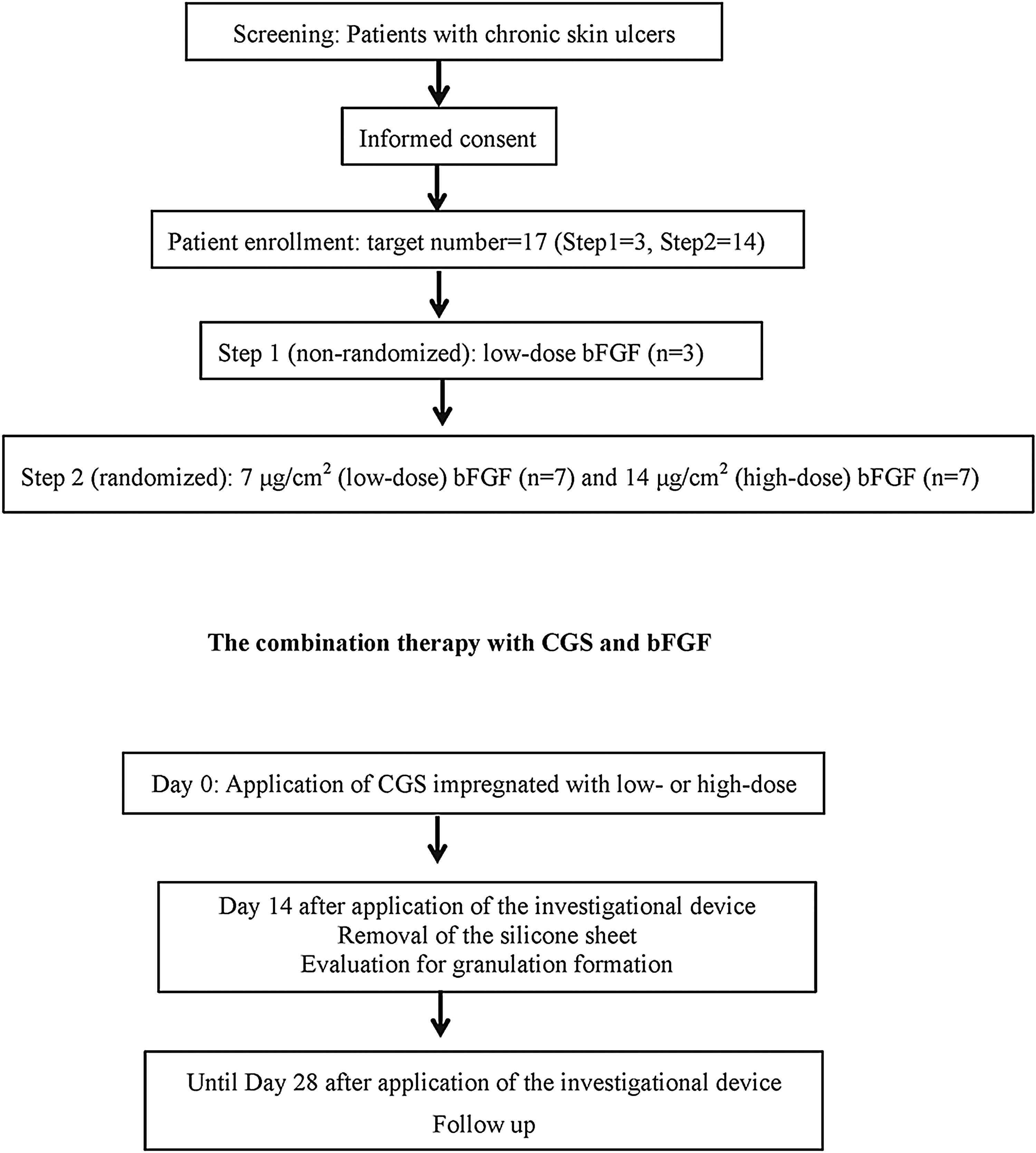
Schema of the study. After enrollment, the initial three patients were enrolled in the low-dose group (step 1) and, after confirming the safety of this step, fourteen patients were randomized to the low-dose or high-dose basic fibroblast growth factor (bFGF) group in step 2. In the combination treatment, collagen/gelatin sponge (CGS) impregnated with bFGF was applied after debridement (day 0). After removing the silicone sheet, the wound area and granulation tissue were evaluated (day 14), and then patients were followed up until day 28.
