
Abstract
Akt kinase is a central signal transduction node that integrates extracellular cues that regulate cell migratory, proliferative, and morphological functions during angiogenesis. However, how Akt activity is modulated and contributes to subsequent vessel maturation is unclear. In this study we investigated the role of Akt1 in vessel maturation using human dermal microvascular endothelial cells (HDMVECs) expressing constitutively active and hemiphosphorylated Akt1 epi-alleles with graded kinase activity. HDMVECs expressing Akt1 epi-alleles were analyzed in vivo in a tissue engineering setting using a model of angiogenesis comprising cell-seeded poly-L-lactic acid scaffolds implanted subcutaneously into NOD/SCID murine hosts. The resultant intraimplant microvasculature was quantified for vascular parameters, including vessel diameter, perfusion, vascular density, and pericyte coverage. We found that constitutive Akt1 kinase activity in implanted HDMVECs correlated with loss of neovasculature function. Further, we found that the presence of coimplanted vascular smooth muscle cells (vSMCs) in the implants failed to promote blood vessel growth and maturation in a graded, Akt1 kinase activity-dependent manner. These results indicate that constitutive Akt1 activity disrupts the normal blood vessel growth and maturation. Therefore, we suggest that a downregulation of Akt1 activity is necessary for vSMC-induced maturation of newly formed blood vessels to occur.
Introduction
T
We endeavored to investigate the consequence of constitutive Akt1 activation in primary human ECs on angiogenesis and vessel maturation in a tissue engineering setting. We further wished to explore the potential contributions of the two phosphorylation sites of Akt1 to the angiogenic process. We used an in vivo tissue engineering model of angiogenesis to investigate determinants of EC–vSMC interactions in angiogenesis in vivo. 16 Human ECs and vSMCs coseeded into poly-L-lactic acid (PLLA) scaffolds form a functional human vasculature within 2 weeks as subcutaneous implants, allowing the functional evaluation of altered endothelial signal transduction in an in vivo tissue engineering setting (Fig. 1). We employed this approach to evaluate the role of EC Akt1 activity in vSMC-induced vessel maturation.

Overview of experimental approach. Phoenix retroviral packaging cells were transfected with myrAkt1 plasmids containing an IRES/GFP marker. Retrovirus produced by the Phoenix cells was used to transduce human dermal microvascular endothelial cells (HDMVECs) to stably express myrAkt1. Transduced HDMVECs were sorted by FACS based on GFP fluorescence, to isolate successfully infected cells. Cells were cultured and seeded in poly-L-lactic acid (PLLA) scaffolds, either HDMVECs alone or in coculture with wild-type pulmonary artery smooth muscle cells (PaSMCs). Scaffolds were implanted subcutaneously on the flanks of NOD/SCID mice, and retrieved after 1 or 2 weeks. The resultant engineered vasculature was examined by histology. Color images available online at www.liebertpub.com/tea
Materials and Methods
Vector production
IRES-GFP and MyrAkt1 retroviral plasmids have been described previously.17,18 Point mutations were introduced into the phosphorylation sites of the MyrAkt1 plasmid to make these sites nonfunctional, exchanging threonine (T) for alanine (A) or serine (S) for alanine, producing the myrAkt1(T308A) and myrAkt1(S473A) mutants, respectively. Point mutations were introduced in the plasmid using the QuikChange® Site-Directed Mutagenesis Kit (Stratagene), following the manufacturer's instructions. Primers used were as follows: T308Akt, 5′ GGTGCCACCA TGAAGGCCTT TTGCGGC ACA CCTG 3′; S473Akt, 5′ CCCACTT CCCCCAGTTC GCCTACTCGG CCAGCGGC 3′.
Cells
Human dermal microvascular endothelial cells (HDMVECs, single donor lot; Lonza) were grown in EGM-2 MV medium (Lonza). Human pulmonary artery smooth muscle cells (PaSMCs, single donor lot; Lonza) were grown in SmGm medium (Lonza). Phoenix A retroviral packaging cells (Dr. Gary Nolan, Stanford University) were grown in Dulbecco's modified Eagle's medium, 4500 mg/mL glucose (Sigma-Aldrich) supplemented with 10% fetal bovine serum (Euro Clone/PAA), 5% penicillin–streptomycin (Sigma-Aldrich), and 5% L-glutamine (Sigma-Aldrich).
Retroviral transduction
Phoenix A retroviral packaging cells were transfected according to Swift et al. (1999). 19 Briefly, subconfluent Phoenix A cells were transfected by CaCl2 precipitation in the presence of chloroquine (Sigma-Aldrich). Virus was harvested in EGM-2 MV medium 48 h post-transfection and added to subconfluent HDMVECs with 5 μg/mL protamine sulfate (Sigma-Aldrich) for 16 h. Transduced HDMVECs were purified by flow cytometric sorting on an FACSAria Cell Sorter (BD Biosciences) based on GFP-marker expression.
Cytohistochemistry
Cells were cultured on IBIDI u-Slide eight-well plates (IBIDI). When subconfluent, cells were washed with ice-cold phosphate-buffered saline (PBS) and fixed in 4% PFA/PBS for 25 min at room temperature (RT). Subsequently, cells were washed 3× with PBS and permeabilized with PBS/0.5% Triton X-100 (Sigma-Aldrich) at RT for 5 min. Blocking was done in PBS/1% bovine serum albumin (BSA) at RT for 30 min. Samples were incubated with Phalloidin-Alexa488 (No. A22287; Life Technologies) at 1:30 ratio in PBS/1% BSA for 20 min at RT. Before imaging, cells were stained with Hoechst (No. B2261; Sigma-Aldrich) nuclear stain at 1:2500 in PBS. Samples were imaged using a Zeiss LSM 510 META confocal microscope (Carl Zeiss AG).
Phospho-flow analysis
Cells were split on ice with 10× Trypsin-EDTA (Sigma-Aldrich), and fixed by addition of PFA (16% EM-grade; Electron Microscopy Sciences) to a final concentration of 2%. Cells were incubated for 10 min at 37°C and centrifuged for 5 min at 700g. Thereafter, cells were incubated for 1 min on ice before addition of 90% methanol (MeOH; Sigma-Aldrich) to permeabilize the cells. Samples were stored at −20°C in 90% MeOH solution until staining.
