
Abstract
Islet transplantation is a promising treatment for human type 1 diabetes mellitus. Transplantation requires systemic immunosuppression, which has numerous deleterious side effects. Islet antigen-specific regulatory T cells (Tregs) have been shown to protect islet grafts from autoimmune destruction in the nonobese diabetic (NOD) model when co-localized in the kidney capsule. An extra-hepatic transplant site was established by transplanting islet-loaded microporous poly (lactide-co-glycolide) (PLG) scaffolds into abdominal fat. This study examined an autoimmune transplantation model and determined whether co-localized Tregs could protect islet grafts in an extra-hepatic and extra-renal transplant site. Normoglycemia was restored, and co-transplanted Tregs extended graft survival, including several instances of indefinite protection. Transplanted Tregs were replaced by recipient-derived Tregs over time, indicating that islet antigen-specific Tregs induce tolerance to islet grafts through host-derived Tregs. Thus, Tregs provided protection against a diverse repertoire of autoreactive T-cell-receptor specificities mediating diabetes in the NOD model, possibly through a phenomenon previously described as infectious tolerance. Interestingly, the infiltration by Tregs protected a second islet transplant, indicating systemic tolerance to islet antigens. In summary, PLG scaffolds can serve as an alternative delivery system for islet transplantation that allows for the co-localization of immunomodulatory cells within islet grafts and induces long-term graft survival in an autoimmune diabetes model. This method of co-localizing immunomodulatory cells with islets in a clinically translatable transplant site to affect the immune system on a local and systemic level has potential therapeutic implications for human islet transplantation.
Introduction
Regulatory T cells (Tregs) present a potential method for controlling autoimmunity in T1DM, and their activity has been implicated in tolerance induction. Tregs maintain homeostasis and self-tolerance in the immune system through their suppressive capabilities of numerous key cell types. 5 Tregs function by suppressing T-cell and dendritic-cell (DC) maturation and activation through the secretion of immunosuppressive factors, such as IL-10 and TGF-β1, and through direct cell-cell interactions.6,7 Tregs have been shown to prevent diabetes by inhibiting effector T cells, which are CD4+ or CD8+ cells that autoreactive to antigens on islet β-cells, locally in autoimmune diabetes models. 8 Tregs are known to suppress both local and systemic immune activation in autoimmune models 9 ; in transplantation models, immunoprotection by Tregs requires localization to the graft site and subsequent migration to the draining lymph node (dLN).10,11 However, additional aspects of Treg function remain open, including whether Tregs remain in the graft site or dLN, provide local and/or systemic protection, or reduce effector T cell numbers or activity.
In this article, we investigate the mechanisms by which Tregs provide long-term graft protection by co-delivery of islets and Tregs on microporous poly (lactide-co-glycolide) (PLG) scaffolds implanted at a clinically translatable site. 12 Clinical islet transplantation is performed by islet delivery into the liver sinusoids, and Tregs delivered simultaneously may not co-localize. Furthermore, extrahepatic transplantation on scaffolds can avoid the instant blood-mediated inflammatory reaction and first-pass exposure to diabetogenic immunosuppression, while also presenting signals to promote engraftment.13–15 The nonobese diabetic (NOD) mouse model, which spontaneously develops autoimmune diabetes similar to human T1DM, was used to investigate the ability of antigen-specific Tregs to protect islet grafts from autoimmune destruction when co-transplanted with islets. Islet-antigen specific Tregs were obtained by isolating and culturing T cells from an NOD transgenic strain, NOD.BDC2.5, which produces T cells that solely express the T-cell receptor (TCR) against an islet antigen derived from Chromogranin A (termed BDC2.5 mimotope or peptide). BDC2.5 naïve CD4+ T cells cultured in the presence of antigen-presenting cells (APCs), BDC2.5 peptide, and TGF-β1 differentiate and expand into CD4+CD25+Foxp3+ Tregs in vitro.11,16 These Tregs maintain expression of the BDC2.5 TCR and exert immunosuppressive functions in the presence of their antigen. The local and systemic impact of Tregs after co-localization with islets on scaffolds was investigated by characterizing infiltration, differentiation, and localization of immune cell types into the scaffold and the phenotype and function of effector T cells in the dLN and spleen. Finally, we investigated local and systemic immune responses to islet antigens for co-delivery of Tregs with islets on scaffolds. Identifying the actions of Tregs can facilitate their application to immunoprotection in a transplant setting.
Results
Tregs prolong islet graft survival when colocalized on PLG scaffolds
PLG scaffolds, a platform for extrarenal and extrahepatic islet transplantation, were investigated as a method to co-localize Tregs with islets to provide graft protection. Donor islets were transplanted on PLG scaffolds into the abdominal fat of diabetic female NOD recipients without Tregs (−Treg), with 3×106 Tregs injected intravenously, and with 3×106 Tregs co-localized in the scaffold (+Treg). The transplanted islets function in all conditions, indicated by normalized blood glucose levels compared with hyperglycemia observed in diabetic mice before transplantation. Without Treg co-transplantation, hyperglycemia develops within 10 days of transplantation, which is consistent with autoimmune destruction of the graft (Fig. 1). In contrast to the −Treg recipients, +Treg PLG scaffold islet grafts have extended euglycemia, indicating delayed or avoidance of rejection. However, for Tregs delivered systemically, no protection was observed. Thus, Tregs co-localized on scaffolds can protect islets from autoimmune destruction to maintain euglycemia.
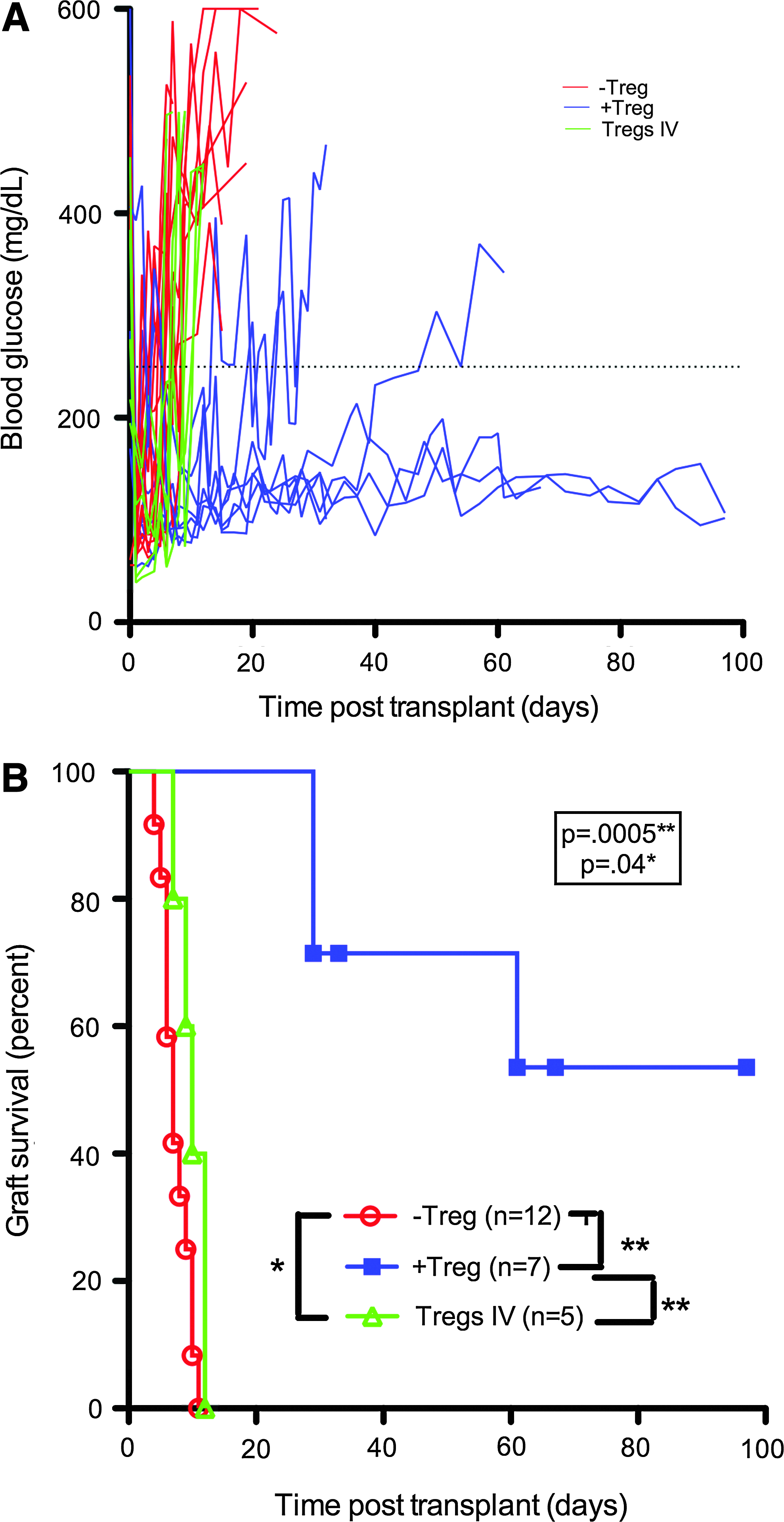
Islet graft survival when transplanted on PLG scaffolds is prolonged by Treg co-localization within the scaffold.