The main inclusion criteria were patients aged 20 years or older, who had given informed consent, with the presence of chronic skin ulcers as follows: not healing for at least 4 weeks with conventional treatments; skin graft was not expected to take place; and could be completely covered by a 70×100 mm CGS. If chronic skin ulcers were present on the lower extremities, the skin perfusion pressure had to be≥30 mmHg at a site proximal or distal to those ulcers. The exclusion criteria were as follows: uncontrolled diabetes mellitus; requiring continued use of oral corticosteroid therapy; having a history of malignant tumor; or having a history of allergy to porcine-derived products. The details were described in our previous article. 24 If a patient had multiple intractable skin ulcers, CGSs were applied to all these ulcers for study therapy. The largest eligible ulcer was selected and evaluated for efficacy and safety, and the others were evaluated only for safety.
Preparations of CGS impregnated with bFGF
The CGS was a modified version of the conventional bilayered artificial dermis (Pelnac®; Gunze Co., Ltd., Kyoto, Japan) and consisted of an upper silicone sheet (0.12 mm in thickness) and a lower sponge (3 mm in thickness).20,25,26 In this study, the size of the CGS was 82×120 mm. The investigator or sub-investigator prepared CGS impregnated with bFGF at 7 μg/cm2 (low dose) or 14 μg/cm2 (high dose) in the operating room just before application (Fig. 2).
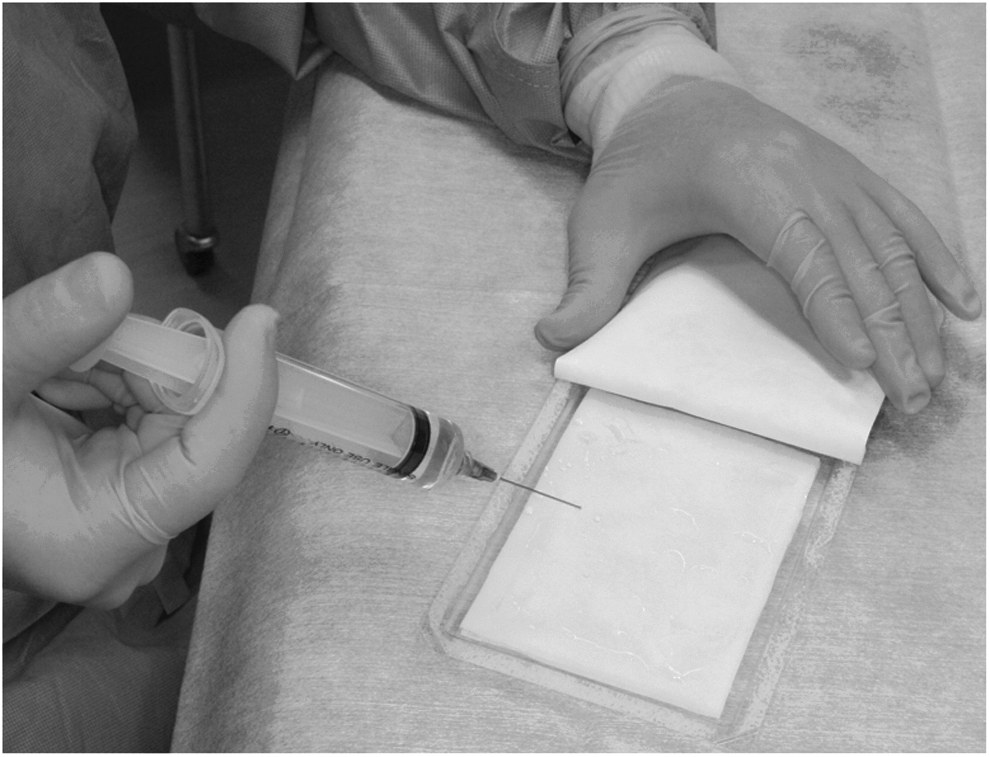
Impregnation of bFGF solution to CGS. After applying bFGF solution to CGS, CGS was left at room temperature for more than 10 min.