Cells were washed in incubation buffer (IB; PBS/0.5% BSA) and blocked in IB for 10 min at RT. Cells were incubated with primary antibodies pAkt S473 (No. 4060; Cell Signaling Technology) at 1:100 or pAkt T308 (No. 9275; Cell Signaling Technology) at 1:50 for 1 h at RT on an agitator. Cells were washed once in IB and incubated with secondary antibody Alexa 647 goat anti-rabbit IgG (No. A21244; Invitrogen) at 1:200 for 30 min at RT on an agitator. Background control samples were incubated with secondary antibody only. Cells were washed once, resuspended in PBS, and kept on ice until analysis. Cells were analyzed on a BD Accuri™C6 flow cytometer (BD Biosciences) and processed with the FlowJo7.6 software.
Western blot
Cells were lysed with a cell scraper on ice using NP-40 lysis buffer with Complete Mini Protease Inhibitor Cocktail (Roche) and phosSTOP Phosphatase Inhibitor Cocktail (Roche). Total protein concentration in lysates was measured using a Pierce BCA protein assay kit (Thermo Scientific) following the manufacturer's instructions. The 10% NuPAGE® Bis-Tris precast gels (Invitrogen) were loaded with 30 μg of protein in each well in Laemmli sample buffer. A 1:1 mix of MagicMark™ XP Western Protein Standard (Invitrogen) and SeeBlue® Plus 2 Pre-Stained Standard (Invitrogen) was used as protein standard. Gels were run at 50 V for 20 min and then at 100 V for 1 h 30 min. Blotting was done on ice for 1 h 30 min at 100 V onto PVDF membranes (Hybond-P; GE Healthcare). Membranes were washed in TBS/0.1% Tween-20 (TBS-T), and blocked in TBS-T/5% BSA for 1 h at RT. Primary antibodies were added at 1:1000 (phospho-Akt [Thr308] antibody [No. 9275; Cell Signaling Technology], phospho-Akt [Ser473] antibody [No. 9271; Cell Signaling Technology], and phospho-[Ser/Thr] Akt substrate antibody [No. 9611; Cell Signaling Technology]) or 1:5000 rabbit anti-actin (No. A5060; Sigma-Aldrich) in TBS-T/5% BSA, and membranes were incubated overnight at 4°C. Next day, membranes were washed three times in TBS-T, and incubated in secondary antibody goat anti-mouse IgG (H+L) HRP conjugate (No. 172-1011; BioRad) or goat anti-rabbit IgG-HRP (No. 65-6120; Invitrogen) at 1:5000 in TBS-T/5% milk for 45 min at RT. Membranes were then washed three times in TBS-T. Detection was performed using enhanced chemiluminescence (ECL) kit (Biorad) using a Molecular Imager ChemiDoc™ XRS (BioRad).
Western blots were quantified using the ImageLab 3.0 software (BioRad). Mean pixel intensity was measured for each band, background was subtracted, and samples were normalized to actin loading control. All quantitative WB data are represented as fold change relative to control (IRES-GFP-expressing HDMVECs), where control is set to 1.
Apoptosis assay
Apoptosis was assessed on live cells using an Annexin V apoptosis kit for flow cytometry (No. V35113; Invitrogen). Experiments were performed according to manufacturer's instructions. Briefly, HDMVECs were split on ice and washed with ice-cold PBS. Cells were resuspended in annexin binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4)—1×106 cells/mL, 100 μL per assay. Five microliters of annexin V-APC conjugate (No. A35110; Invitrogen) and 1 μL of propidium iodide (No. P3566; Invitrogen) were added to each assay, and incubated for 15 min at RT. Control samples were treated with only PI, annexin V-APC, or left unstained. Four hundred microliters of binding buffer was then added to each sample, and the samples were kept on ice and analyzed immediately by flow cytometry (BD Accuri™C6; BD Biosciences) and processed with the FlowJo7.6 software.
Experimental animals
For all experiments, nonobese mice with severe combined immunodeficiency disease (NOD/SCID; Gades Institute/Taconic Farms) aged 6–8 weeks were used. All experiments were approved by the Norwegian Animal Research Authority and conducted according to the European Convention for the Protection of Vertebrates Used for Scientific Purposes.
Scaffold preparation
PLLA scaffolds were produced by a solvent-casting particulate-leaching technique, previously described by Nor et al. 20 and Hegen et al. 16 Briefly, 1 g of PLLA (Resomer L 206 S; Boehringer Ingelheim) was dissolved in 20 mL chloroform (Sigma). About 3.45 g of NaCl (Fisher Scientific) with a pore size ≥450 μm was distributed in silanized glass beakers. The NaCl was mixed with 5% PLLA solution and the solvent was left to evaporate. Thereafter, scaffolds were leached for 48 h with double-distilled water to wash out the NaCl, and dried and cut into 6×6×1 mm3 pieces. Scaffolds were sterilized in a descending alcohol series from 100% to 70% EtOH and stored in sterile PBS until implantation.
Implantation of scaffolds
The operations were performed as previously described by Hegen et al. 16 Briefly, sterile scaffolds were seeded with a total of 1×106 cells in 36 μL of 50:50 EGM-2 MV and growth factor reduced matrigel (BD), and left at 37°C for 30 min for the matrigel to solidify. Scaffolds were seeded with 1×106 HDMVECs alone or 1:4 (200,000 HDMVECs:800,000 PaSMCs) ratio. For each experimental group, six mice were implanted with two scaffolds each. NOD/SCID mice were anesthetized with an intramuscular injection of 20 μL 1:2 concentration of Rompun (Xylazin, 20 mg/mL; Bayer Health Care) and Narketan (Ketamin, 100 mg/mL; Vetoquinol) in the thigh muscle. A 2.5-cm incision was made on the back of the mouse and the scaffolds were implanted in skin flaps at the flanks. After 7 or 14 days, the mice were sacrificed by cervical dislocation after deep anesthesia (Isoflurane; Schering-Plough), and scaffolds were recovered for fixation and histology.
