
Abstract
Achieving adequate vascularization within implanted engineered tissues is a significant obstacle to maintaining viability and functionality. In vitro prevascularization of engineered tissues has been explored as a potential solution to this challenge. The traditional paradigm of in vitro prevascularization is to implant an engineered tissue with a preformed vascular network that is perfused after anastomosis with the host circulation. We investigated the efficacy of this strategy by implanting cell-dense prevascularized tissues created via cell-mediated contraction and composed of collagen and a collagen-fibrin mixture into dorsal window chambers surgically prepared on immunocompromised mice. We found that host-implant anastomosis takes place in 2–6 days and that perfusion of vessels within the implants is subsequently restricted by thrombosis. However, by day 7, a functional vascular network composed of host and implant vessels developed. Prevascularization enhanced intra-implant pO2 significantly as early as 2 days postimplantation, reaching a maximum of 55 mmHg by day 8, which was significantly greater than the maximum within cellularized control tissues (18 mmHg). By day 14, collagen tissues supported ∼0.51×109 implanted and host-derived cells per mL. Our findings elucidate key features of in vitro prevascularization that can be used toward the design of larger and more functionally complex engineered tissues.
Introduction
T
Several strategies have been developed to create a functional vascular network within implanted engineered tissues. Perhaps the most explored strategy entails the induction of angiogenesis from host vessels into an implant. This can be done via delivery of pro-angiogenic growth factors either exogenously or via gene transfer.11–13 However, this process can take several days, and the tight spatiotemporal control required to deliver these factors is complex.5,14 Alternative strategies include the use of custom-fabricated or patterned matrices and decellularized native tissues that are seeded with cells of interest and cultured in vitro before implantation.15–19 These strategies mark progress toward creating engineered tissues with vascular networks that better reflect the density and hierarchy of native vascular networks. However, the in vivo performance of engineered tissues with custom-fabricated or patterned matrices has not been extensively studied, and there remains uncertainty regarding how to best introduce new cells into decellularized native matrices at densities high enough to mimic native tissue. 20
In vitro and in vivo prevascularization are promising methodologies for creating engineered tissues that quickly form a functional vascular network after implantation.21–24 We previously showed that fibrin-only engineered tissues which have been prevascularized in vitro and implanted into dorsal window chambers surgically prepared on immunodeficient mice will anastomose with the host circulation in as short as ∼20 h after implantation. 25 However, thrombosis can occur rapidly (<1 h) after anastomosis, resulting in nonfunctional vasculature. Furthermore, the density of cells was ∼3×106 cells/mL, far less than that found in metabolically active native tissue (∼108 cells/mL).26,27 Such tissues likely do not accurately reflect the metabolic and functional characteristics of native tissue.
The goals of our current study were fourfold: (1) employ prevascularized tissues that had undergone fibroblast-mediated contraction to increase cell density and, thus, metabolic demand to a level more representative of native tissue; (2) determine whether early anastomosis and thrombosis can enhance the oxygen tension within the implants; (3) assess whether, and over what time frame, the prevascularized network of vessels remodel and become functional; and (4) quantify the number of viable cells remaining within the implants by day 14.
Materials and Methods
ECFC-EC isolation
Endothelial colony-forming cell-derived endothelial cell (ECFC-EC) isolation was performed as follows and as previously described.24,25 ECFC-ECs were isolated from human umbilical cord blood that was obtained from the University of California, Irvine Medical Center according to an Institutional Review Board-approved protocol. Mononuclear cells were separated from 15 to 20 mL cord blood using a lymphocyte separation medium (Fisher Scientific, Pittsburgh, PA). The mononuclear cells were then seeded on 1% gelatin (Sigma-Aldrich, St. Louis, MO)-coated tissue culture flasks and fed with a 1:1 mixture of endothelial growth medium (EGM-2; Lonza, Walkersville, MD) and M199 (Invitrogen, Carlsbad, CA), supplemented with 20% fetal bovine serum and 1% endothelial cell growth supplements (Fisher Scientific) for 2–4 weeks. The endothelial outgrowth cells were purified using CD31 (Dako, Carpinteria, CA)-coated magnetic beads (New England Biolabs, Ipswich, MA).
ECFC-EC transduction
ECFC-ECs were transduced using a lentivirus to express enhanced green fluorescent protein (EGFP) as follows. HEK293T cells were seeded on day 1 into a six-well plate at a density of 0.5×106 cells per well. 0.5 mL of Dulbecco's modified Eagle's medium (DMEM) without sodium pyruvate and with 10% fetal bovine serum (Invitrogen) was added to each well. Cells were kept at 37°C in 100% humidified air containing 5% CO2.
On day 2, media were replaced with 1 mL of fresh media per well. In addition, the following was performed for each well: 3 μg of plasmid DNA was added to 250 μL of opti-MEM (Invitrogen). The plasmid DNA consisted of 1.5 μg pRRLSIN.cPPT.PGK-GFP.WPRE, 0.75 μg pMDLg/pPRE, 0.3 μg pRSV-Rev, and 0.45 μg pMD2.G (Addgene, Cambridge, MA). The resulting plasmid DNA solution was allowed to incubate for 25 min at room temperature. Further, 7.5 μL of Lipofectamine 2000 (Invitrogen) was added to 250 μL of opti-MEM and the mixture was allowed to incubate for 5 min at room temperature. These solutions were then combined and added dropwise to each well of HEK293T cells.
On day 3, media in each well were replaced with 2 mL of fresh media. On day 4, the viral supernatant in each well was collected and centrifuged to remove cell debris. Supernatant was then frozen at −80°C before use.