The combination therapy with CGS and bFGF
In this study, human recombinant bFGF (FIBRAST SPRAY; Kaken Pharmaceutical) was used. In the impregnation of the low-dose group, 1000 μg of bFGF was dissolved in 14.4 mL of distilled water and 10 mL of this solution was applied to CGS. In the impregnation of the high-dose group, 2000 μg of bFGF was dissolved. About 10 min were needed for impregnation.
After surgical debridement by a single operator, CGS impregnated with bFGF at 7 or 14 μg/cm2 and cut according to the shape of the wound was applied and sutured to the surrounding skin. Reapplication of the device within 3 days of initial application was allowed if necessary because of hematoma formation or dislocation of the device.
After the application of CGS, dressings were changed as necessary. Patients were hospitalized until day 7 to ensure stabilization of the applied CGS and for the safety assessment of study participants. On day 14 after application, the sutures and silicone sheet of CGS were removed.
The additional use of bFGF or a collagen-based artificial skin was prohibited until day 28. After day 29, no particular restrictions were imposed.
Evaluation of treatment and endpoints
Using a digital camera (Canon EOS Kiss Digital X; Canon, Inc., Tokyo, Japan), digital images of the wounds were taken with a calibrator (CASMATCH®; BEAR Medic Corp., Tokyo, Japan) placed on the skin adjacent to the wound. The color and size of images were adjusted using CASMATCH and image editing software (Adobe Photoshop; Adobe Systems) to assess the wound and granulation areas under a standardized procedure. As with the primary endpoint, the baseline wound area, the wound area on day 14, and the granulation area on day 14 were independently measured by blinded reviewers in a central review.
The primary endpoint was wound bed improvement. Granulation tissue is wound connective tissue, which forms at the beginning of wound healing.27,28 This highly fibrous tissue is usually pink because of the invasion of numerous small capillaries to supply oxygen and nutrients. The appearance of granulation tissue is a good sign of healing because, when a wound starts granulating, it means that the healing process of the wound has started.27–29 The area of granulation tissue was measured as the granulation formation area in this study. An unhealed area was defined as an area with no epithelialization and no granulation formation. In this study, the percentage of wound bed improvement was defined as the value (%) calculated from the sum of the granulated and epithelialized areas on day 14 divided by the baseline wound area after debridement on day 0 multiplied by 100, and the patient was diagnosed with wound bed improvement if the wound bed improvement indicator was 50% or higher. 24 The use of 50% or more as the cutoff for the wound bed improvement indicator refers to the definition of the pressure ulcer healing assessment scale by the Japanese Society of Pressure Ulcers30,31 because 50% or more granulation formation indicates the beginning of the wound healing process of retarded chronic ulcers and pressure ulcers.
The secondary endpoints were adverse reactions and serious adverse reactions, percentage of wound bed improvement, percentage of wound area reduction (defined as the value (%) calculated from the wound area of the ulcer on day 14 divided by the baseline wound area after debridement on day 0 multiplied by 100), and percentage of granulation area (defined as the value [%] calculated from the granulation area divided by the wound area on day 14 multiplied by 100). Adverse reactions and serious adverse reactions were documented according to the Medical Dictionary for Regulatory Activities (MedDRA), version 14.0.
Sample size
This study was conducted to determine whether CGS impregnated with bFGF is promising for the treatment of chronic skin ulcers, as evaluated by wound bed improvement as the primary endpoint. The primary analysis was conducted using all data treated with CGS in step 1 and step 2. Since debridement and conventional therapies rarely lead to wound bed improvement in this patient population, the null hypothesis tested in this study was that the proportion of patients with wound bed improvement was 10% or less. This null hypothesis was also supported by previous trials.32–34 Considering the minimum clinically important difference, the expected proportion of patients with wound bed improvement in this study was set to 50% or more. For exact testing based on a one-sample binomial distribution conducted with a one-sided significance level of 2.5% and a statistical power of 90% or higher, the required number of subjects was 14. Allowing for a drop-out rate of 20%, the total number of patients for registration was 17.