Immunohistochemistry
Scaffolds were fixed in 10% formalin overnight at 4°C and subsequently paraffin embedded for sectioning. Five-micrometer sections were deparaffinized with xylene and rehydrated in decreasing concentrations of ethanol to water. Microwave antigen retrieval was performed in target retrieval solution (pH 6.0; Dako). Sections were pretreated with dual endogenous enzyme-blocking reagent (Dako). Monoclonal mouse anti-human CD-31 (M0823, Clone JC70A; Dako) diluted 1:50 was applied for 60 min at RT, followed by horseradish peroxidase anti-mouse EnVision (Dako). CD-31 was visualized with 3-amino-9-ethylcarbazole peroxidase as substrate. CD31 monostained sections were counterstained with hematoxylin (DakoREAL Haematoxylin S2020; Dako). Monoclonal rat anti-mouse CD-34 (ab8158; Abcam) diluted 1:50 was applied for 60 min at RT followed by alkaline phosphatase goat anti-rat diluted 1:50 (Sc-3824; Santa Cruz Biotechnology) for 30 min at RT. CD-34 was visualized with Ferangi Blue™ Chromogen Kit (Biocare Medical). Monoclonal mouse anti-human smooth muscle cell actin (SMCA; M0851; Dako) diluted 1:200 was applied for 30 min at RT followed by alkaline phosphatase rat anti-mouse IgG2a (Santa Cruz Biotechnology). SMCA was visualized with Ferangi Blue Chromogen Kit (Biocare Medical). Antibody specificity was validated using negative control sections containing murine vascular tissue and positive control sections containing human vascular tissue.
Image analysis
To quantify vessel diameter, histological sections of scaffolds were examined with a light microscope (Leica). Photos were taken and, using the image-processing program AnalySIS, the outlines of anti-hCD31-positive vessels in five fields of view of a section were encircled manually at 200× magnification. Vessel diameter was calculated from the circumference measured. Vessels were then subdivided into five size categories based on diameter: <10, 10–20, 20–30, 30–50, and >50 μm, which are characteristic for capillary, intermediate size (arterioles and venules), and large microvessels, respectively.21,22 Graphs shown represent the average percent of vessels in each size group from five fields of view.
The total number of vessels formed in five fields of view from each section was also counted, and the average number of vessels formed per mm2 scaffold was calculated. Vessel number was normalized to the amount of ECs implanted (average number of vessels/mm2/250,000 ECs). Further, vessels were examined for intralumenal red blood cells, and vessel perfusion was quantified as earlier. All graphs represent the average number from five fields of view from one section of each scaffold.
To examine colocalization of engineered blood vessels with mural cells, three sections from each group of 2-week coculture experiments were double stained with anti-hCD31 and anti-SMCA. The whole scaffolds were examined by light microscopy, and the number of colocalized and noncolocalized CD31-positive vessels was calculated. Outliers were identified and removed by ROUT analysis (Prism; GraphPad), and the data are represented as total number of colocalized (CD31+/SMCA+) and noncolocalized (CD31+) vessels per field of view.
Three sections from each group of 2-week coculture experiments were H&E stained (Dako) and independently evaluated by a skilled clinical pathologist for the presence of thrombosis.
Statistics
Statistical significance of the data was evaluated using an unpaired, two-tailed, two-sample Student's t-test assuming unequal variance in Microsoft Excel v.14.1.0 or Prism (GraphPad). Statistically significant differences are denoted (*p<0.05). Number (n) in each experiment refers to the number of scaffolds analyzed. All values in bar diagrams are presented as mean±standard error of mean.
Results
Akt1 phosphorylation epi-alleles have graded downstream activity in ECs
To investigate the role of Akt1 in angiogenesis and vessel maturation, we prepared constitutively active Akt1 retroviral constructs comprising an N-terminal myristoylation signal, 23 and alanine substitutions at the two phosphorylation sites of Akt1. Expression of the Akt1 epi-alleles by retroviral transduction [myrAkt1, myrAkt1(T308A), and myrAkt1(S473A)] in HDMVECs yielded wild-type (dual) phosphorylated and hemiphosphorylated Akt1 protein expression. The phosphorylation status of Akt1 in the transduced HDMVECs was analyzed by flow cytometry (Fig. 2A, B) and western blot (Fig. 2C, D). Expression of myrAkt1 in HDMVECs led to constitutive phosphorylation of both T308 and S473 residues. HDMVEC/myrAkt1(S473A) displayed T308 phosphorylation, while the myrAkt1(T308A) protein was phosphorylated only at S473. To gain an overview of the relative downstream activity of hemiphosphorylated myrAkt1 mutants, we probed a western blot with phospho Akt-substrate (RXRXXS/T) antibody. HDMVECs expressing the three myrAkt1 constructs were compared with control cells expressing endogenous Akt1. Overall, the staining profile was similar between the cell types with an overall change in total downstream substrate phosphorylation (Fig. 2E). Several bands show a differential regulation among the different Akt1 mutant cells and the control. Quantification of total staining intensity (all bands) in each lane showed that the general downstream activity increased in a graded manner from the control to the myrAkt1(T308A)-, myrAkt1(S473A)-, and the myrAkt1-expressing mutants, respectively, forming an epi-allelic series of Akt1 activity (Fig. 2F). Several bands, most notably a 48-kDa protein, consistent with Akt substrate glycogen synthase kinase-3β (GSK-3β), displayed an abrupt increase in phosphorylation between the myrAkt1(T308A) and myrAkt1(S473A) mutants to the myrAkt1 mutant. A different effect is observed for a 51-kDa species, congruent with Akt1-target GSK-3α, which is selectively nonphosphorylated in the myrAkt1-expressing cells. Hence, Akt1 epi-alleles show unique graded downstream substrate phosphorylation.