ECFC-ECs at passage 3–5 were transduced by adding 2 mL of viral titer and 12 μL of 10 mg/mL polybrene (Millipore, Billerica, MA) to ∼40% confluent ECFC-ECs in 150 cm2 flasks along with 18 mL of EGM-2 (Lonza). ECFC-ECs were then cultured at 37°C in 100% humidified air containing 5% CO2 for 2 days, after which media and viral supernatant were aspirated and cells were used as described earlier. Transduction efficiency was greater than 93% for all cells used.
Preparation of prevascularized tissues
Prevascularized tissues were composed of ECFC-ECs and normal human lung fibroblasts (NHLFs) suspended in a matrix consisting of either type I rat tail collagen (n=9) or 50:50 collagen-fibrin (by mass) (n=9). Here, we define prevascularized tissues as any tissue in which embedded endothelial cells have formed an interconnected network of tubules. Before tissue preparation, ECFC-ECs and NHLFs were cultured in EGM-2 (Lonza) and fibroblast growth medium (FGM-2; Lonza), respectively. Media were changed every 2–3 days. ECFC-ECs were used from passages 3–5, and NHLFs were used from passages 3–6. During collagen tissue preparation, cells were trypsinized and resuspended in 4 mg/mL type I rat tail collagen (BD Biosciences, San Jose, CA) diluted in double-distilled water, 10×PBS (Invitrogen), and 1N NaOH (Fisher Scientific) as per the manufacturer's instructions. The final cellular concentrations were 1×106 ECFC-ECs/mL and 2×106 NHLFs/mL. These cellular concentrations have been previously shown to result in in vitro vessel formation and anastomosis with the host after implantation.24,25 During collagen-fibrin tissue preparation, cells were trypsinized and resuspended in a 50:50 mixture of 4 mg/mL bovine fibrinogen (Sigma-Aldrich) dissolved in serum and phenol red-free DMEM (Invitrogen) and 4 mg/mL type I rat tail collagen prepared as described earlier. The final cellular concentrations were identical to the pure collagen tissues.
For both matrix formulations, 50 μL of cell suspension was pipetted onto a 12-mm circular glass cover slip with an affixed polydimethylsiloxane (PDMS) retaining ring. The PDMS retaining rings had a diameter of 8 mm and a height of 0.8 mm. For collagen-fibrin gels, the pipetted cell suspension was then mixed with 2 μL of 50 U/mL bovine thrombin (Sigma-Aldrich). Tissues were allowed to polymerize for 20 min at 37°C, then suspended in EGM-2, and maintained at 37°C in 100% humidified air containing 5% CO2 for the next 7 days. Media were changed every 2–3 days. Cell-mediated contraction of the tissues resulted in the formation of tissues that were 1.54±0.23 mm in diameter during in vitro culture which were roughly spheroidal in shape, thus giving an average volume of 1.91±0.29 μL assuming sphericity. Representative images of collagen and collagen-fibrin tissues with EGFP-expressing ECFC-ECs on day 7 of in vitro culture can be seen in Supplementary Figure S1 (Supplementary Data are available online at www-liebertpub-com.web.bisu.edu.cn/tea).
Acellular control tissues (n=9) were created as described earlier; however, after 7 days of in vitro culture, the tissues were kept in an air-tight container for 7 days to induce apoptosis in the embedded cells. Before implantation, the acellular control tissues were placed in a solution of 70% ethanol for 1 h to ensure no living cells remained within the implants. The absence of living cells was confirmed using live-dead staining (Invitrogen) (data not shown).
Collagen NHLF-only control tissues (n=9) were created using the same steps described earlier; however, a cellular density of 3×106 cells/mL was used to maintain a consistent number of cells within each tissue. As with the prevascularized tissues, NHLF-only control tissues were suspended in EGM-2 and maintained at 37°C in 100% humidified air containing 5% CO2 for 7 days after their creation.
Animal model
All in vivo experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee at the University of California, Irvine. Titanium dorsal window chambers were surgically installed onto the dorsal area of ICR-SCID (Taconic Farms, Oxnard, CA) mice (n=36) under anesthesia (50 mg/kg ketamine and 5 mg/kg xylazine administered via i.p. injection). Additional anesthetics were administered during surgery as needed. Preparation was performed as follows.25,28,29 Animals' eyes were first lubricated with a sterile ophthalmic ointment to prevent corneal dehydration. Dorsal hair was removed using electric clippers, followed by application of a commercial depilatory cream (Nair; Church & Dwight Co., Inc., Princeton, NJ) to remove fine hair. The dorsal skin was then pulled up and transilluminated using a white light source to enable visualization of the surgical field and location of blood vessels. One half of the window chamber frame was used as a template to find the appropriate placement. Three 16-gauge needles were pushed through the bolt holes of the window chamber frame and underlying dorsal skin, creating a channel through which bolts could be pushed. The needles were then removed, and bolts were pushed through the three bolt holes of the window chamber frame. Spacers were threaded onto each of the three bolts, and the remaining half of the window chamber frame was placed over the points of the inserted bolts. The window chamber assembly was then secured by screwing three nuts onto the frame bolts, followed by suturing the frame and dorsal skin together via suture holes at the four corners of the window chamber frame. The skin framed by the window chamber was then pinched upward using forceps, and a roughly 1 mm-long incision was made into the skin. This incision was used to remove one full thickness of skin, revealing the dermis of the underlying skin. A small (∼0.5 mm) border of skin around the frame opening was left to prevent the leakage of fluid from the window chamber. Fine-tipped forceps and microscissors were then used to remove fascia from the exposed skin.
Three engineered tissue were then placed into the window chamber, in direct contact with the exposed subdermal tissue. Care was taken to avoid placing the tissues in direct contact with arterioles or venules, as doing so can confound accurate quantification of blood flow and pO2. To enable quantification of pO2, ∼100 μL of 5×10−6 M Oxyphor G4 dissolved in sterile saline was injected into the window chamber. 30 The chamber was closed by placing a 12 mm cover slip into the window of the frame. Following the surgical procedure, buprenorphine was administered (0.1 mg/kg administered via i.p. injection). Mice were imaged once per day for 14 days. After all imaging was completed, mice were sacrificed via pentobarbital overdose.