Histological examination of PLG scaffold-transplanted islet grafts with Tregs
The distribution of islets and Tregs was examined by histology in +Treg and −Treg islet recipients. The scaffolds were removed on day 7 for both −Treg and +Treg conditions, day 25 for −Treg, and later than day 25 (>25) for +Treg. In −Treg recipients, insulin-producing cells were detected at day 7 but were completely absent at day 25 (Fig. 2a, c). No Foxp3+ cells were observed within the scaffolds or near the islets before or after rejection. In contrast, for the +Treg condition, insulin-producing cells were detected in short (day 7) and long-term grafts (day >25, a time point postrejection without Treg intervention), and the islet architecture was preserved at all +Treg conditions (Fig. 2b, d). Foxp3+ cells were detected in close proximity to insulin-producing cells in all +Treg recipients. The number of Foxp3+ cells within the graft was quantified and compared between −Treg and +Treg mice at day 7 and day >25 (Fig. 2e). In both cases, significantly more Foxp3+ cells were present in grafts that were cotransplanted with Tregs than in their absence (p=0.02 at both day 7 and >25).
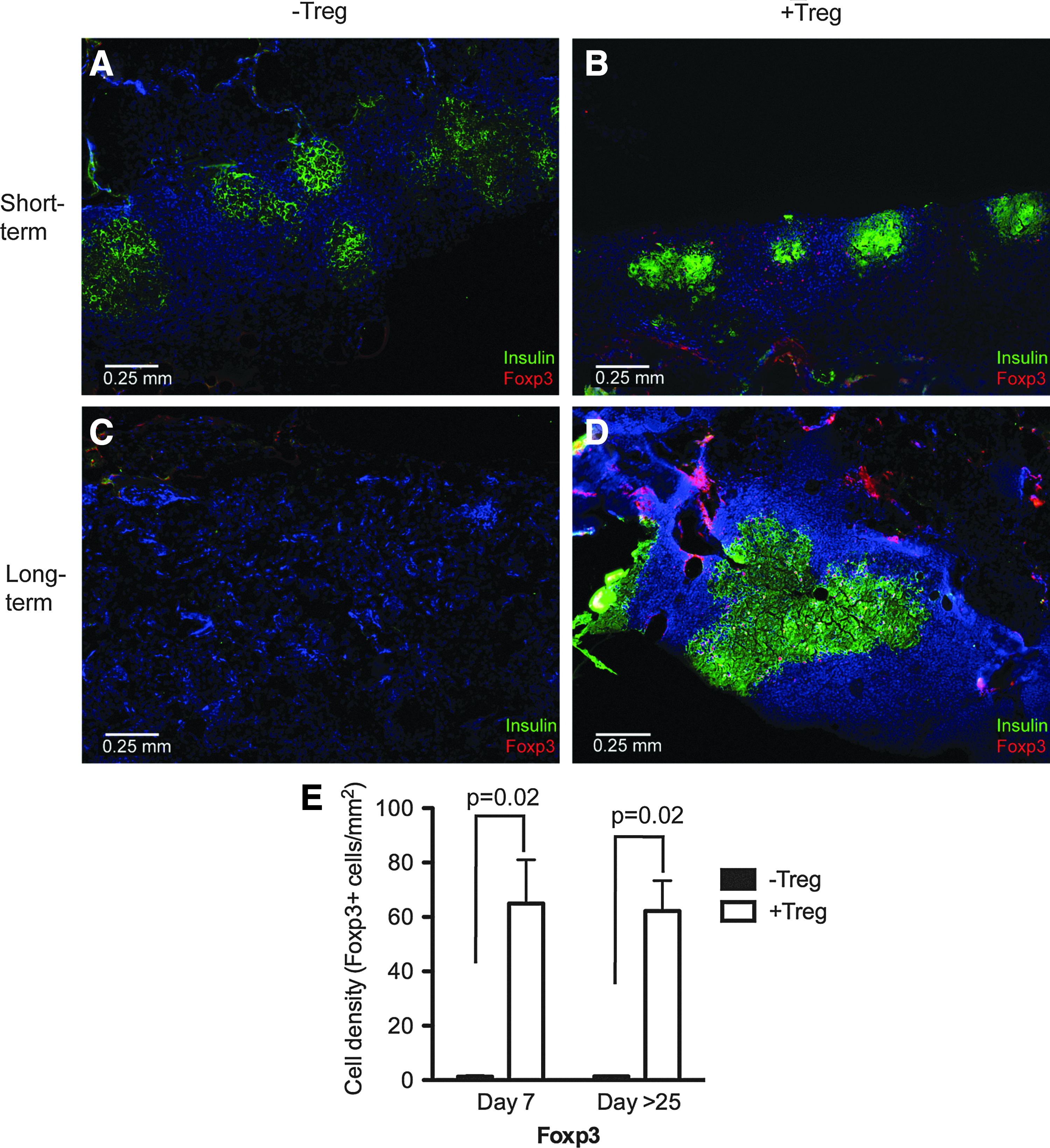
Protection of PLG scaffold transplanted islets by Tregs is associated with robust insulin production and Foxp3+ Treg co-localization around islets. Grafts from day 7 [both−Treg
Grafts were examined for the presence and localization of additional immune cells that were important in the protection and destruction of islet grafts. F4/80 (macrophage) and CD11c (DC) staining, which identify cells that would usually present antigens to activate an immune response, was not observed around the islets, and no obvious differences in staining were observed in either −Treg or +Treg conditions (Fig. 3a). DCs and macrophages were observed on the polymer surface. On quantification of immune cells within the islet containing regions of the scaffold, the density of CD11c+ and F4/80+ cells were similar (Fig. 3b, c). Both CD4 and CD8 T cells were present within the pores of the scaffold and localized around and infiltrated islets both in −Treg and +Treg conditions at day 7 (Fig. 4a). CD4 and CD8 T cells were present in similar densities in islet-containing areas of the scaffold graft (Fig. 4b, c).