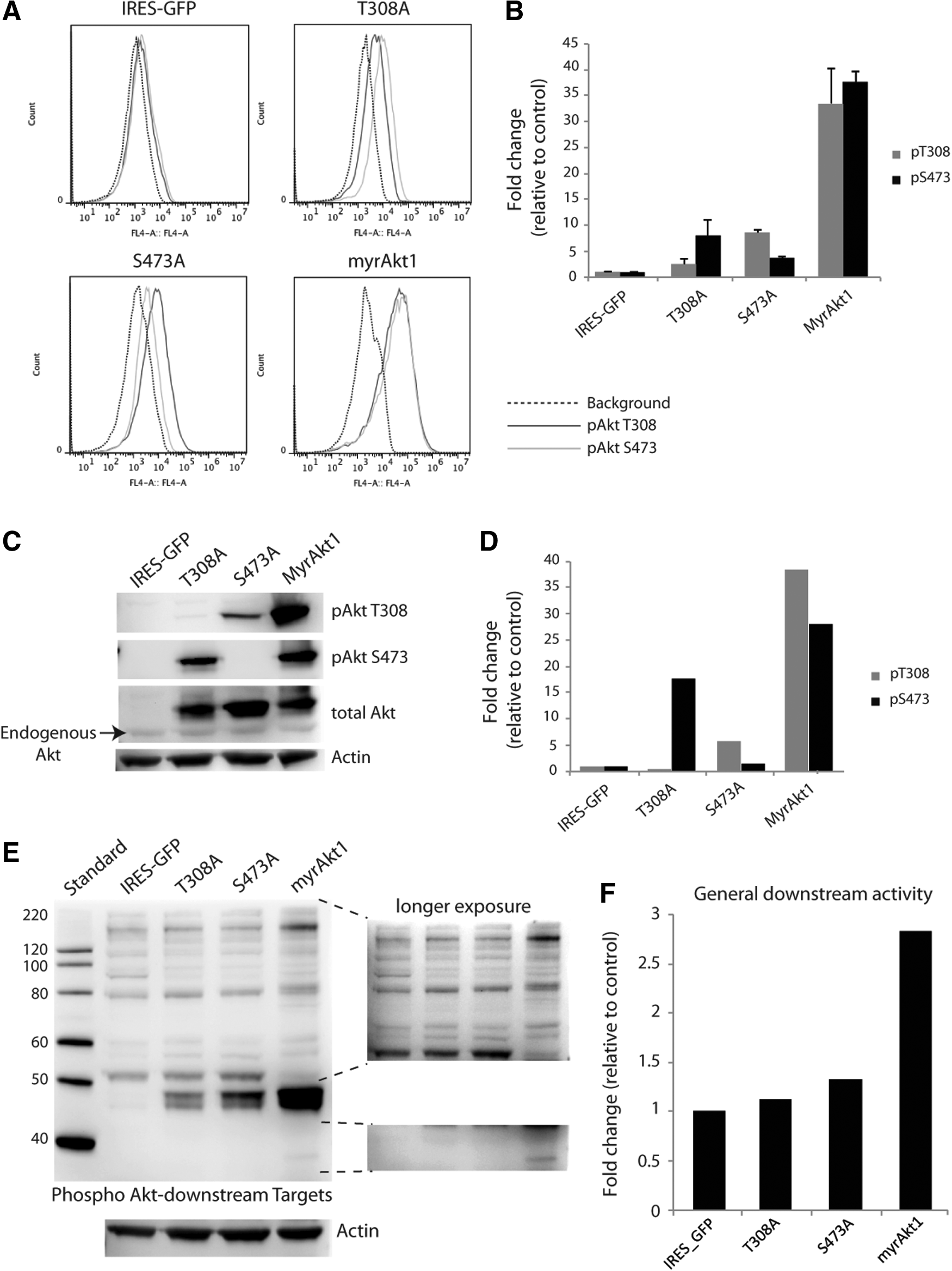
Myristoylated Akt1 constructs give increased Akt1 phosphorylation and a gradual increase of activity downstream of Akt1 in HDMVECs. HDMVECs stably expressing myrAkt1 constructs were analyzed for Akt1 phosphorylation at T308 and S473 by flow cytometry
Akt1 epi-alleles confer graded morphological effects on ECs
We next evaluated effects on morphological features of HDMVECs that express Akt1 epi-alleles. HDMVECs show a predominantly elongated morphology in cell culture (Fig. 3A). MyrAkt1-expressing HDMVECs displayed dramatic morphological differences. They were notably larger with multidirectional outgrowths (Fig. 3D). MyrAkt1(T308A)- and MyrAkt1(S473A)-expressing cells appeared more rounded than control HDMVECs, but distinct from myrAkt1-expressing cells (Fig. 3B, C). The actin cytoskeleton in cells expressing myrAkt1 was altered (Fig. 3D, right panel), while HDMVEC/myrAkt1(T308A) and myrAkt1(S473A) cells were similar to control cells (Fig. 3A–C, right panel).
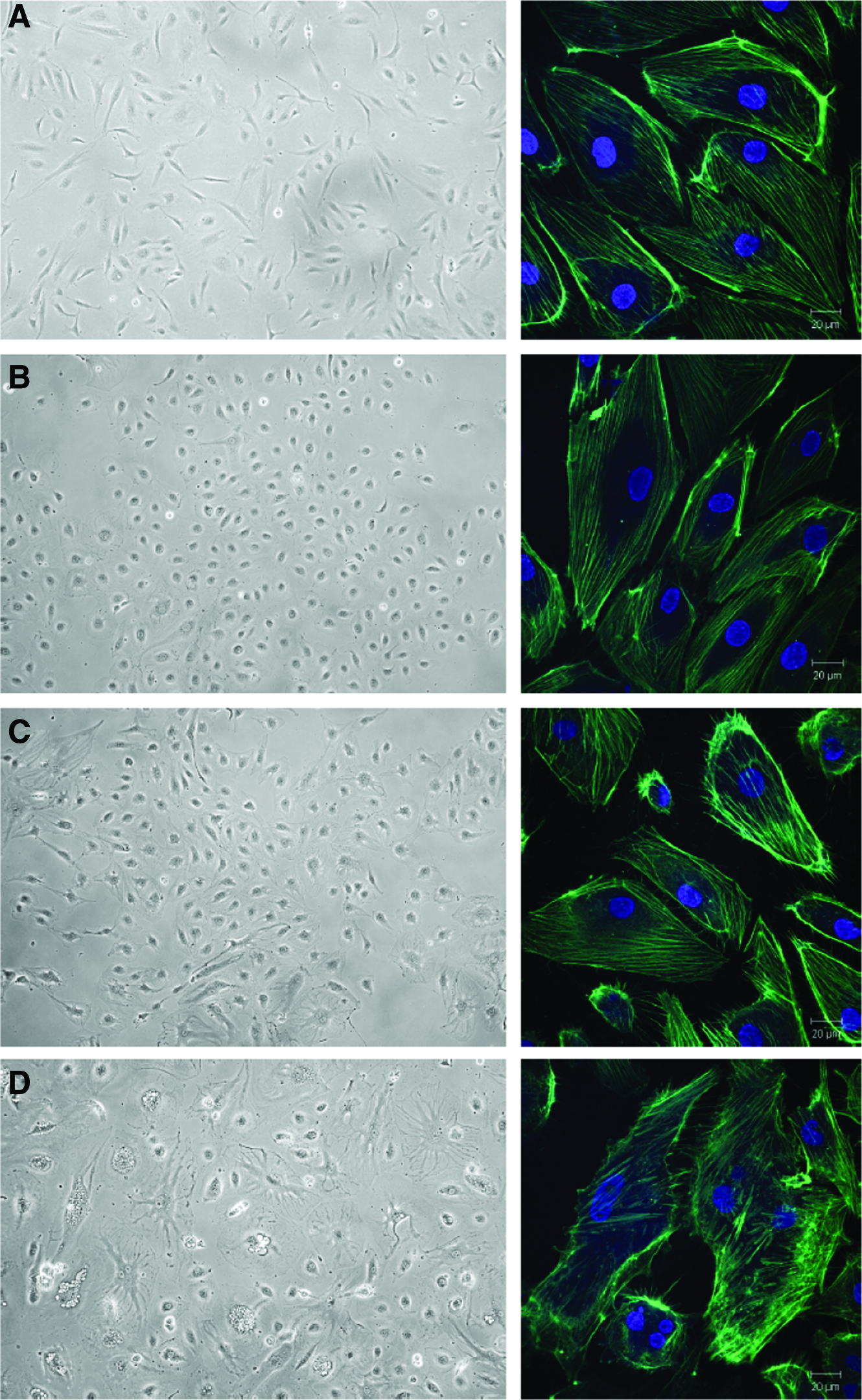
Expression of myrAkt1 constructs alters cell morphology. HDMVECs stably expressing myrAkt1 constructs were imaged using light microscopy (left panel) and the actin cytoskeleton was stained and imaged by confocal microscopy (right panel). Control cells were spindle shaped and had an organized actin cytoskeleton
To our surprise, myrAkt1-expressing HDMVECs were very challenging to culture. They grew slowly, never reaching confluency, and the cell yield from each culture flask was very low compared with the growth of control cells (data not shown). Notably, we also observed a large number of intracellular vacuoles in the HDMVEC/myrAkt1 (Fig. 3D, left panel). This was not observed in any of the other mutants. The formation and fusion of vacuoles in ECs is an important step in the lumenalization of newly formed vasculature in vivo. 24 However, intracellular vacuole formation could also be an indication that myrAkt1 expression in HDMVECs might induce apoptosis. 