Imaging system
All in vivo imaging was performed on a Nikon Diaphot TMD microscope (Nikon, Melville, NY). During imaging, mice were anesthetized using 1.5% isoflurane (IsoFlo; Abbott Laboratories, Abbott Park, IL) with balance oxygen. Color images were acquired at 1×, 4×, 10×, 20×, and 40× magnification using a color charge-coupled device camera (Grasshopper 2; Point Gray Research, Inc., Richmond, BC, Canada). Laser speckle imaging (LSI) images were acquired at 4× magnification using a monochrome charge-coupled device camera (Nuance; Caliper Life Sciences, Woburn, MA).
Laser speckle imaging
LSI was used to create maps of the relative flow of blood in and around implanted tissues.25,31–33 These maps were used to compute the functional vascular density (FVD) within and around implanted tissues. During LSI, the dorsal window chamber was transilluminated using a helium-neon laser (Newport, Irvine, CA), and 10 images were acquired at three camera exposure times (T): 10, 100, and 1000 ms. Multiple exposure times were used to compensate for the fact that the linearity and sensitivity of LSI depends on proper choice of exposure time in relation to the speed of blood movement within an image.31,34–37 Use of longer exposure times increases sensitivity to the relatively slow flow in small-caliber vessels.36,38 Exposure times of 1000 ms enable detection of flow speeds as slow as 15 μm/s (unpublished data). Laser intensity was adjusted for each exposure time using a polarizer such that the entire dynamic range of the CCD camera was utilized without overexposure. Speckle contrast images were then computed from the collected raw images by performing the following computation using a 7×7 sliding window algorithm:
where K is speckle contrast, σ is the standard deviation of the pixel values within the sliding window, and <I> is the average intensity of the pixel values within the sliding window. Care was taken to assure that, on average, each imaged speckle was sampled by at least two CCD pixels to satisfy the Nyquist criterion.
39
The 10 speckle contrast images computed for each exposure time were then averaged together to reduce noise, and the resultant image was used to compute a speckle flow index (SFI) map using the following simplified speckle imaging equation at each pixel
40
:
SFI is assumed to be inversely proportional to the correlation time at each pixel and thus proportional to the speed of blood flow.40,41 All image processing was performed using MATLAB (MathWorks, Natick, MA).
Calculation of vascular density and FVD
Vascular density (VD) (length of blood-filled vessels, but not necessarily exhibiting flow, per unit area) and FVD (length of blood-filled vessels exhibiting flow per unit area) were computed within tissues in a manual fashion using ImageJ (US National Institutes of Health, Bethesda, MD).25,36 A single individual blinded to the sample grouping was used for all computations to avoid inter-observer variability. For VD calculations, 4× color images of implanted tissues were used to first compute the total length of all blood-filled vessels within a given tissue. VD values were then computed by dividing this value by the area of the tissue. FVD was similarly computed; however, 4× SFI maps acquired using LSI were used for this computation, because SFI maps display only blood vessels exhibiting flow.
Phosphorescence lifetime measurements
pO2 values were acquired using phosphorescence lifetime measurement and a phosphorescent oxygen-sensitive probe (Oxyphor G4). 30 The probe was introduced into the tissue microenviroment during dorsal window chamber preparation, and subsequent pO2 measurements using the probe were performed on the same microscope setup as described earlier. During pO2 measurements, the probe was excited using a helium-neon laser (wavelength of 632.8 nm) pulse 10 μs in duration. Pulsing was achieved using an acousto-optic modulator (23090-1-LTD; Gooch and Housego, Melbourne, FL). Phosphorescence immediately after the end of the laser pulse was collected using a 10× objective and filtered using a longpass filter (ET780lp; Chroma, Bellows Falls, VT). During measurements, the beam spot was centered within the implanted tissues to acquire a single pO2 value from the interrogated volume. In addition, at least 15 min was allowed to pass between induction of gas anesthesia and phosphorescence lifetime measurement, as isoflurane has been shown in induce a transient alteration in pO2 level that recovers within 10 min of continued administration. 42
The size of the region interrogated using the 10× objective was estimated by measuring the full-width at half-maximum of the emission spot size produced by interrogating a tissue-simulating phantom with 2.5×10−6 M Oxyphor G4 added. The tissue-simulating phantom was composed of a 2-mm thick layer of 2% Intralipid (Baxter Healthcare, Deerfield, IL) diluted in saline. This type of phantom has been previously shown to have a reduced scattering coefficient of 1.2 mm−1 and an absorption coefficient of 0.03 mm−1 at 632.8 nm, similar to that of 1–2 mm of skin.43,44 The emission volume was imaged using a charge-coupled device camera (Grasshopper 2; Point Gray Research, Inc.).
In vivo phosphorescence intensity was measured using a photomultiplier tube (R928; Hamamatsu, Bridgewater, NJ). After each pulse of the excitation source, phosphorescence intensity was measured for 1.2 ms at a sampling rate of 1.25 MHz. The process of excitation and phosphorescence intensity measurement was repeated 50–150 times, and the results were averaged together to achieve an appropriate signal-to-noise ratio.
As expected, the phosphorescence intensity of the Oxyphor G4 followed an expected exponential decay, and the decay time (τ) was related to the partial pressure of oxygen (pO2) by the Stern–Volmer relationship
30
:
where τ0 is the phosphorescent lifetime in the absence of oxygen and kq is the quenching constant. The kq value of 222 mmHg−1s−1 was used as this value corresponds to a temperature of 27°C, the average temperature of the dorsal window chamber skin during imaging. 30 The value of τ0 was determined to be 243 μs under our experimental conditions using a solution of Oxyphor G4 into which nitrogen gas was bubbled for 20 min.