Statistical analysis
Patients who had been registered for the study and had undergone investigational device application at least once were included in all statistical analyses. These patients became the statistical unit of analysis. The primary analysis was conducted using an exact test based on binomial distribution with a null proportion of 10% and a one-sided significance level of 2.5%. The 95% confidence interval of the proportion of patients with wound bed improvement was calculated using an exact method based on binomial distribution. The frequency/incidence of adverse reactions and serious adverse reactions that could be causally related to the investigational device was summarized by reaction and severity.
Results
From May 2010 to June 2011, 17 patients (10 patients in the low-dose bFGF group and 7 patients in the high-dose bFGF group) were enrolled in this study. In step 2, 14 patients were randomized to the two dose groups. All 17 patients were included in the FAS and safety analysis set. In patients with multiple chronic skin ulcers, the largest ulcer (n=17) was analyzed for safety and efficacy and the other ulcers (n=8) were used only for safety. Baseline characteristics are shown in Table 1. All patients underwent the application of CGS impregnated with bFGF. One lesion was located on the right middle finger and the other lesions on the lower extremities. One patient enrolled in step 1 discontinued the study 4 days after reapplication because of infection, but the others completed the study.
bFGF, basic fibroblast growth factor.
Regarding the primary endpoint, wound bed improvement was achieved in 16 patients among the 17 (Table 2A). The proportion of patients with wound bed improvement within the total treatment population was 94.1% (95% CI: 71.3%–99.9%), which was significantly superior to the null hypothesis of 10% (p<0.001) (Table 2A). Significant subgroup effects were not found for diabetes, collagen disease, and trauma (results not shown). The outcomes in step 2 and for the total population are shown in Table 2B. We compared subgroup effects between the low-dose group (n=7) and the high-dose group (n=7) according to the randomized comparison in step 2. Among the randomized patients in step 2, significant difference was not seen between the low-dose group and the high-dose group (p=1.00). As for the secondary endpoint of efficacy, the percentage of wound bed improvement in the total treatment population was 86.3% (95% CI: 71.2%–100%) and, in step 2, the percentage in the low-dose group (97.4%, 95% CI: 94.0%–100%) was higher than that in the high-dose group (88.1%, 95% CI: 78.8%–97.5%) (p=0.04). The percentage of wound reduction in the total treatment population was 87.3% (95% CI: 76.5%–98.0%) and, in step 2, there was no difference between the low-dose group (77.5%, 95% CI: 60.2%–94.8%) and the high-dose group (88.2%, 95% CI: 69.2%–107.2%) (p=0.33). The percentage of granulation area in the total treatment population was 86.3% (95% CI: 73.7%–98.9%) and, in step 2, there was no difference between the low-dose group (96.6%, 95% CI: 92.0%–100%) and the high-dose group (85.4%, 95% CI: 73.6%–97.3%) (p=0.052).
Regarding the safety, overall, 13 (76%) of 17 treated patients, comprising 9 (90%) of 10 in the low-dose group and 4 (57%) of 7 in the high-dose group, had at least one adverse reaction. In addition, 6 (60%) in the low-dose group and 2 (29%) in the high-dose group had a reaction that was considered to have a possible, probable, definite, or unknown relationship to the study medication; these reactions are summarized in Table 3. These adverse reactions in the low-dose group were infection (2 reactions), wound complications including wound pain (5 reactions) and wound itching (one reaction), laboratory test abnormality including aspartate aminotransferase increase (one reaction), alanine aminotransferase increase (one reaction), cholinesterase increase (one reaction), and hematoma formation under CGS (one reaction). Adverse reactions in the high-dose group were laboratory test abnormality including hemoglobin decrease (one reaction), C-reactive protein test increase (one reaction), and white blood cell increase (one reaction). One patient in step 1 of the low-dose group discontinued the study because of moderate infection. No serious adverse reactions were reported. Adverse reactions with a clear causal relationship to the study treatment were mild and patients quickly recovered from them.
Data are n (%).
CGS, collagen/gelatin sponge.