25 We therefore assessed the basal level of apoptosis in the Akt1 epi-allelic cells. Flow cytometry analysis of HDMVEC/myrAkt1 demonstrated a significantly increased percentage of dead and apoptotic cells relative to both control, T308A- and S473A-expressing cells (Fig. 4A, B). Although Akt has a prominent antiapoptotic role, 26 this result suggests that expression of constitutively active myristoylated Akt1 in HDMVECs contributes spontaneous apoptosis in these cells.
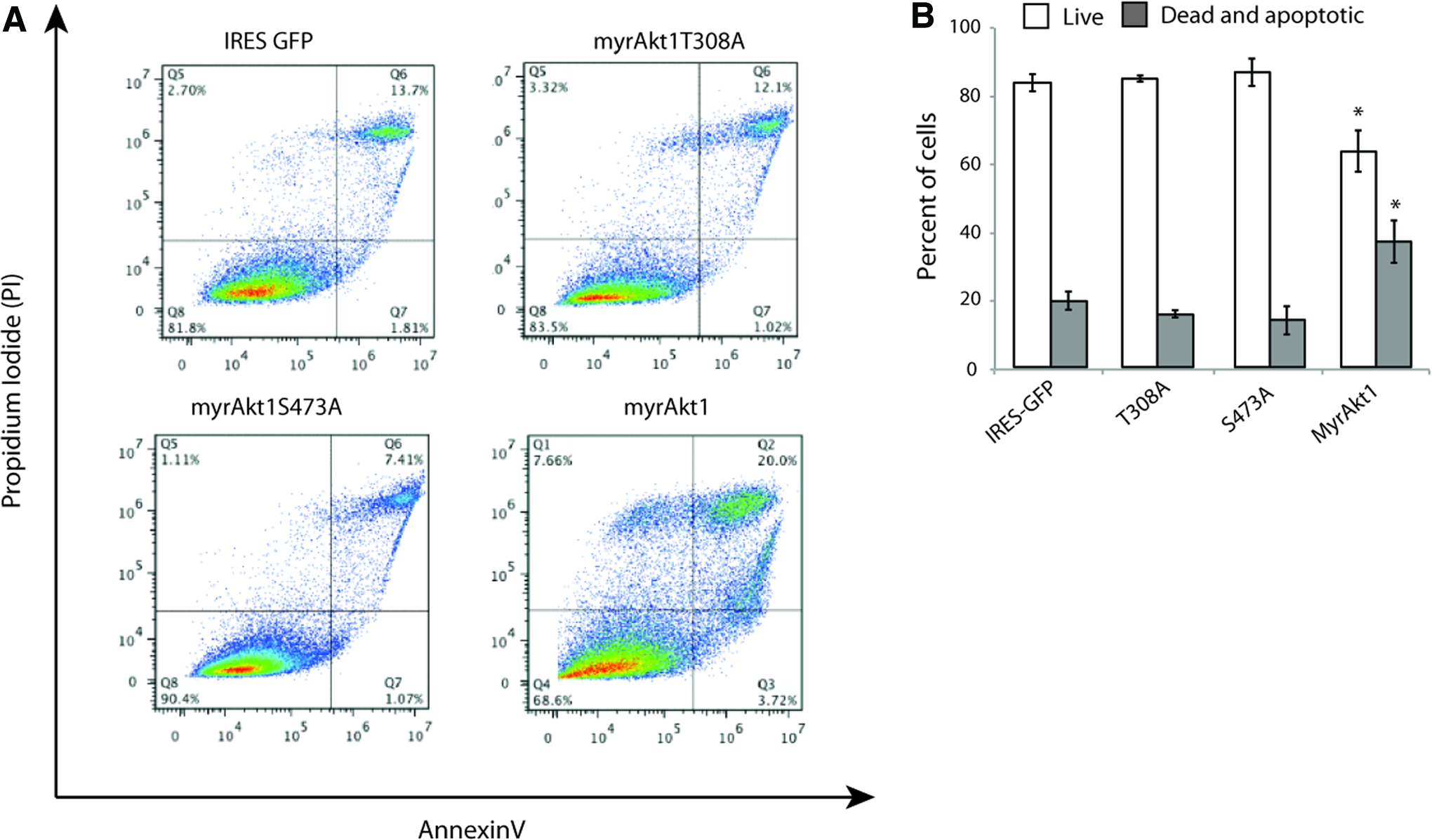
MyrAkt1-expressing HDMVECs were apoptotic. HDMVECs stably expressing myrAkt1 constructs were stained for annexin V-APC and PI. Representative scatter plots had increased apoptosis and cell death in myrAkt1-expressing HDMVECs
Increased Akt1 kinase activity induces formation of small-diameter vessels in vivo
To investigate the functional consequence of increasing levels of constitutive Akt1 kinase activity on blood vessel formation and maturation in vivo, we employed a tissue engineering model system of angiogenesis previously described by Hegen et al. 16 Control HDMVECs or HDMVECs expressing myrAkt1, myrAkt1(T308A), and myrAkt1(S473A) constructs were seeded into a matrigel-supplemented PLLA scaffold either in monoculture (1×106 cells in total) or in coculture with PaSMCs (1:4 ratio, 1×106 cells in total), and implanted subcutaneously in NOD/SCID mice. After 1 or 2 weeks, scaffolds were retrieved, and stained with anti-human CD31 to identify human blood vessels formed by the implanted HDMVECs in the TE scaffolds (Fig. 5). We have previously shown that the presence of mural cells in the scaffold promotes vessel maturation, accelerating formation of a functional perfused microvasculature comprising mainly vessels of <30-μm diameter within the first 2 weeks. 