Fluorescence imaging of intravascular fluorescent dextran
To visualize the location of functional mouse- or human-derived blood vessels, a tetramethylrhodamine isothiocyanate–dextran (Sigma-Aldrich) or fluorescein isothiocyanate-dextran (Polysciences, Warrington, PA) solution was injected retro-orbitally. These fluorescent dextrans had an average molecular weight of 155 and 150 kDa, respectively, and were dissolved in sterile physiological saline to a final concentration of 10 mg/mL before injection.
Histology
After host sacrifice, implanted tissues were removed with adjacent host skin and fixed in 10% formalin. Samples were embedded in paraffin and sagittal sections that were 4 μm in thickness were obtained. Adjacent sections were alternatively labeled using either hematoxylin and eosin or antibodies for human (rabbit anti-human CD31; Abcam, Cambridge, MA) and mouse (Alexa Fluor 488-conjugated rat anti-mouse CD31; Biolegend, San Diego, CA) endothelial cells. Immunofluorescent staining was performed as follows.23–25 After deparaffinization and rehydration, tissue sections were blocked with a 2% bovine serum albumin (Sigma-Aldrich) and 0.1% Tween 20 (Sigma-Aldrich) solution for 1 h. Sections were then incubated for 1 h with the primary antibody, diluted 1:200 (anti-human CD31) or 1:100 (anti-mouse CD31) in a 0.1% Tween 20 solution. Sections were then washed four times for 1 min, followed by incubation for 1 h with an Alexa Fluor 555-labeled goat anti-rabbit antibody (Invitrogen) diluted 1:500 in a 0.1% Tween 20 solution. Sections were then washed four times for 1 min, followed by incubation in a 200 nM DAPI (Invitrogen) solution for 20 min.
To estimate the number of live cells within collagen tissues, hematoxylin and eosin-stained sections from the center of six tissues were analyzed. Within each section, the area fraction of cell nuclei to total implant area was computed. In addition, the average length of the short and long axis of each implant was recorded. Finally, the lengths of the short and long axis of 100 cell nuclei were measured and used to compute an average nucleus size. The average tissue and nucleus volume was then computed using the averaged short and long axis dimensions by modeling these objects as oblate spheroids. The approximate number of live cells within collagen tissues was then computed by multiplying the average volume of the tissues by the average area fraction of nuclei, followed by dividing the result by the average nucleus volume. Average cell density was then computed by dividing the result by the average implant volume. It was assumed that hematoxylin-stained nuclei represented viable cells.
Statistical analysis
Average values are expressed as mean±standard deviation. Comparisons and changes in FVD, VD, and pO2 values were analyzed statistically using the Mann–Whitney test. p-Values<0.05 were considered significant.
Results
Measurement of the phosphorescence emission spot size in a tissue phantom
The imaged emission spot intensity distribution had an approximately Gaussian profile. A nonlinear least squares fit through a cross-section drawn through the center of the spot to a Gaussian resulted in an R value of 0.98. The full-width half-maximum of the Gaussian fit was 1.28 mm. The data and the Gaussian fit can be seen in Supplementary Figure S2. This was considered an appropriate size for measuring pO2 within the implanted tissues, which had a diameter of ∼1–2 mm on implantation.
Prevascularized tissues anastomose with the host circulation
Microvasculature within both collagen and collagen-fibrin prevascularized tissues anastomosed with the host circulation after implantation. Anastomosis was identified by the presence of blood-filled vessels within prevascularized tissues. The time required for prevascularized tissues to anastomose with the host was 3.8±1.2 days for collagen tissues and 3.9±0.7 days for collagen-fibrin tissues. Anastomosis was frequently accompanied by perivascular hemorrhaging of blood as has been previously noted. 25 Hemorrhaged blood was absorbed by the host in 2–3 days. To ensure that these blood-filled vessels within the implants were of human origin and not due to angiogenesis of host vessels, fluorescence microcopy was used to determine whether such vessels could be co-localized with EGFP fluorescence from implanted EGFP-expressing ECFC-ECs. The results can be seen in Figure 1. Figure 1A shows a color image of a collagen-based tissue after anastomosis on day 5. The corresponding fluorescence image (Fig. 1B) shows that the blood-filled vessels within the tissue are of human origin. Similarly, Figure 1D and E shows a color image and a corresponding fluorescence image of a collagen-fibrin tissue on day 5 showing that blood-filled vessels within the tissue are of human origin. The finding that the initial influx of blood into the prevascularized tissues was due to host-implant anastomosis was consistent through all samples.

Clotted blood within implanted collagen and collagen-fibrin tissues after host-implant anastomosis is localized within human ECFC-EC containing vessels.
Thrombosis occurs within human-derived vessels
After anastomosis with the host circulation, human-derived vessels were perfused with host blood. However, thrombosis occurred within all collagen and collagen-fibrin tissues, resulting in cessation of blood flow in affected vessels within 24 h.
Clotting was identified by a lack of blood flow as determined using LSI. Representative results for a prevascularized collagen and collagen-fibrin tissue are shown in Figure 1. The SFI maps correspond to the blood-filled vessels previously described (Fig. 1A, D), and demonstrate that blood flow is only present in host vessels and that the human-derived blood vessels are clotted, as indicated by the lack of SFI signal (Fig. 1C, F). All clotted vessels were composed, at least in part, of implanted ECFC-ECs.