Case presentation
Case 1: a case of ulcer related to collagen disease of the right leg (step 1, low-dose group)
A 35-year-old female had been suffering from rheumatoid arthritis for more than 20 years. She had been treated with oral corticosteroid therapy (7.5 mg/day prednisolone equivalent) and methotrexate. She had a chronic ulcer after trauma on the anterior surface of the leg for 1 month (Fig. 3A). Skin perfusion pressure (SPP) at a site 1 cm proximal to the ulcer was 73 mmHg. After debridement, CGS impregnated with bFGF at 7 μg/cm2 was applied and sutured (Fig. 3B, C). On day 14, silicone sheets were removed. The percentage of wound bed improvement was 92.8%. The formed granulation area was evaluated as 93.2% and the ulcer was judged as improved. Three months later, the ulcer was mostly epithelialized (Fig. 3E) and no recurrence was observed 6 months after application (Fig. 3F).

A case of ulcer related to collagen disease of the right leg (step 1, low-dose group).
Case 2: a case of venous ulcer of the left leg (step 2, high-dose group)
A 77-year-old female had a venous ulcer on her left leg for 1 year after undergoing ligation two times (Fig. 4A). SPP at a site 1 cm proximal to the largest ulcer that was evaluated for safety and efficacy was 65 mmHg. After debridement, CGS impregnated with bFGF at 14 μg/cm2 was applied and sutured (Fig. 4B, C). On day 14 after application, the silicone sheets were removed. The wound area and formed granulation area of the largest ulcer were evaluated and the formed granulation area was evaluated as 100%. This ulcer was judged as improved. On day 16, skin grafts were applied on the formed granulation tissues and they took completely (Fig. 4D, E). Two months after application, the ulcers had healed and no recurrence was observed (Fig. 4F).
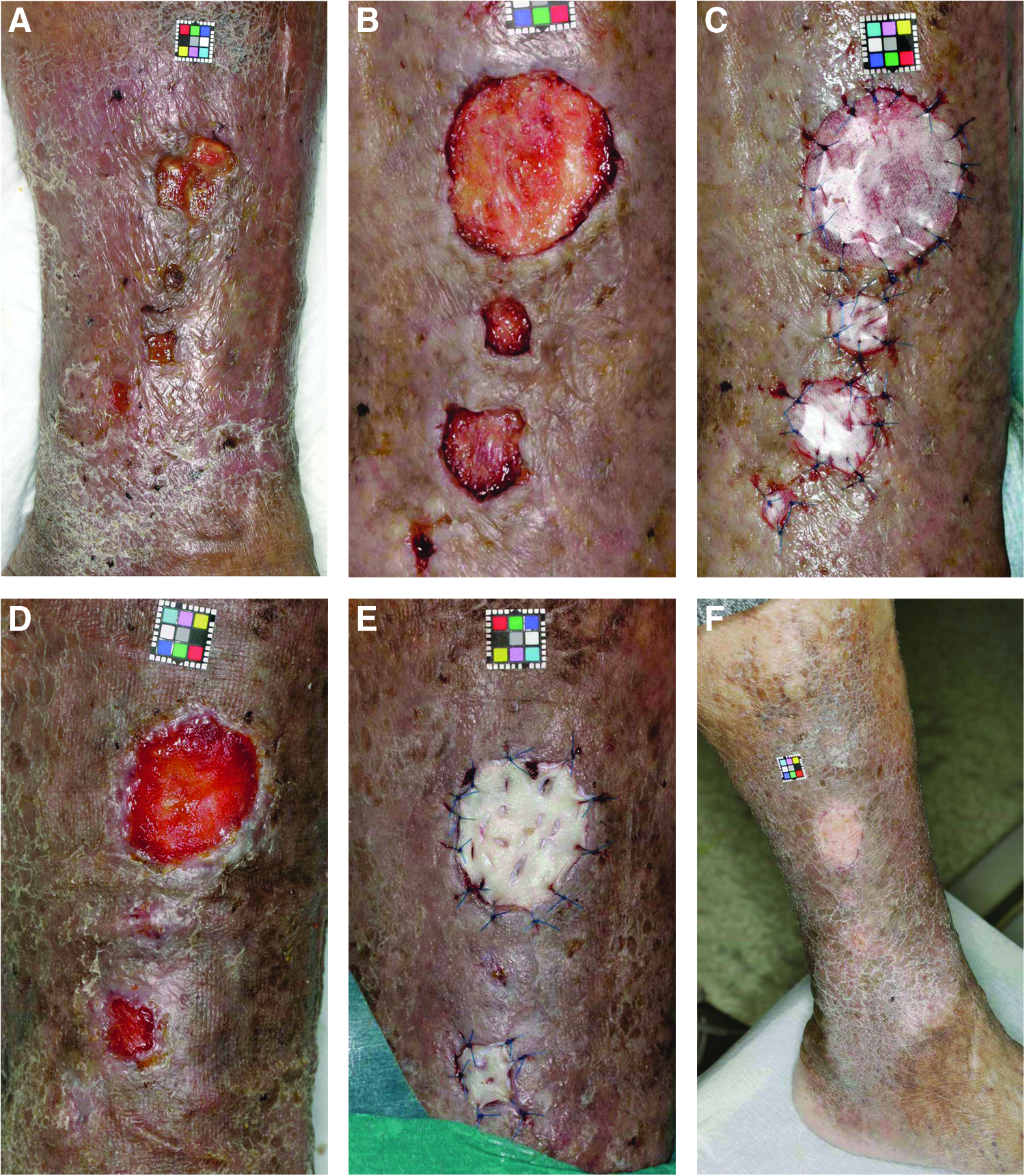
A case of venous ulcer of the left leg (step 2, high-dose group)
Case 3: a case of pressure ulcer of the left leg (step 2, high-dose group)