PLG scaffold transplanted islets are infiltrated with dendritic cells and macrophages in both −Tregs and +Tregs conditions and primarily localize on the scaffold surface.
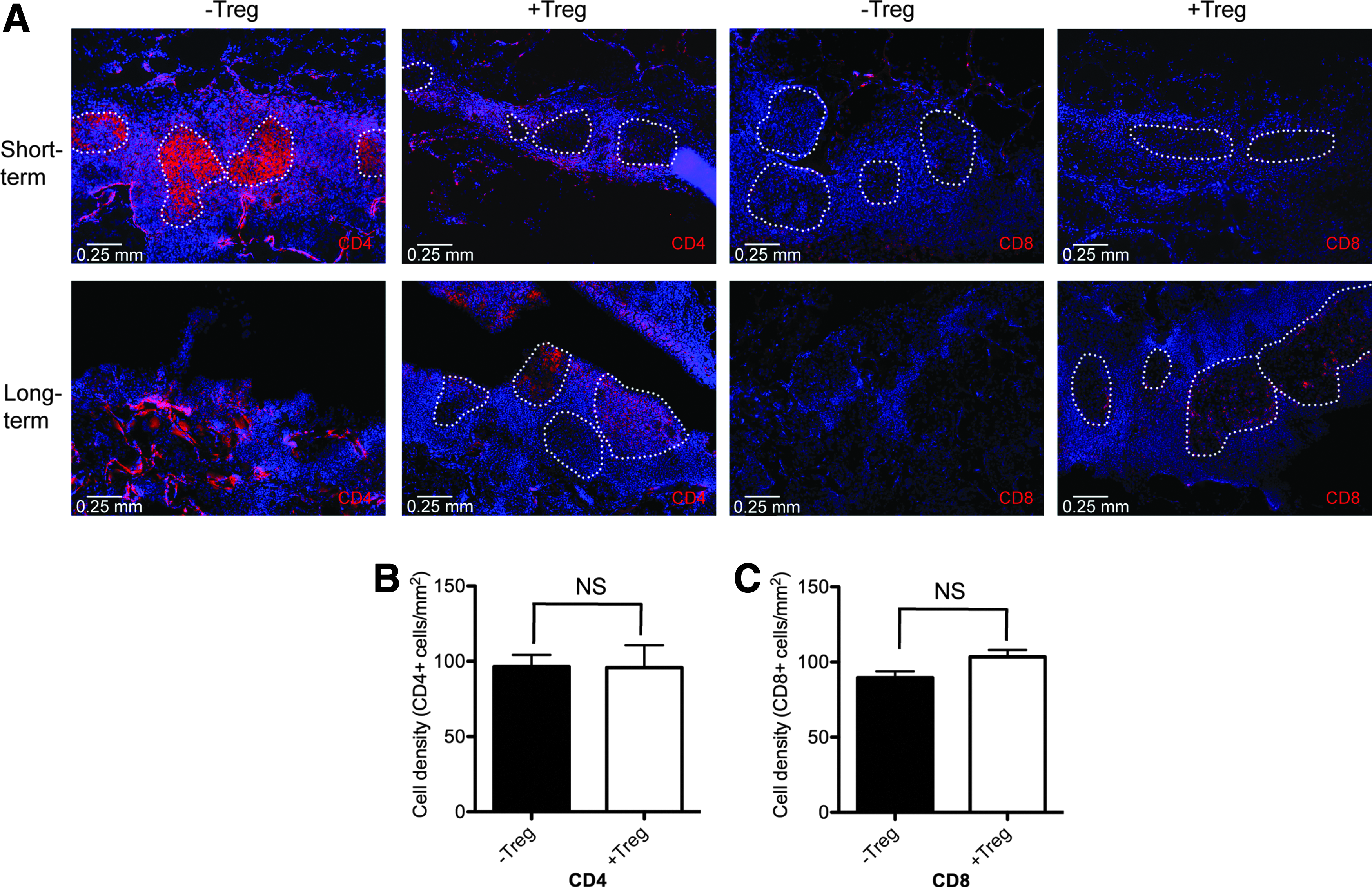
PLG scaffold transplanted islets are infiltrated with CD4 and CD8 T lymphocytes that localize around islets in both −Treg and +Treg conditions.
Systemic or local protection
BDC2.5 Tregs were investigated as suppressors of islet-specific effector cells, which are indicative of graft protection. A previous work has demonstrated that BDC2.5 T cells proliferate significantly in the pancreatic lymph node, but not nondraining lymph nodes (ndLN), likely because antigens from islet β-cells are presented by DCs in the draining pancreatic lymph node in autoimmune destruction of grafts. 17 To verify the in vivo activity of BDC2.5 Tregs, naïve BDC2.5 CD4+ cells were labeled (Carboxyfluorescein succinimidyl ester [CFSE]) and intraveneously transferred into −Treg and +Treg scaffold islet recipients (Fig. 5). BDC clonotype-positive cells were identified using an antibody specific for the Vβ4 subunit of the TCR, the sole TCR expressed by BDC2.5 mice. A few BDC clonotype+ cells were present in the ndLN, and most had not diluted CFSE (i.e., not proliferated) 3 days post-transplantation in both −Treg and +Treg conditions. In contrast, in the dLN of −Treg transplant recipients, clonotype+ T cells accumulated and underwent one or more divisions, as indicated by lower CFSE fluorescence. In the dLN of +Treg transplant recipients, Vβ4+ T cells accumulated, but the fraction of cells undergoing division was significantly lower (p=0.03), which was similar to ndLN (Fig. 5b). Therefore, BDC2.5 Tregs limit the proliferation of islet-specific naive cells in vivo when transplanted with islets on scaffolds despite there being similar numbers of CD4+ cells in the dLN of both conditions (Fig. 5c). Given the similar percentage of CD25+Foxp3+ cells in the dLN of both −Treg and +Treg conditions (Fig. 6), the more profound suppression by Tregs suggests either an increase of Treg potency or enriched Treg specificity in the dLN.