Prevascularized tissues develop a functional vascular network after thrombosis
The presence of blood-filled vessels within implanted tissues resulted in an increase in VD over time (Fig. 2A). The increasing number of clotted blood-filled vessels present within collagen and collagen-fibrin prevascularized tissues is reflected by the increase in VD starting on day 3. Starting at day 4, VD within collagen and collagen-fibrin tissues was significantly higher than their respective day 1 values. Groups of clotted vessels were often found to be discontinuous, suggesting that multiple anastomoses with the host circulation occurred at different time points. Small increases in VD were seen in NHLF-only and acellular control tissues. This was due to the ingrowth of host vessels. The VD within NHLF-only and acellular control tissues never significantly increased over their respective day 1 values. Representative color images of all tissue types on day 1 and 14 can be seen in Supplementary Figure S3.
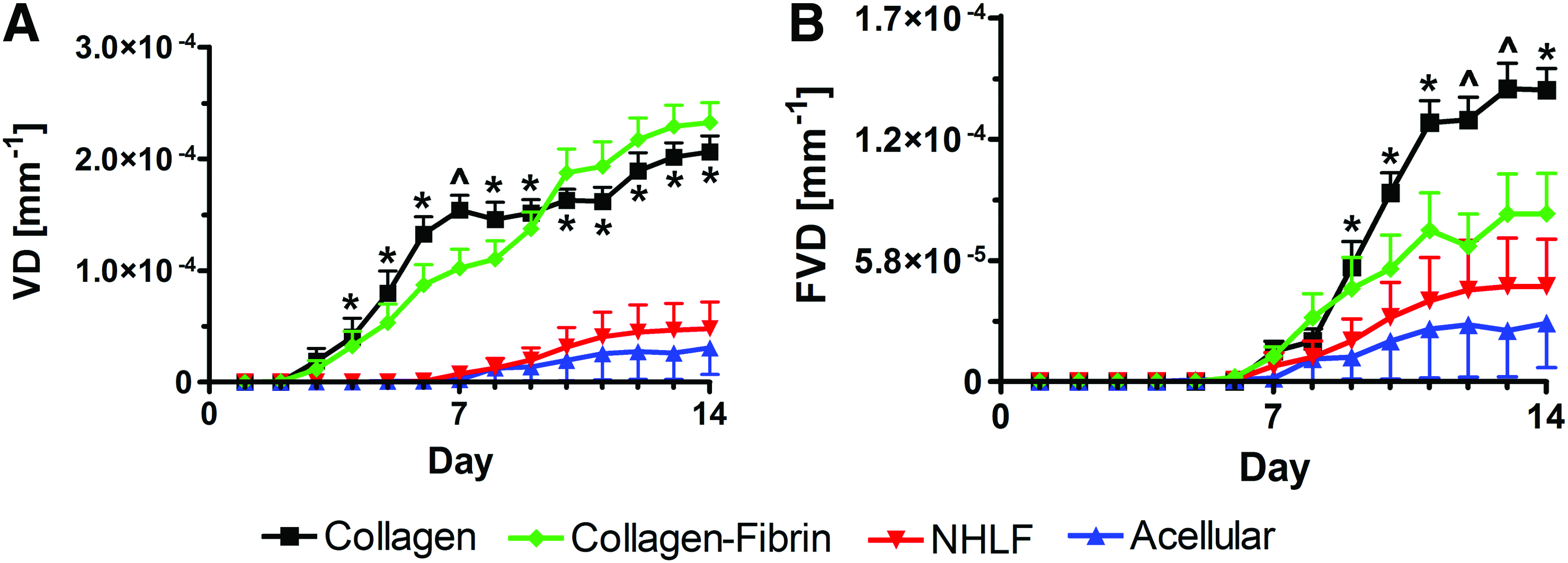
VD
The greatest increase in FVD occurred in prevascularized collagen tissues (Fig. 2B), and it was significant from day 7 onward. The FVD in prevascularized collagen tissues was significantly greater than that in NHLF-only and acellular control tissues from day 9 onward, and greater than the FVD within collagen-fibrin tissues on days 12 and 13. Since prevascularized collagen tissues exhibited greater FVD as compared with prevascularized collagen-fibrin implants, thus creating a microenvironment more amenable to adequate oxygen delivery, subsequent control experiments were performed using collagen tissues. The increase in FVD in NHLF-only and acellular control tissues was concomitant with the increase in VD, both of which resulted from the ingrowth of host vessels. However, this increase was not significant compared with day 1 values.
Functional vasculature in prevascularized tissues is composed of host- and human-derived vessels
To determine the origin (host vs. human) of the functional vasculature that developed within prevascularized tissues, fluorescence microscopy was used to determine whether fluorescent dextran injected into the host vasculature co-localized with EGFP fluorescence from ECFC-ECs. Fluorescence from the injected fluorescent dextran (red) often was not co-localized with fluorescence from ECFC-ECs (green) at the border of the implanted tissues, indicating the ingrowth of host vessels (Fig. 3A). Thus, it was frequently observed that the initial functional vasculature observed within prevascularized tissues was host-derived vasculature resulting from angiogenesis. However, co-localization of fluorescent dextran and ECFC-EC fluorescence was frequently visible past the border of the implant, indicating the presence of functional human-derived vessels (Fig. 3B). The earlier findings were present in both collagen and collagen-fibrin tissues.

Prevascularized tissues develop functional vasculature composed of both host (mouse) and implanted (human) vessels.
The finding that functional human-derived vessels were present within the core of the implant was confirmed using histology (Fig. 3C–E). This representative histological section was taken from the center of a collagen tissue on day 14 that no longer contained visible clotted vessels (see Supplementary Video S1). All vessels containing host platelets (green) stained positive for anti-human CD31 (red), indicating that these vessels are derived from ECFC-ECs.
Functional vasculature develops from the outside to the inside of implanted tissues
Functional vasculature was always initially found to be present around the periphery of implanted tissues, gradually developing toward the center of the tissues (Fig. 4). Although six of the nine collagen tissues contained functional vasculature that reached the center of the implant, this degree of vascularization occurred on days 10–14 and was preceded by development of functional vasculature around the border of the implant.