16 Implanted scaffolds seeded with myrAkt1 epi-allelic HDMVECs with or without PaSMCs were retrieved after 7 or 14 days, and the resultant intrascaffold vasculature was quantified by histomorphometric analysis after staining the vessels for anti-human CD31. The human vessels that formed were counted and binned into five size categories based on vessel diameter. After 1 week, the majority of vessels formed within the scaffolds by control HDMVECs were of large caliber (>30 μm). There was no significant difference between HDMVEC monoculture and HDMVEC/PaSMC coseeded scaffold vessels formed at this time point (Fig. 6A). However, after 2 weeks, the size distribution of the vessels shifted toward a higher percentage of intermediate- to small-caliber vessels (<20 μm) (Fig. 6B). At this time point, the average percentage of vessels in the 10–20- and 0–10-μm-size groups was significantly higher in HDMVEC/PaSMC coseeded scaffolds compared with HDMVECs alone, indicating that the presence of PaSMCs at the time of implantation allowed the vasculature to reach maturation faster. In contrast, the vessel distribution pattern observed in scaffold monocultures of HDMVECs expressing the hemiphosphorylated myrAkt1(T308A) and myrAkt1(S473A) constructs showed a major distribution toward small-caliber (0–10 and 1–20 μm) vessels already after 1 week (Fig. 6C, E, respectively). A surprisingly high percentage of these vessels was of the smallest caliber (0–10 μm). Notably, HDMVEC/myrAkt1(T308A) and myrAkt1(S473A) had a similar vascular size distribution independent of the presence of PaSMCs. After 2 weeks, a high percentage of small- to intermediate-caliber vessels persisted, but an increased percentage of large-caliber (>30 μm) vessels was also apparent (Fig. 6D, F). This pattern is opposite to the control HDMVECs. However, some significant differences were observed between mono- and cocultures at this time point in both cell types, indicating that PaSMCs do have some effect on vascular diameter in these cells.
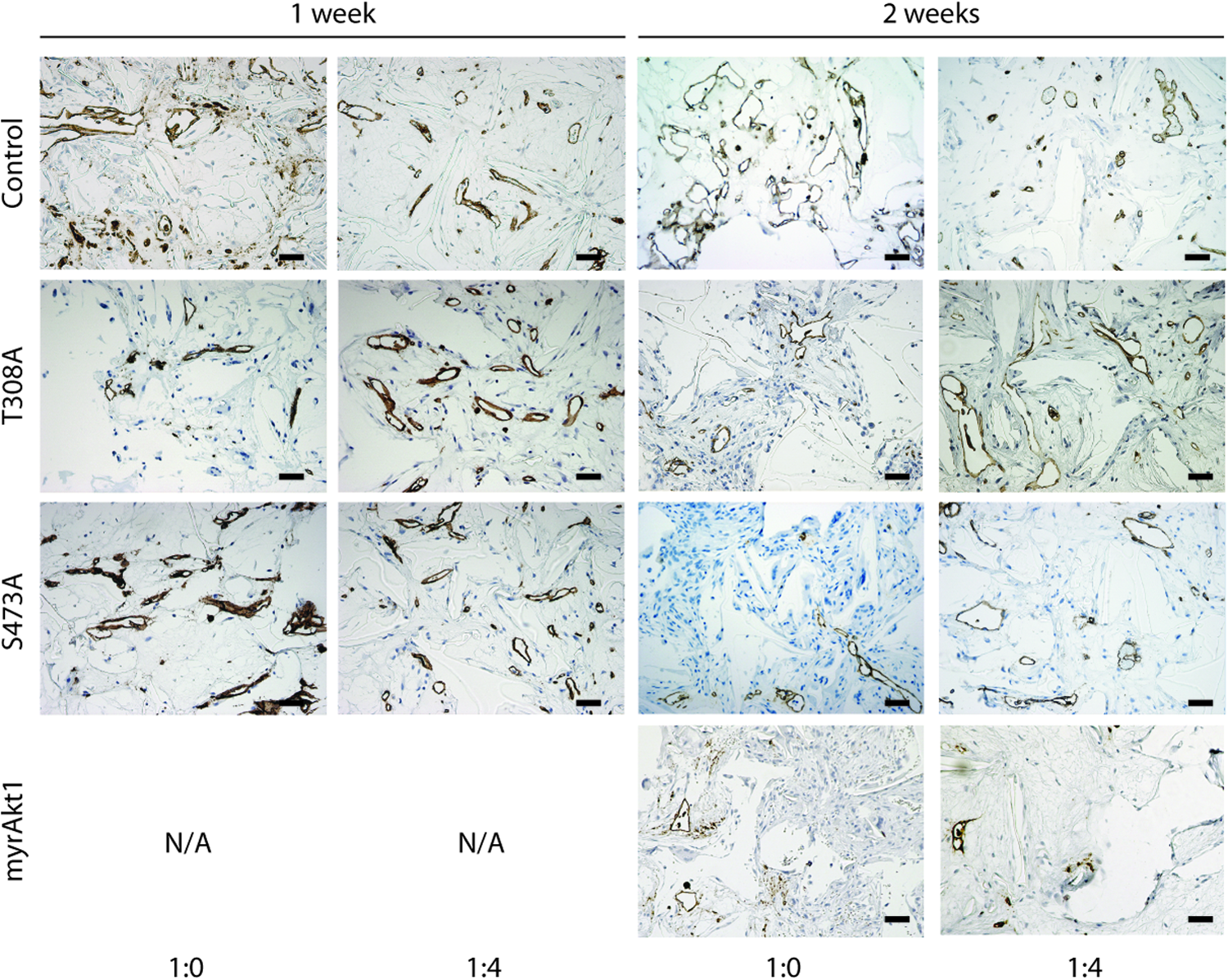