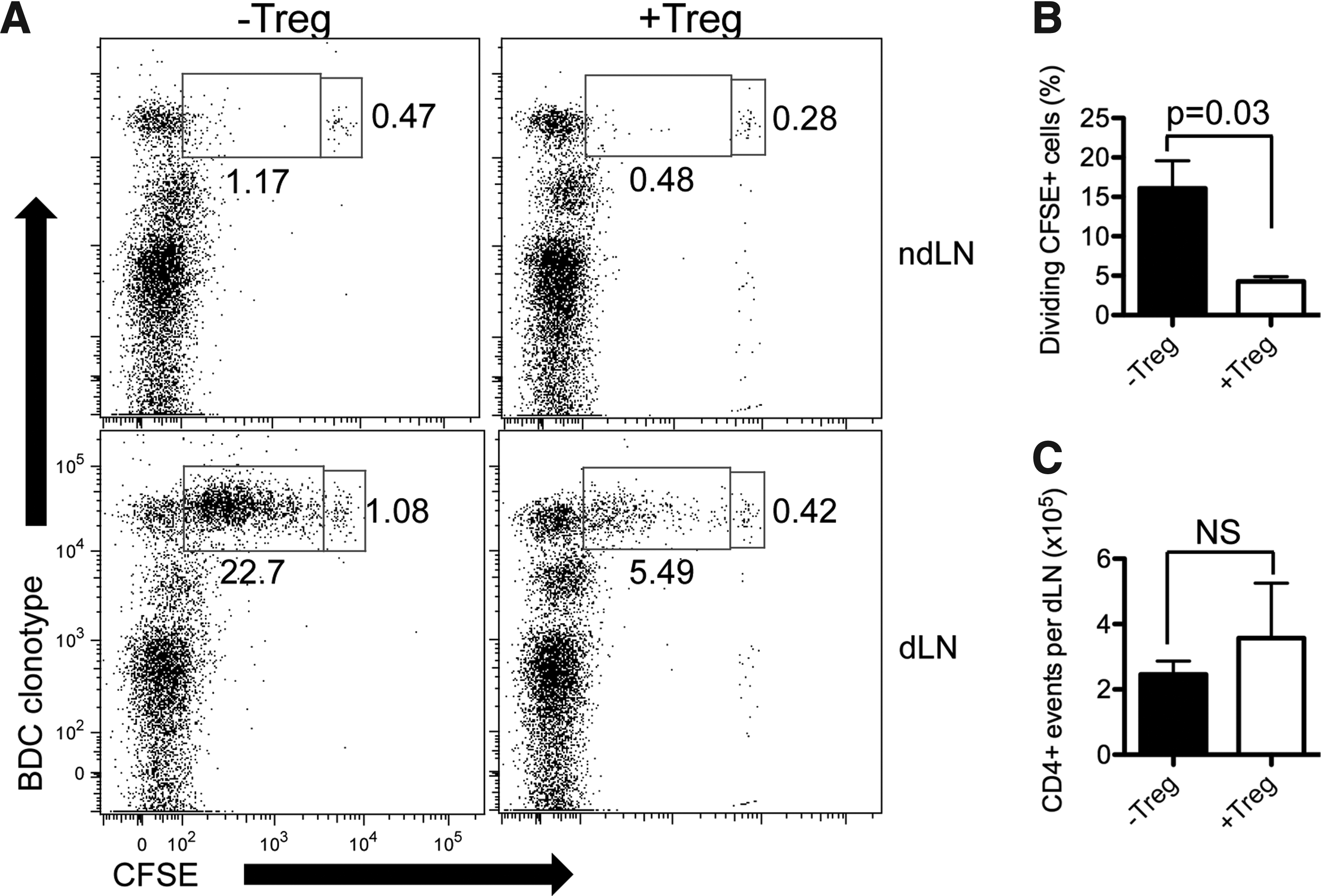
PLG scaffold transplanted islets +Tregs prevent proliferation of BDC2.5 naïve CD4+ in vivo.
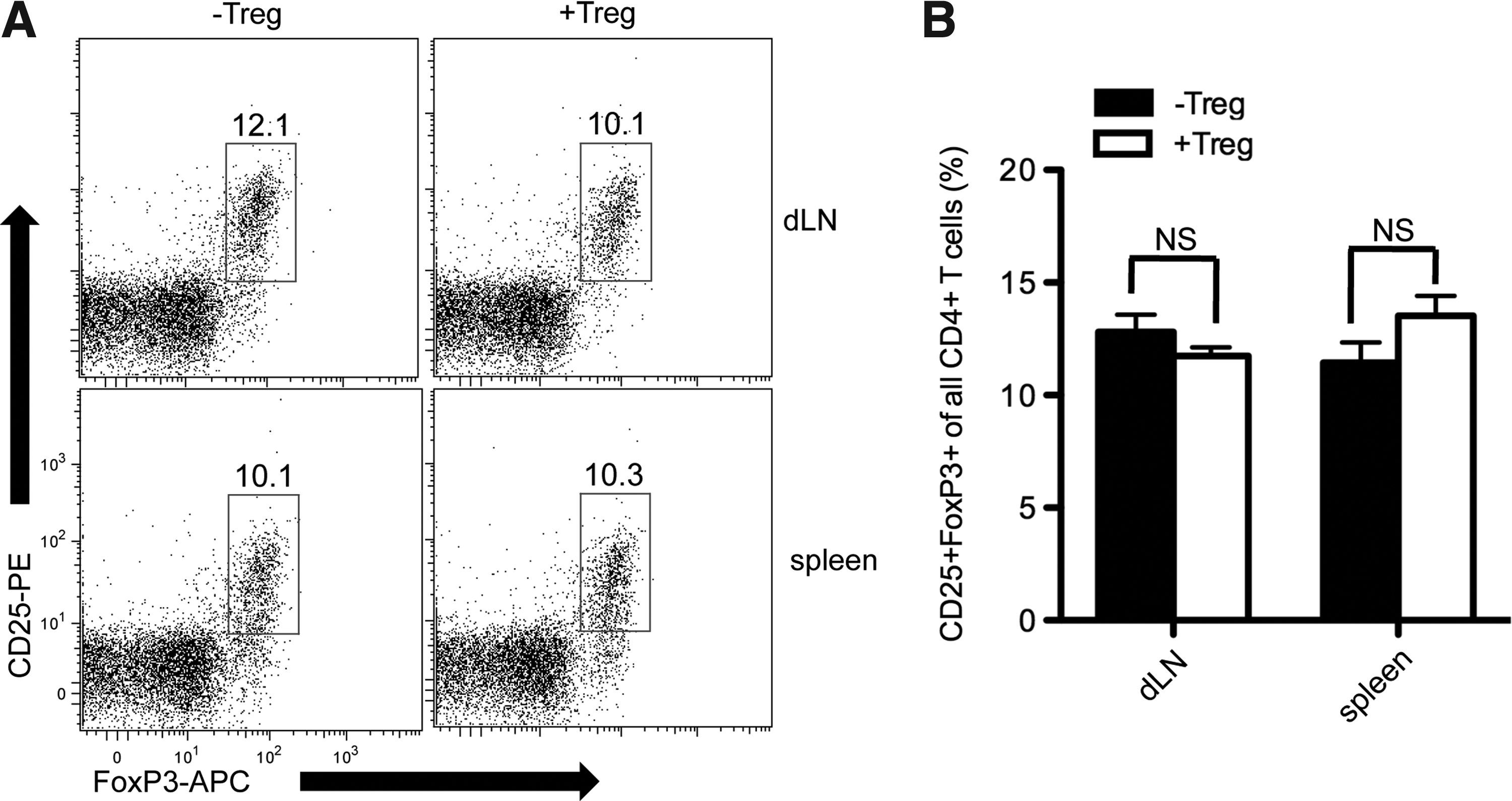
Protection of PLG scaffold transplanted islets by Tregs is not associated with peripheral up-regulation of total Treg population. CD25+Foxp3+ Treg cells were assessed in the spleen and the graft dLN on day 25 (−Treg, n=5) and day 33 (+Treg, n=3).