Development of functional vasculature within prevascularized tissues progresses from the periphery.
pO2 is significantly higher in prevascularized tissues as compared with controls
For days 2–14, the pO2 within prevascularized collagen tissues (Fig. 5A) was significantly greater than the day 1 value. It was also significantly greater than the value within collagen-fibrin prevascularized tissues on days 2 and 7 and significantly greater than the value within NHLF-only and acellular control tissues at varying time points between days 1–14. Starting at day 4, the pO2 within prevascularized collagen-fibrin tissues (Fig. 5B) was significantly higher than the day 1 value. pO2 within NHLF-only tissues was significantly greater than the day 1 value from day 2 onward (Fig. 5C). pO2 within acellular tissues never significantly increased above the day 1 value (Fig. 5D).

Average pO2 within collagen
Quantification of live cells within collagen tissues using histology
The estimated number of live cells within six collagen tissues at day 14 was 0.21±0.16×106. The average lengths of the short and long axes of the collagen tissues were 0.53±0.14 and 1.21±0.19 mm, respectively. This resulted in an average day 14 volume of 0.41±0.17 μL and an average live cell density of 0.51±0.43×109 cells/mL. A representative tissue section can be seen in Supplementary Figure S4. Although thin histological sections are prone to error in computing nuclear volume because nuclei are sectioned in varying planes, the use of 100 nuclei to compute average volume and the presence of more than 400 nuclei per section when computing area fraction likely mitigated any bias. In addition, although the cellular density varied within collagen implants depending on the portion of the tissue sampled, quantification was performed on sections through the center of implants because this region better reflects the vascular network's capacity to deliver oxygen and maintain cellular viability as compared with the periphery of the implants, which may benefit to a greater degree from oxygen diffusion.
Discussion
In this study, prevascularized tissues composed of either collagen only or a 50:50 mixture of collagen and fibrin were implanted into surgically prepared dorsal window chambers on SCID mice. Optical analysis over the 14 days after implantation was used to quantify VD, FVD, and pO2 in the tissues, as well as to identify the role of the host and human-derived vessels in forming a vascular network. In addition, histological analysis of excised collagen tissues was used to estimate the number of live cells that could be supported. Our major finding is that, despite rapid clotting after initial anastomosis between the host and implanted vessels occurring at 1–6 days postimplantation (Fig. 1), the human vascular network within prevascularized tissues progressively remodels and becomes functional by day 7 (Figs. 2 and 3). The rapid anastomosis and remodeling of the human vascular network within collagen tissues results in enhanced VD as early as day 4 and enhanced oxygen levels as early as day 2 after implantation (Figs. 2 and 5). As a result, the viability of ∼0.21×106 cells at a cell density of ∼0.51×109 cells/mL can be supported by day 14. The viable cell density is similar to cell dense in vivo tissue (∼108 cells/mL), and the findings aid in determining the upper limit of implant cellular density that this technique can support.
Collagen was chosen as a matrix component in this study, as opposed to fibrin-only prevascularized tissues used in previous studies, because significant resorption of fibrin-only matrices can limit the observation period of implanted tissues.23–25 Collagen serves as a nonprovisional and more permanent matrix that can be used to create longer-lasting implantable tissues. A 50:50 ratio of collagen and fibrin was also investigated, because this ratio has been shown to be effective for developing blood vessel networks in vitro. 45 In addition, fibrin can elicit an intrinsic angiogenic response. 46 These positive attributes contribute to this matrix combination receiving increased usage as scaffolding for engineered tissue constructs.47–49
Contracted tissues were chosen for use in this study, because they potentially offer several advantages compared with the disk-shaped implants previously investigated. 25 First, the tissues created in this study initially were relatively compliant before contraction, which began at 2–4 days after tissue creation. It has been shown that endothelial cell-containing matrices with lower concentrations (2–2.5 mg/mL) of pure fibrin or fibrin and collagen result in a greater vessel density and length compared with higher-density (3–10 mg/mL) matrices.45,50 However, low-density matrices are fragile and thus difficult to handle, limiting their clinical utility. By allowing the tissues used in this study to contract after vessel formation has taken place, the density of the matrices increases by a factor of ∼15, resulting in tissues that can be easily handled in addition to resulting in a cell density which is more consistent with native tissue. In addition, we hypothesized that the increased cellular density resulting from matrix contraction, in combination with the denser and thus less permeable matrix, may elicit greater metabolic demand than the noncontracted disk-shaped implants after implantation. As a result, this increased metabolic demand may stimulate the release of pro-angiogenic growth factors, potentially reducing the time required for anastomosis with the host to occur. In addition, use of contracted tissues spatially limits the density of vessel bifurcations, which may be advantageous toward maintaining continued blood flow, as an abnormally high bifurcation density within prevascularized tissues may contribute to abnormally low intravascular shear rates, which promotes thrombosis. 25 Lastly, contracted tissues undergo directed self-assembly and may serve as modular building components for larger and more complex engineered tissues. 51
Acellular and NHLF-only controls were used to determine the degree of implant vascularization that would result from only inherent pro-angiogenic properties of the extracellular matrix or cytokines released from embedded cells, respectively. It should be noted that although acellular controls likely contained cytokines remaining from the apoptosis of embedded cells, creation of tissues with a matrix density comparable to the experimental groups (∼50 mg/mL) required the use of embedded NHLFs to contract the matrix, as collagen at this higher concentration is not currently commercially available. In addition, NHLF-only controls were used in place of NHLF and ECFC-EC-containing nonprevascularized tissues, because creation of contracted tissues required 7 days of incubation, during which time prevascularization of ECFC-EC- and NHLF-containing tissues cannot be avoided. However, as with the acellular control, the protein expression of a monoculture NHLF-only tissue differs from a co-culture tissue, which could potentially affect the degree of vascularization after implantation.