A 73-year-old male with type II diabetes had been suffering from left-sided paresthesia after cerebral infarction for 9 years. He had a pressure ulcer on the left lateral malleolus for 6 months. The wound was debrided and treated with negative pressure wound therapy (VAC therapy®, KCI KK, Tokyo, Japan) for 3 weeks, but granulation tissue formation was insufficient and the tendon was still exposed (Fig. 5A). SPP at a site 1 cm proximal to the ulcer was 58.5 mmHg. After debridement, CGS impregnated with bFGF at 14 μg/cm2 was applied (Fig. 5B, C). On day 14, this ulcer was judged as improved (Fig. 5D). On day 23, a skin graft was applied on the formed granulation tissues and it took mostly (Fig. 5D, E). Six months after application, no recurrence was observed (Fig. 5F).
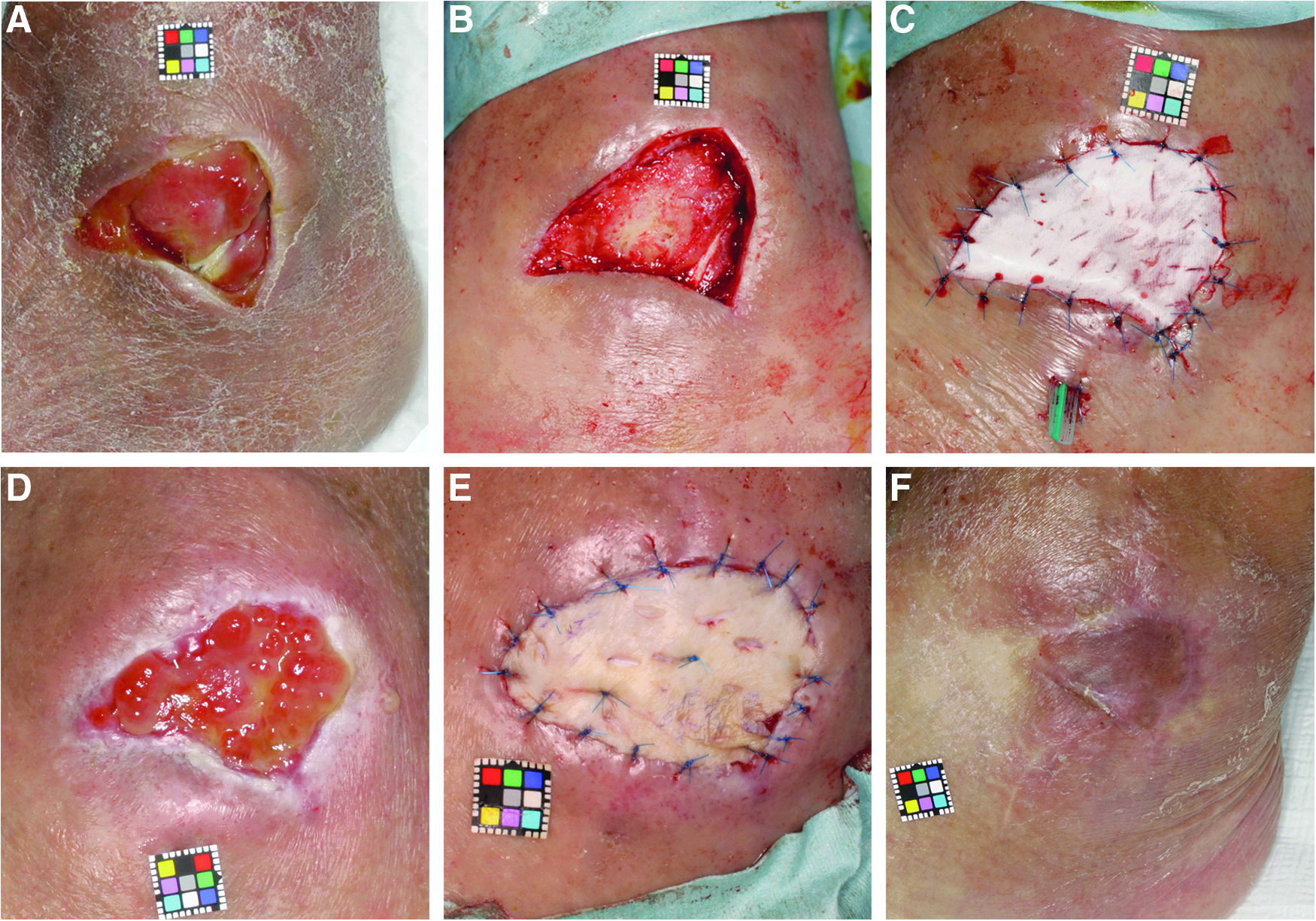
A case of pressure ulcer of the left leg (step 2, high-dose group)
Discussion
This study is the first clinical trial to evaluate the efficacy and safety of a novel artificial dermis that can perform sustained release of bFGF in the treatment of chronic ulcers.
A bilayered artificial dermis, composed of an upper silicone sheet and a lower collagen sponge, has been used in full-thickness skin defects after injuries or tumor removal. 25 After application, its collagen sponge is biodegraded and replaced by host dermal tissue, so-called dermis-like tissue, in 2 or 3 weeks.25,26 Secondary split-thickness skin grafting is performed to complete epithelialization on that dermis-like tissue if necessary. When artificial dermis was applied to chronic wounds in this study, dermis-like tissue was not generally expected to be formed because of insufficient blood flow from the wound bed or infection. Therefore, living cell therapy or growth factors will be required for the treatment of chronic wounds. This is the reason why the novel artificial dermis, CGS, which can exhibit sustained release of bFGF, was developed.
Regarding the primary endpoint, wound bed improvement, that of the total treatment population was significantly improved. This primary endpoint was set because it was necessary to evaluate whether or not chronic wounds start to undergo the wound healing process after debridement with our CGS and bFGF treatment. However, in this disease area, a standardized method of evaluating