Implanted HDMVECs form human blood vessels in tissue engineering implants. Tissue engineering scaffolds implanted with control HDMVECs and HDMVECs expressing the three mutant Akt1 constructs in monoculture (1:0) or coculture (1:4) were stained with anti-human CD31, and imaged at 200× magnification, as demonstrated by representative histological images. Left panel: 1-week experiments, right panel: 2-week experiments. Scale bar=50 μm. Color images available online at www.liebertpub.com/tea
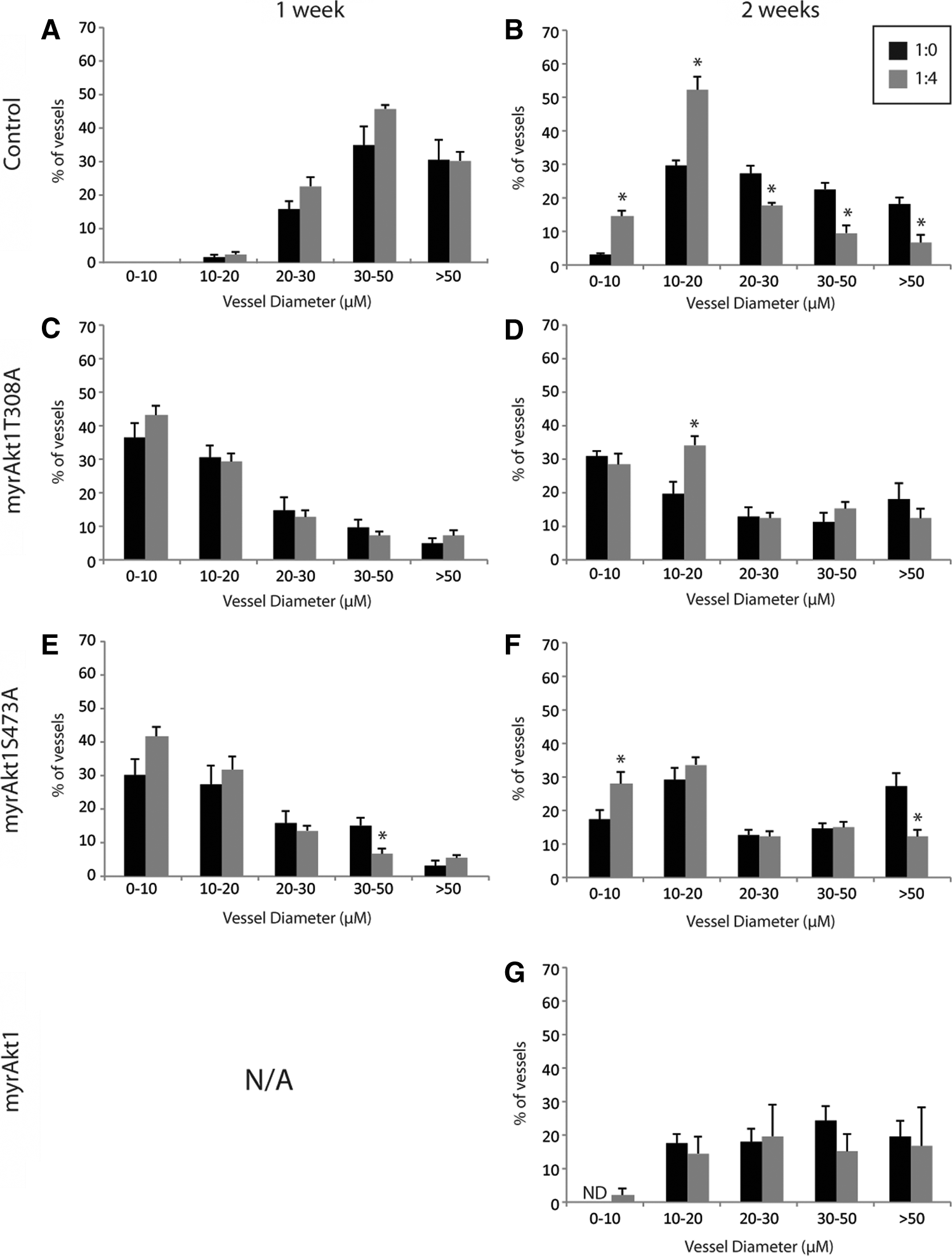
Expression of Akt1 mutant constructs in HDMVECs induces formation of vessels with a heterogeneous size distribution. Histomorphometric analysis of CD31-positive intrascaffold vasculature formed by monocultured HDMVECs (black bars) or HDMVECs cocultured with PaSMCs in a 4:1 ratio (gray bars) 1-week postimplantation
Generally, the results obtained with myrAkt1(T308A)- and myrAkt1(S473A)- expressing cells were strikingly similar, indicating that a moderate upregulation of Akt1 kinase activity in this setting has a similar effect, independent of the hemiphosphorylation state.
Since myrAkt1-expressing cells had poor viability, growing a sufficient number of cells for in vivo experiments was challenging and very time consuming. Therefore, only 2-week in vivo experiments were performed with these cells, as this was the time point we expected a mature vasculature to form, based on our previous results 16 and results found using control cells in this study. We found that the few vessels formed by these cells, both in mono- and coculture, displayed a heterogenous size distribution (Fig. 6G), except a lack of the smallest-caliber vessels (<10 μm). Further, there was no significant difference between monoculture and coculture, in strong contrast to the results obtained with control cells. These results indicate that constitutive Akt1 kinase activity interferes with mural cell–EC crosstalk in vivo.