Limited CD4+ proliferation in the dLN with Tregs motivated subsequent studies to investigate whether Treg protection of islets was local or systemic. On day 97 post-transplantation of islets and Tregs on scaffolds, a second islet graft was implanted into the contralateral kidney capsule (KC) without Tregs. On day 99, the scaffold was removed. Blood glucose was monitored for evidence of graft rejection for 43 days post-KC transplant, which corresponds to day 140 after the initial transplantation (Fig. 7a). The mice remained euglycemic until day 140 when a nephrectomy was performed, at which time the mice became hyperglycemic, indicating the KC graft was maintaining euglycemia. Histological staining of scaffold (day 99) and KC grafts (day 140) indicated that Foxp3+ cells were in close proximity to insulin-producing cells in both scaffold and KC grafts, which was consistent with long-term immunoprotection of scaffold grafts and systemic protection of islet grafts at distal locations (Fig. 7b, c).
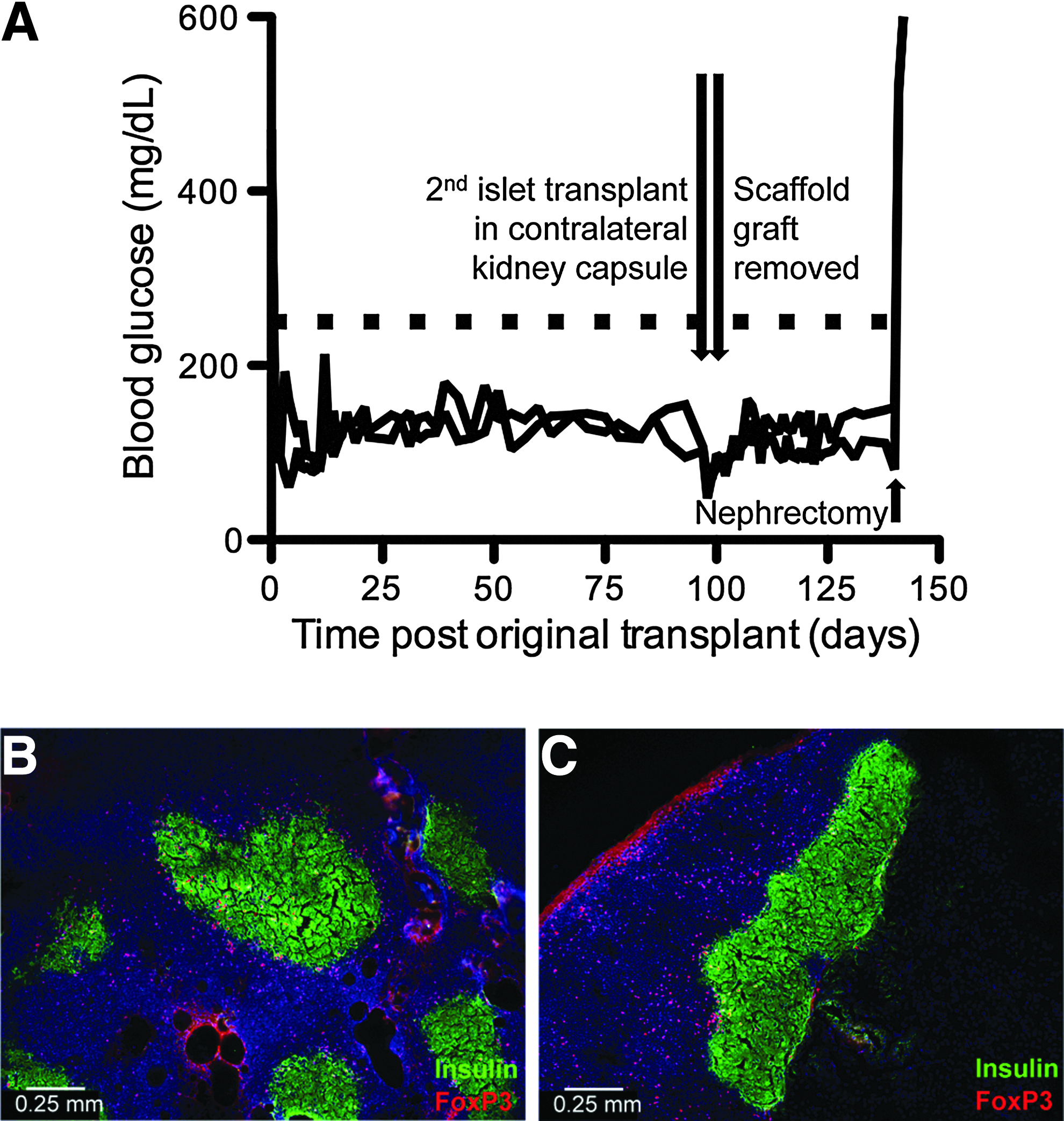
PLG scaffold transplanted islets +Tregs induce systemic tolerance to islet grafts.
Scaffold and KC grafts were subsequently analyzed to determine the Treg source: originally transplanted Tregs or endogeneously recruited. BDC2.5 Tregs exclusively express the Vβ4 TCR; thus, all Tregs originally transplanted with islets on scaffolds were Foxp3+Vβ4+. Foxp3+Vβ4+ cells were observed at day 7. However, day 33, 99, and a second KC transplant contained predominantly Foxp3+Vβ4− cells, indicating that BDC2.5 Tregs induce endogenous Treg infiltration and/or differentiation into scaffold islet grafts. Vβ4+ Tregs were not observed in islet grafts after day 7, indicating that BDC2.5 Treg did not persist at the graft, yet long-term graft protection was obtained (Fig. 8).
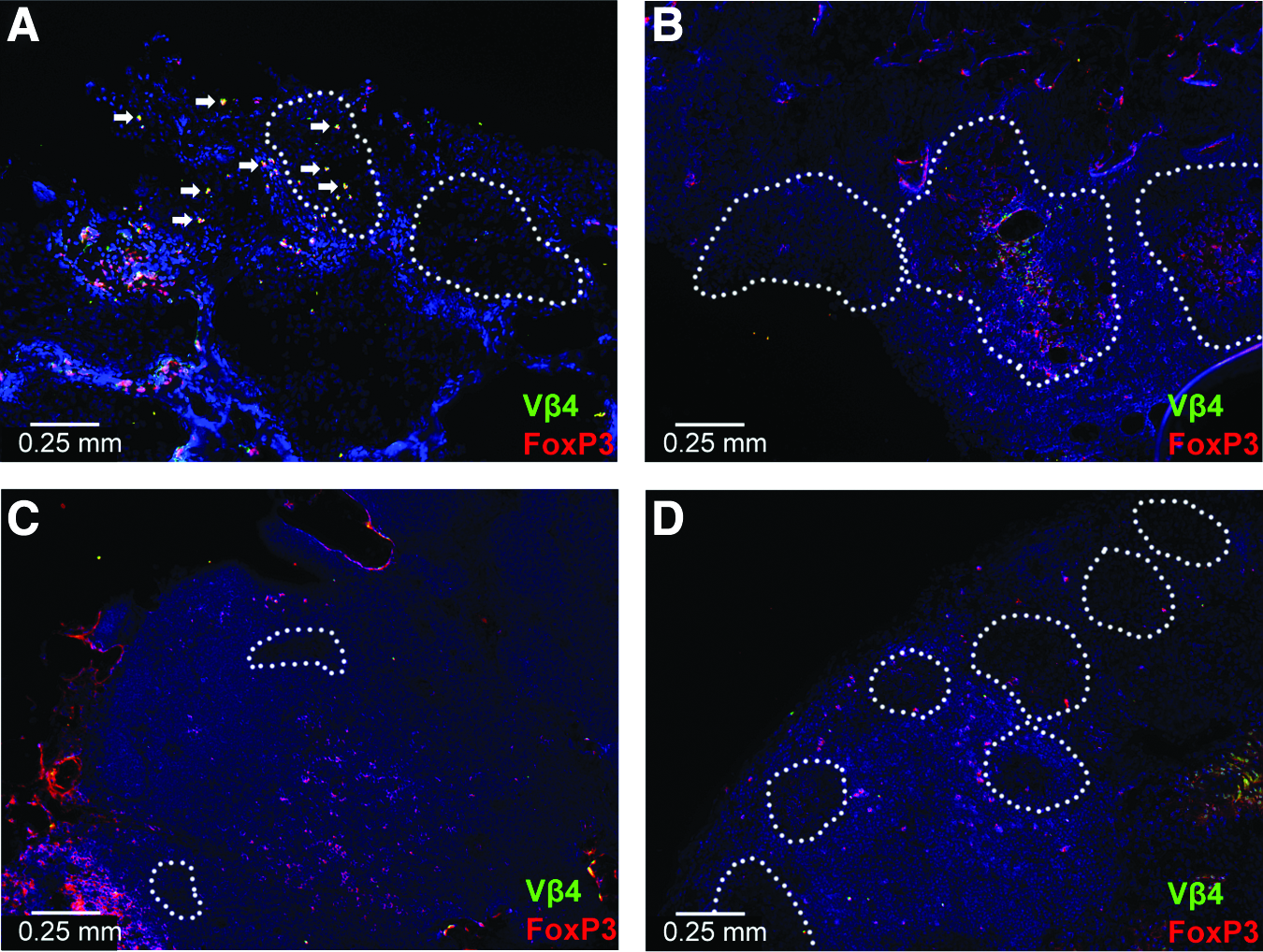
Islet grafts transplanted with BDC2.5 Vβ4+ Tregs are infiltrated with Vβ4−Foxp3+ Tregs over time.