whether a wound begins to heal or not has not been established. The wound area reduction and granulation tissue formation after CGS treatment were focused on because wound area reduction within the first 4 weeks of treatment was reported to predict the likelihood of healing at 16 weeks, and granulation tissue formation was the first step of wound healing.28,35 Therefore, the primary endpoint was set on the basis of wound reduction and granulation tissue formation measured by central review by three wound healing specialists to evaluate whether the wound started to heal or not. In this study, 14 patients were followed up until 16 weeks after CGS application. It was found that 6 wounds (42.9%) within 12 weeks and 9 wounds out of 14 wounds (64.3%) within 16 weeks were completely healed. In clinical trials using growth factors or bioengineered skin substitutes, PDGF-BB gel for the treatment of lower extremity ulcers, the complete healing rates within 20 weeks were from 44% to 50% compared with 28% to 36% in the control group. 12 The healing rates of diabetic foot ulcers were 56% upon treatment with a bilayered bioengineered skin substitute (Apligraf®; Organogenesis, Canton, MA) and 38% in the control group within 12 weeks.11,36 In the treatment of venous leg ulcers, 63% of the Apligraf-treated patients achieved complete wound healing in comparison with 49% of the control group at 6 months.3,11 The healing rates of diabetic foot ulcers were 30% upon treatment with human fibroblast-seeded dermal substitute (Dermagraft®; Smith and Nephew, Largo, FL) and 18.3% in the control group at 12 weeks.11,37 Compared with these trials, our CGS treatment was considered to be effective for the treatment of chronic skin ulcers. In this trial, 16 patients out of 17 were judged to show wound bed improvement, that is, the wound area was reduced and covered with granulation tissue. However, the primary endpoint could not entirely predict complete wound closure. This might be because our CGS could sustain release of bFGF for only 2 weeks, so repeated application of CGS, every 2 or 3 weeks, might be necessary for complete wound closure.