We found that both collagen-fibrin and pure collagen prevascularized tissues anastomosed with the host circulation and required similar amounts of time to do so (3.9±0.7 and 3.8±1.2 days, respectively). This timeframe is comparable to prevascularized fibrin-only disks implanted subcutaneously and within dorsal window chambers previously studied (3.1±1.4 and 3.5±2.1 days, respectively).24,25 However, the shortest time required for host-implant anastomosis in this study was 2 days, which is 1 day more than in previous studies.24,25 This is potentially due to the approximately fivefold increase in implant matrix density in this study, which may have increased the time required for endothelial cell migration. We have previously shown that the density of co-transplanted fibroblasts, as well as the choice of endothelial cell source will impact the time required for anastomosis. 24 Other factors that may impact the rate of anastomosis include the addition of pro-angiogenic and pro-migratory cytokines, such as VEGF, to the wound site on implantation.
Also consistent with previous findings was the fact that blood clotting occurred in prevascularized implants after host-implant anastomosis. 25 Clotting was isolated to vessels containing ECFC-ECs. Clotting may be due to shear rates within blood vessels dropping below a critical threshold (∼100 s−1), resulting in longer residence times of clotting factors and initiation of blood coagulation.25,52,53 Such low shear rates may result from the fact that prevascularized tissues created using NHLFs and ECFC-ECs have been previously shown to generate vascular networks that violate Murray's law and thus fail to conserve shear rate at bifurcations. 25 An abnormally long residence time of clotting factors may also result from the implanted vascular network's inability to conduct blood flow. These issues may be mitigated by perfusion of the vessel network within the prevascularized tissues in vitro before implantation. It has been demonstrated that perfusion encourages vessel maturation and that perfusion of small capillary-like vascular networks can be achieved in vitro.54,55
Administration of anti-coagulants is a common strategy for limiting thrombosis. We have previously investigated the utility of the thrombin inhibitor dabigatran etexilate toward eliminating thrombosis in implanted prevascularized tissues. However, the permeability of implanted vessels is high, as evidenced by the extravasation of blood after anastomosis with the host circulation. This results in the frequent exposure of extravasated blood to tissue factor, which has been shown to be expressed by surrounding fibroblasts and vessel walls.56,57 Tissue factor is a potent initiator of blood coagulation, and it has therefore been suggested that increased vessel permeability can promote thrombosis. 58 Providing an anticoagulant dosage that is high enough to overcome this effect not only limits the ability to effectively image the implanted tissue (Supplementary Fig. S5), but also poses life-threatening bleeding risks. 59 As such, reduction of thrombosis may be better accomplished by decreasing implanted vessel permeability via further in vitro vessel maturation, rather than administration of anticoagulants.
Although the observation of blood clotting was isolated to the dorsal window chamber microenvironment, similar studies performed within other physiological environments show similar results. Lesman et al. 60 observed that prevascularized tissues composed of fibrin and containing a mixture of human foreskin fibroblasts and human umbilical vein endothelial cells anastomosed with the host circulation and filled with host blood after implantation into a full-thickness defect in the abdominal wall of nude mice. Human-derived vessels within these engineered tissues were subsequently shown to be nonfunctional, a result that may be explained by clotting within these vessels.
Clotting within prevascularized implants was followed by the development of functional vasculature containing both mouse and human vessels. Significant day-to-day variations in the vascular networks within the implants made analysis of single vessels difficult. As such, determining whether these functional vessels were derived from the breakdown of thrombi within clotted vessels versus the creation of new blood vessels from ECFC-EC migration from clotted vessels was not possible. However, the likelihood of ECFC-ECs within the implant migrating toward functional vessels to initiate the extension of functional vasculature into the bulk of the implant is supported by a study performed by Cheng et al., 61 in which nonprevascularized collagen tissues containing human endothelial cells and implanted into dorsal skinfold window chambers underwent host-vessel ingrowth at the border of the implant followed by development of a predominantly human-derived functional vascular network within the center of the implant.
Thrombosis will likely compromise the viability of prevascularized tissues by limiting nutrient delivery. However, thrombosis may, nonetheless, have beneficial qualities that increase vascularization. For example, thrombin, a key enzyme in the clotting cascade, has been shown to induce angiogenesis via several different mechanisms. Thrombin increases expression of vascular endothelial growth factor receptor by endothelial cells, in addition to increasing their proliferation and migration.62,63 In addition, it may induce release of vascular endothelial growth factor, sphingosine 1-phosphate, and angiopoietin-1 from platelets, thus facilitating angiogenesis and vessel maturation.64–66 Importantly, these factors are endogenous to the host, potentially mitigating dosing, delivery, and regulatory complications related to their exogenous delivery.
Angiogenesis mediated by thrombosis may also explain why prevascularized collagen-fibrin tissues developed less functional vasculature than collagen tissues. The density of clotted vessels within collagen-fibrin tissues, represented by the quantity (VD − FVD), was lower than that of prevascularized collagen tissues from days 1 through 9. Thus, the concentration of pro-angiogenic factors resulting from clotting may have been lower in collagen-fibrin tissues as compared with collagen tissues. It is also possible that fibrin resorption within collagen-fibrin tissues results in a more dynamically changing extracellular environment as compared with collagen tissue, which may not be as conducive to the development of mature functional vasculature. The fact that prevascularized collagen tissues demonstrated the highest density of clotted vessels at early time points, yet later exhibited the highest pO2 and density of functional vessels, suggests that a limited degree of thrombosis within implanted prevascularized tissues may subsequently support increased vascularization and oxygenation.