Finally, the presence of autoreactive cells in mice transplanted with islets and Tregs was investigated to determine whether systemic deletion occurred with Treg transplantation. Splenocytes isolated from −Treg and +Treg scaffold islet graft recipients at post-transplant day 33 were adoptively transferred via an intravenous injection into normoglycemic male NOD.scid mice. Mice receiving splenocytes from −Treg recipients became diabetic by day 29, while diabetes induction was significantly delayed with splenocytes from +Treg donors, including 40% of the recipients from the +Treg donor group that remained euglycemic for the study duration (Fig. 9). Taken together, these results indicated that autoreactive cells were present in mice which had protected islet grafts by Tregs, but were lower in number or had their effector function controlled by Treg cotransplantation.
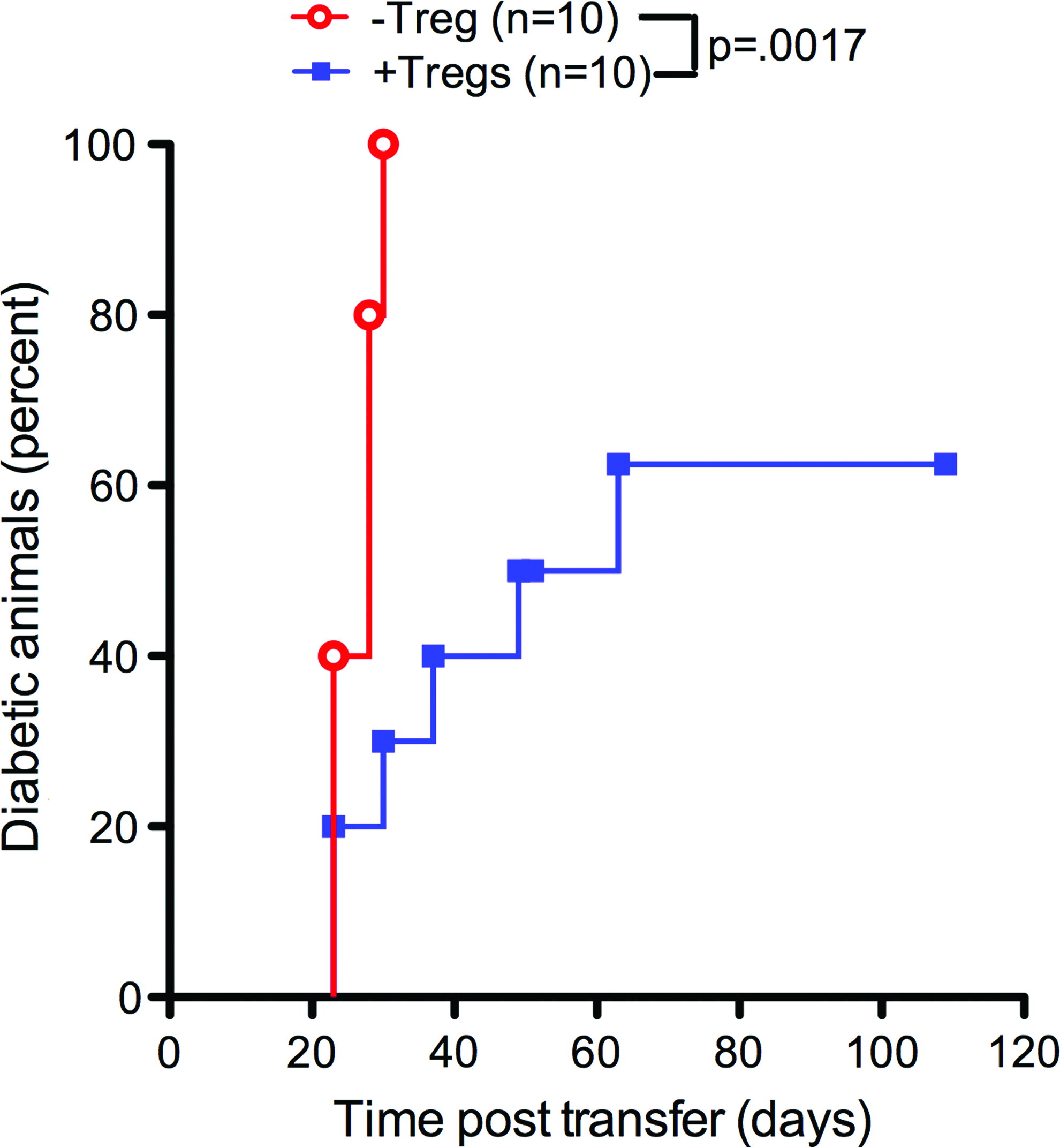
NOD.scid recipients of splenocyte adoptive transfer from PLG scaffold transplanted islets. +Tregs became diabetic at a slower rate than PLG scaffold transplanted islets −Treg. p=0.0017. N=10 for both conditions. Color images available online at www.liebertpub.com/tea
Discussion
PLG scaffolds provided the means to co-localize islets and Tregs in a clinically translatable site. Current clinical islet transplantation for the treatment of T1DM relies on delivery via intra-portal infusion and lodging of the islets in the portal sinusoids. Co-localization of islets and Tregs bypasses the need for Tregs to home to the graft site, directly placing Tregs in their location of initial action immediately. Manipulating islets or Tregs to promote co-localization, or the use of nontranslatable transplantation sites has limited translation of Treg technology.11,18 Co-delivery of Tregs and islets into the liver sinusoids is not expected to result in co-localization needed for protection of islets because of the nontargeted large distribution of cells in this delivery method. The design of the scaffolds allowed for the effective seeding of Tregs seeded into the pores of the scaffold. PLG scaffolds provide a localized space for islet transplantation that avoids issues associated with the hepatic graft site, such as the instant blood-mediated inflammatory response and first-pass exposure to diabetogenic immunosuppression therapy, which combine to create a nonideal transplant environment that may contribute to the loss of islets and insulin independence.2,19,20 While many biomaterial-based approaches to islet transplantation involve encapsulation to isolate islets from immune attack, porous scaffolds encourage cell infiltration and integration with the host, leading to revascularization of the transplanted islets. 21 Furthermore, the microenvironment of the porous scaffold can be readily engineered to enhance islet survival and engraftment.13,14 In summary, scaffolds provide an extra-hepatic transplant platform that allows for the co-localization of islets and Tregs while addressing the shortcomings of current clinical transplantation methods.