As for the safety, no serious adverse reactions were reported and other adverse reactions were mild and patients quickly recovered from them. The incidence of reactions with a clear relationship to the study treatment in the high-dose group (29%) was lower than that in the low-dose group (47%). This indicated that the higher dosage of bFGF did not affect the safety of this treatment. As for the dosage of bFGF, our preclinical studies using chronic wounds on diabetic mice showed that the effect in the high-dose group was superior to that in the low-dose group; on the other hand, other studies using skin defects on healthy mice indicated the opposite results.21,23 In step 2, the percentage of cases of wound bed improvement in the low-dose group was greater than that in the high-dose group. These results showed that the low dosage of bFGF was fully effective and the high dosage could be used if necessary.
This study was the first trial to evaluate the efficacy of CGS impregnated with an appropriate concentration of bFGF for the treatment of chronic ulcers. Various bioengineered skin substitutes that provide growth factors secreted by living cells have been reported to be effective for chronic skin ulcers, although they are costly and access is limited because they contain living cells. Both CGS and bFGF are freeze-dried and can be stored off-the-shelf at room temperature. Our CGS impregnated with bFGF can exhibit sustained release of bFGF in a controlled manner; therefore, the effectiveness of CGS could either equal or surpass that of existing approaches, as our results show.
In this study, bFGF was used in combination with CGS because bFGF can be clinically used in Japan. The acidic gelatin contained in CGS can sustain the release of positively charged growth factors such as PDGF-BB and transforming growth factor-β, which are the dominant growth factors in platelet-rich plasma (PRP).38–40 PRP can be prepared from a patient's blood or from donated platelets. When bFGF cannot be used, combination therapy involving CGS and PRP can be an alternative.
Because of ethical and clinical considerations, this study did not have a control group with conventional treatment and randomization was performed between the two bFGF treatment groups. In our clinical practice, artificial dermis is not usually applied for the treatment of chronic ulcers because it is easily infected in wounds with insufficient blood flow from the wound bed. Accordingly, a control group treated with conventional artificial dermis or CGS without bFGF impregnation could not be set. In this study, only the single application of CGS was allowed and the etiologic causes of ulcers were not specified because there were no clinical data of its efficacy. On the basis of the results of this study, it is desirable to explore the efficacy for a specific disease, such as diabetic ulcer or venous ulcer, or the efficacy of multiple applications of CGS, with a control group.
Conclusion
This study is the first clinical trial to evaluate the efficacy and safety of a novel artificial dermis that can sustain the release of bFGF in the treatment of chronic ulcers. We applied this in 17 patients with chronic ulcers and, 2 weeks after implantation, the wound bed was improved in 16 of them. No serious adverse reactions were observed. The combined therapy with CGS and bFGF could be a promising treatment that is comparable to skin substitutes containing living cells in terms of cost and usability.
Authors' Contributions
S.S. was the PI of this trial. N.M., K.Y., M.N., H.T., S.T., and T.M. wrote the protocol and designed the trial. K.Y. and S.T. did the statistical analysis. M.N. and H.T. did the quality control of the trial. N.M., R.A., J.F., N.K., and S.S. recruited and managed the patients in the study.
Footnotes
Acknowledgments
This work was supported by a grant from the Japan Science and Technology Agency. We thank A. Kinoshita for data management; M. Higashi for monitoring; R. Ishizuka for management of investigational new drug; N. Yoshioka, H. Naito, and H. Toichi for the central committee; and Y. Horiguchi, N. Inagaki, Y. Yamawaki, and J. Arata for the data and safety monitoring committee.
Disclosure Statement
The authors declare no conflicts of interest.