Our observation that prevascularized collagen tissues had a significantly higher pO2 compared with both controls and collagen-fibrin tissues is likely due to enhanced development of oxygen-delivering functional vasculature. It should, however, be noted that the pO2 within collagen and collagen-fibrin tissues may have been affected by differences in oxygen consumption resulting from varying degrees of cellular replication or metabolism. Consistent with this concept is the higher average pO2 within acellular tissues (18±6.3 mmHg) compared with NHLF-only tissues (11±4.7), likely due to the presence of metabolically active cells.
The rationale for the multiple peaks in pO2 within the collagen tissues may be elucidated by the changing (F)VD within these tissues. Specifically, the initial sharp increase in pO2 on day 8 occurs soon after the VD within the collagen tissues reaches an initial peak (day 7). However, as evidenced by the minimal concurrent increase in FVD in the collagen tissues at this time, the majority of the vessels causing this increase are nonfunctional. As such, the subsequent decrease in pO2 in the collagen tissues may be caused by the oxygen supply from clotted blood that these nonfunctional vessels are depleting. The pO2 in the collagen tissues then increases from days 11 through 14. Concurrent with this increase in pO2 is the FVD within the collagen tissues reaching a maximal plateau. As such, the pO2 may reach its maximum value during this time due to the increased presence of functional oxygen-delivering vasculature.
The maximum pO2 value within collagen tissues (55 mmHg) was comparable to pO2 values near normal vessels adjacent to tumors implanted into rat dorsal window chambers (51–72 mmHg).67,68 The pO2 within NHLF-only tissues was also comparable to that measured within tissues implanted into the dorsal window chamber in previous studies. For example, LS174T human colon adenocinoma tumors implanted into the dorsal window chambers of SCID mice had an average pO2 of ∼15 and 7 mmHg at 17 days and 27 days postimplantation, respectively. 9 In addition, the average pO2 within a tumor composed of 4T1 mammary carcinoma tumor cells implanted into the dorsal window chamber of a nude mouse was ∼8 mmHg. 69
It is noteworthy that, unlike the control tissues, the prevascularized collagen tissues reached pO2 values above what is generally considered hypoxic. Although the pO2 required to induce hypoxia varies depending on cell type, hypoxia-inducible factor DNA-binding activity and protein levels increase exponentially for pO2 values less than ∼45 mmHg in HeLa cells, with a peak response at 4 mmHg. 70 A range of pO2 values from 8 to 20 mmHg has been frequently described as hypoxic.71–74 Thus, while prevascularizing collagen tissues results in early anastomosis and thrombosis, it may, nonetheless, ameliorate hypoxic conditions within the implant, and thus improve the overall likelihood of cell viability while functional vasculature develops.
Finally, it was found that prevascularized collagen tissues supported the viability of an estimated 0.21±0.16×106 cells by day 14, slightly greater than the number of cells the tissues were seeded with at the start of in vitro culture (0.15×106 cells). This average cell density (0.51±0.43×109 cells/mL) was comparable to or greater than many native tissue types, such as adult rat heart (0.05–0.1×109 cells/mL), human skeletal muscle (0.64×109 cells/mL), human articular cartilage (0.02×109 cells/mL), and human bone (0.12×109 cells/mL).26,27,75,76 However, the metabolic demands of native tissues such as cardiac or skeletal muscle are likely much greater than the prevascularized tissues used in this study. It should be noted that the cell density was significantly affected by the decrease in average matrix volume (1.91±0.29 μL on day 1 vs. 0.41±0.17 μL on day 14). It is unknown whether this was due to matrix degradation or further matrix contraction.
Although the number of viable cells within the prevascularized tissues includes mouse cells which have migrated into the implants (Fig. 3), the presence of EGFP-expressing ECFC-ECs within the implants at day 14 indicates that implanted cells can remain viable while functional vasculature develops. While the prevascularized implants used in this study most likely cannot be increased significantly in scale due to the increase in volume that should be progressively vascularized, our findings provide key details regarding the vascular dynamics, oxygenation, and number of cells that can be supported using in vitro prevascularization, thus enabling more effective utilization of this strategy.
Conclusion
Difficulty in achieving adequate nutrient delivery, particularly oxygen, within implanted engineered tissue currently limits their viability, functionality, and thus broad clinical application. Prevascularization of engineered tissues may aid in overcoming this limitation by expediting the development of a functional and nutrient-delivering vascular network within implanted tissues. In this study, we found that cell-dense prevascularized tissues were capable of consistent anastomosis formation with the host vasculature within as few as 2 days after implantation, resulting in perfusion and subsequent clotting in the implant vessels. This process was followed by the development of a functional vascular network within the implanted tissues from day 7 onward. This process resulted in a significantly higher pO2 within prevascularized tissues as compared with controls as early as day 2. Although our observations deviate from the paradigm of prevascularized tissues experiencing rapid development of a functional microcirculation, they suggest that prevascularization significantly increases implant oxygenation while functional vasculature develops, and that physiologically relevant implant cell densities of ∼0.51×109 cells/mL can be supported.
Sources of Funding
This work was supported in part by the Arnold and Mabel Beckman Foundation (B.C.), the National Science Foundation through a Graduate Research Fellowship to Sean White, a Dean's Triumvirate Grant from the UC-Irvine Institute for Clinical and Translational Science (UL1 TR000153, B.C., S.C.G., and C.C.W.H.), grants from the National Heart Lung and Blood Institute (R21 HL104203, S.C.G. and C.C.W.H.), the National Institute for Child Health and Development (R01 HD065536, B.C.), and the National Institutes of Health Laser Microbeam and Medical Program (LAMMP, a P41 Technology Research Resource, B.C.). C.C.W.H is supported by the Chao Family Comprehensive Cancer Center through grant P30A062203 and from NCI. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.