Treg co-transplantation delayed or prevented islet graft rejection without systemic immunosuppression with delivery on a scaffold, which is consistent with a previous report in the KC. 11 Scaffold implantation leads to a foreign body response, a nonspecific inflammatory response, which could complicate islet engraftment and strategies to promote immune protection. 22 The process of implantation and the biomaterial recruits host APCs, such as macrophages and DCs, which can, in turn, induce secretion of inflammatory cytokines at the injury site. The localization of APCs along the pores of the scaffold matched previous reports.23,24 APCs did not localize to islets in either the +Treg or −Treg condition. Our previous work with syngeneic islet transplantation in mice demonstrated the potential to reverse diabetes with a minimal islet mass, suggesting that the inflammatory response did not significantly impair islet function. 13 However, the inflammatory environment could further activate APCs to allow effective antigen presentation to autoreactive T cells infiltrating the graft; or alternatively, these APCs could travel to the dLN to present antigens to T cells.25,26 The presence of APCs in the graft site may be necessary for the systemic immunoprotection observed; tolerogenic APCs may be presenting multiple antigens in the presence of co-localized Tregs to induce “infectious tolerance.”27,28 Despite the presence of immune cells, Treg therapy was highly effective in protecting scaffold-transplanted islets in the NOD T1DM model. Although graft protection was not permanent in some recipients, results matched previous observations in the KC transplant location, and are thought to be caused by the heterogeneous nature of autoimmune diabetes in this model system. 11
Scaffolds create a space in which Tregs can be co-localized with islets, which are necessary for Treg-mediating systemic immunosuppression. At the graft, a robust accumulation of Foxp3+ cells was observed that was localized around insulin-positive cells in the +Treg protected scaffold islet graft. Although nonregulatory CD4 cells have been shown to transiently express Foxp3 on activation, we observe minimal Foxp3 expression in the −Treg condition and robust Foxp3 expression in the +Treg condition, implying a regulatory phenotype of the Foxp3+ cells present in the protected graft. 29 The Tregs observed in the graft at day 7 were those initially transplanted (Vβ4+ BDC2.5 Tregs). However, at later time points, the Treg population that is localized around islet grafts shifts to Vβ4−Foxp3+ Tregs. This result indicated that BDC2.5 Tregs induce and recruit host Tregs for long-term graft protection, most likely islet antigen-specific, including specificity for antigens causing autoimmune diabetes. BDC2.5 Tregs provide protection against a diverse repertoire of autoreactive TCR specificities mediating diabetes in the NOD model, possibly through a phenomenon previously described as “infectious tolerance” or “linked suppression.”27,30,31 Transplanted Tregs localized in scaffold islet grafts remained protective despite the plasticity of these cells in inflammatory environments and were able to protect islets from destruction by infiltrating CD4 and CD8 T cells.32,33 Treg production of immunomodulatory factors and direct cell-cell interactions may underlie the mechanism of protection, with previous reports indicating Treg control of DC and T-cell maturation and migration to dLN.6,7
Systemically, a second islet graft without Tregs implanted in a distal location ≈100 days after the initial islet transplant with Tregs was protected from autoimmune destruction in transplant recipients in which permanent protection was observed. Robust Foxp3+ cell infiltration was observed in the graft of this second transplant, indicating Treg trafficking to the site. In addition, the Foxp3+ cells localizing to the second islet graft were Vβ4−, indicating that host Tregs are induced by the initial BDC2.5 Treg transplantation and mediate long-term tolerance. The induced Tregs are capable of trafficking to an additional site of antigen-driven inflammation to exert protection, thereby protecting subsequent islet grafts. However, adoptively transferred splenocytes from +Treg mice induced diabetes, although at a slower rate than splenocytes from −Treg controls. Induction of diabetes from splenocytes in tolerized mice indicated that autoreactive T cells remain in the host, yet the slower rate of diabetes onset suggests that these cells were present in smaller numbers and/or were effectively controlled by Treg co-transplantation. Several other possible explanations for this observation include that the persistence of Tregs requires tolerogenic APCs that reside in the original host or that Tregs require the initial local site to turnover.
In conclusion, this article demonstrates effective long-term protection of islet grafts from autoimmune destruction in PLG scaffolds when co-transplanted with antigen-specific Tregs in the abdominal cavity. Scaffold transplanted islets with co-localized Tregs restored euglycemia and prolonged islet graft survival, including permanent protection in a subset of recipients. Protection of these grafts was associated with Treg localization around islets. Initially, these Tregs were those transplanted at the time of islet transplantation, but recipient-derived Tregs replace the transplanted Tregs over time. This result indicated that islet antigen-specific Tregs induce tolerance to islet grafts through host-derived Tregs, likely islet antigen specific as well. Interestingly, the infiltration by Tregs protected a second islet transplant, indicating possible systemic tolerance to islet antigens mechanisms. Nevertheless, autoreactive cells remained in the tolerized mouse. In total, results from this study indicate a “systemic tolerance” that manifests as antigen-specific Tregs homing to a site of inflammation for protection of islet grafts by Tregs when co-transplanted on scaffolds. Tregs control both islet-antigen specific naïve and effector T-cell function on a systemic level when co-localized within islet grafts on scaffolds.
Materials and Methods
Scaffold fabrication
Scaffolds were fabricated from PLG microspheres as described previously. 15 Microspheres were formed by homogenizing a 6% solution of PLG (Lakeshore Biomaterials) in dichloromethane in 1% polyvinyl alcohol. Microspheres were washed, lyophilized, and combined in a 1:30 ratio with NaCl particles (250–425 μm in diameter). Scaffolds were compression molded and gas foamed as previously described. 15
Animals and diabetes determination
NOD, NSG, NOD.scid, and BDC2.5 TCR transgenic mice were purchased from The Jackson Laboratory or bred in house. Female NOD mice were considered diabetic and ready for receiving islet transplants after two consecutive blood glucose measurements over 250 mg/dL. Female NSG mice 10–14 weeks old were used as islet donors. BDC2.5 trangenic mice were used for CD4+ cell isolations between 8 and 12 weeks of age. Male NOD.scid mice were used for adoptive transfer experiments. Mice were used according to institutional guidelines and protocols approved by the institutional Animal Care and Use committee at Northwestern university.
Cell purifications
Splenic DCs were isolated from NOD males as previously described. 34 In brief, spleens from normoglycemic NOD males between the ages of 6 and 10 weeks were digested with collagenase D (Sigma–Aldrich) followed by the enrichment with CD11c antibody-conjugated microbeads (Miltenyi Biotec). DCs were irradiated with 15 Gy before use. Naive CD4+CD25− cells were purified from BDC2.5 spleen and lymph nodes. Cells were enriched by depletion with biotinylated anti-CD8a (53–6.7), anti-CD25 (7D4), anti-Ter-119 (TER-119), anti-Ly6G and anti-Ly-6C (RB6-8C5), anti-CD49b (DX5), B220 (RA3-6B2), and CD11b (M1/70) (all BD Biosciences) and streptavidin microbeads (Miltenyi Biotec).
Cell culture
Cell culture of Tregs was done as previously described. 11 The sequence of the mimetope peptide used (BDC) was RVRPLWVRME. In brief, 2×104 CD4+CD25− BDC2.5 T cells per well were cultured for 7 days with splenic DCs at a 3:1 T/DC cell ratio in 96-well plates with 100 ng/mL BDC peptide, with 2 ng/mL TGF-β1. For transplantation, DCs were removed by CD11c antibody-conjugated microbeads, and CD25+ cells were further enriched by labeling with PE-conjugated anti-CD25 (PC61; BD Biosciences) and anti-PE microbeads.
Mouse islet isolation, scaffold seeding, and transplantation
Islets were isolated from healthy donor female NSG mice using collagenase digestion as previously described. 15 Isolated islets were hand-counted, and 300 islets were seeded per scaffolds. Recipients were anesthetized with an intraperitoneal injection of ketamine (Ketaset) and xylazine (Anased, Lloyd Labs). The abdomen was shaved and sterile-prepped. A midline incision was made, and the right-sided intra-abdominal fat was identified and spread on the abdominal surface. Islet-seeded scaffolds were placed onto the fat, seeded with Tregs using a pipette, wrapped in the fat, and returned to the abdominal cavity. For some islet transplant recipients, a second islet transplant of 300 NSG islets was performed into the contralateral (left) KC as previously described. 35
Assessment of graft function
After transplantation, nonfasting blood glucose measurements were taken between 12:00 and 17:00 using the following schedule: every day during the first postoperative month and every other day until the conclusion of the study. Graft rejection was confirmed by blood glucose measurements above 250 mg/dL on two consecutive days. For some recipients, a second islet transplant was performed into the contralateral KC at post-transplant day 97. Scaffold grafts were removed at post-transplant day 99, and blood glucose levels were monitored daily for 43 days and monitored for return of hyperglycemia. On day 43 post-KC islet transplant, a nephrectomy was performed, and the animal was monitored daily to confirm return of hyperglycemia.
Graft histology and quantification
Immunohistochemistry was performed on tissue sections to analyze cellular infiltration. At the time of sacrifice, day 7 −Treg and day 7 and >25 +Treg mice were normoglycemic (<250 mg/dL) and day 25 −Treg mice were hyperglycemic (>250 mg/dL). At 7, 25, 33, and 99 days post-transplantation, scaffold grafts and day 140 KC grafts were retrieved, frozen, and embedded, sectioned (14 μm thick), and stored at −20C until staining as previously described. 24 Tissue sections were next fixed in 4% PFA and blocked with 10% normal goat or donkey serum. Primary and secondary antibodies used were diluted in PBS containing 1% serum. For detection of Foxp3+ Tregs with islets, tissue was double stained with anti-swine insulin (1:250; Dako A0564) and anti-Foxp3 (1:200; eBioscience 14-5773). Visualization of insulin and Foxp3 was done using DyLight 488 donkey anti-guinea pig (1:400; Jackson Immunoscience 706-485-148) and DyLight 549 Fragment F(ab′)2 donkey anti-rat (1:100; Jackson 712-506-150). To identify a specific subtype of Tregs, tissue was double stained with anti-Foxp3 (1:200, eBioscience) and biotinylated rat anti-vβ4 TCR (1:50; BD Pharmigen, 553364) after blocking endogenous biotin and streptavidin (Vector Laboratories SP-2002). Tissue was also examined for CD4 and CD8 T cells, macrophages, and DCs. Representative sections from each time point were stained with anti-CD4 (1:500; BD Biosciences 553045), anti-CD8 (1:500, BD Biosciences 553027), anti-F4/80 (1:1000; AbD Serotec MCA497GA), and anti-CD11c (1:100; Novus Biologicals NBP1-06651). For visualization of CD4, CD8, and F4/80, AlexaFluor 546 goat anti-rat (1:500; Invitrogen A-11081) was used and for visualization of CD11c DyLight 594, goat anti-armenian hamster (1:100; Jackson 127-515-160) was used. Hoeschst (1:2000; Invitrogen H3569) staining was performed concurrently to identify cell nuclei. All antibodies are anti-mouse unless otherwise noted. Negative controls were performed by eliminating primary antibodies and including secondary fluorescent antibodies.
For quantification purposes, the scaffolds were fully sectioned for each animal, and slides from at least 3 nonadjacent islet-containing regions were selected at random. Sections around the islet-containing region were stained for Foxp3, CD4, CD8, CD11c, and F4/80 as described earlier. Images were captured, and the number of positively stained cells was manually counted in at least three or more sections per animal. A positively stained cell was identified as both positive for the antibody as well as for Hoechst. Cell numbers were normalized to area of the scaffold within the histological section.
Quantification of CD4+CD25+Foxp3+ Tregs
T cells were isolated from the dLN and spleen from −Treg and +Treg islet transplant recipients on post-transplant day 25 and 33, respectively. Cells were blocked with Fc block (2.4G2; BD Biosciences) and stained with V500-conjugated anti-CD4 (RM4-5; BD Biosciences), PE-conjugated anti-CD25 (PC61; BD Biosciences), and APC-conjugated anti-Foxp3 (FJK-16s; eBioscience). Gating was done on live cells followed by CD4+ population.
CFSE labeling and injection
Freshly isolated BDC2.5 CD4+CD25− cells were isolated as earlier. Cells were labeled with CFSE as per manufacturer's protocol (Invitrogen) and injected into the tail vein of mice receiving islet transplants immediately after transplantation. Three days post-transplantation, dLN (lumbar LN) and ndLN (contralateral axillary LN) were removed, and cells were blocked with Fc block (2.4G2; BD Biosciences) and stained with V500-conjugated anti-CD4 (RM4-5; BD Biosciences), biotinylated anti-vβ4 (KT4; BD Biosciences), and APC-conjugated streptavidin (eBioscience). Gating was done on live cells followed by CD4+ cell population.
Adoptive transfer of splenocytes
Cells were isolated from spleens of −Treg and +Treg transplant recipient mice on day 25 and 33 post-transplant, respectively. The spleens were mashed between two sandblasted glass slides, the red blood cells were lysed, and the cells were filtered. The cells were re-suspended to a concentration of 107 cells/200 μL. About 200 μL of the cell suspension was injected into the tail vein of nondiabetic 12-week-old NOD.scid male mice. The recipient mice were monitored for diabetes onset by checking their blood glucose levels twice per week.
Statistical analysis
Results are presented as mean±standard error. The student's t-test was used to determine statistical significance as appropriate. Differences in the number of days for diabetes reversal were compared using the Kaplan–Meier survival analysis (Prism Software). A value of probability (p)<0.05 was considered statistically significant.
Footnotes
Acknowledgments
This project was supported by the National Institutes of Biomedical Imaging and Bioengineering (NIBIB) through grant number R01EB009910 and the Northwestern University Regenerative Medicine Training Program through grant number 5T90DA022881-03.
Disclosure Statement
No competing financial interests exist.