Increased Akt1 kinase activity interferes with recruitment of PaSMCs to the vessel wall
The lack of consistent differences in the vascular size distribution between monocultured and cocultured vessels formed by Akt1-mutant-expressing HDMVECs was consistent with a defect in the mural-cell-induced vessel maturation of the vasculature formed in these scaffolds. This could be due to an abrogated mural cell–EC crosstalk, or incomplete recruitment of mural cells to the vessel wall. To investigate this, histological sections of scaffolds from 2-week coculture experiments were costained for anti-human CD31 and anti-αSMCA, and the number of CD31-positive vessels invested with vSMCs was quantified. In control scaffolds, the implanted mural cells appeared much more organized and tended to colocalize with the vasculature, whereas in all scaffolds implanted with Akt1-mutated HDMVECs, the mural cells were disorganized and spread throughout the scaffold (Fig. 7A). Quantification of the number of colocalized (CD31+/SMCA+) and noncolocalized (CD31+) vessels showed that the scaffolds containing control HDMVECs had a higher ratio of SMC-invested CD31-positive vessels (66.7% on average) than vessels formed by HDMVECs expressing the Akt1-mutant constructs (Fig. 7B). This indicated that constitutive activation of Akt1 in implanted HDMVECs interfered with EC-induced paracrine signaling that stimulates recruitment of mural cells to the vessel wall during angiogenesis.
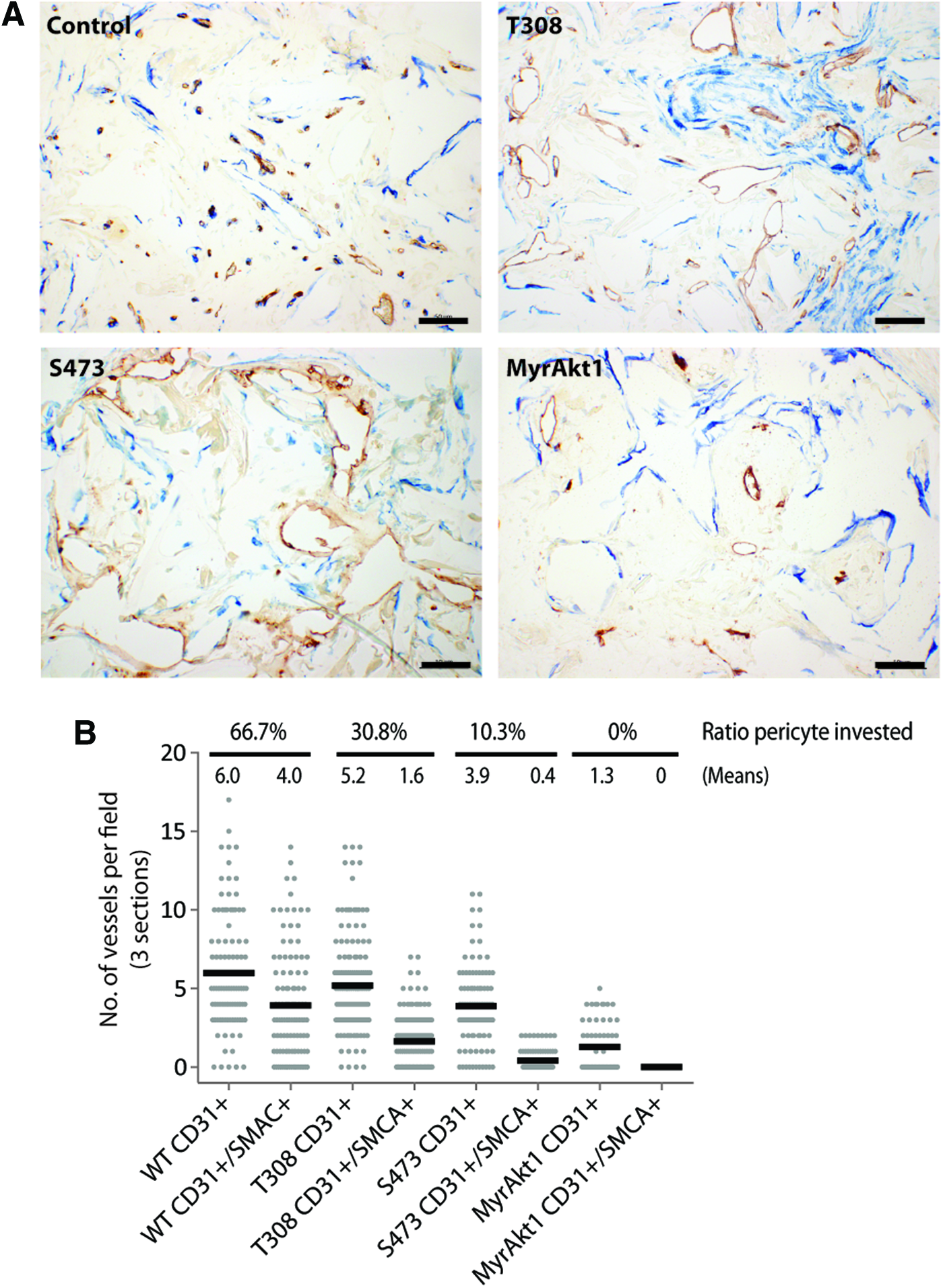
Expression of Akt1 mutant constructs in HDMVECs interferes with mural cell recruitment. TE scaffolds from 2-week coculture experiments were costained with anti-human CD31 (brown) and anti-α smooth muscle cell actin (SMCA) (blue) and imaged at 400× magnification. Representative images show that SMCs colocalized to CD31-positive vessels in control scaffolds
Increased Akt1 kinase activity blocks microvasculature functionality in vivo
Akt is a mediator of EC promigratory and proproliferative signals. 27 Therefore, we hypothesized that the microvascular density within implants seeded with myrAkt1-allele-expressing HDMVECs would be higher than in scaffolds containing control HDMVECs. To assess intrascaffold microvascular density, we quantified the number of CD31-positive vessels per mm2. However, as cocultures contained one-fourth the number of HDMVECs compared with monocultures, we normalized the vessel count to the number of cells implanted (NOV/mm2/250,000 HDMVECs). The functionality of the engineered microvasculature was also assessed, by quantifying the amount of CD31-positive vessels containing intraluminal red blood cells.
We found that the number of intrascaffold vessels formed by control cells was significantly higher than the myrAkt1-allele-expressing HDMVECs, both in monoculture and coculture. Further, there were significantly more vessels formed by cocultured HDMVECs/PaSMCs than monocultured HDMVECs of all types, both in 1- and 2-week experiments (Fig. 8A). These results indicated that the PaSMCs do have an impact on vessel formation in the presence of myrAkt1 epi-allele expression. However, the number of vessels formed by cocultured myrAkt1-expressing HDMVECs and PaSMCs was significantly lower than all other cell types, suggesting that graded constitutive Akt1 activity correspondingly interferes with EC–mural cell crosstalk.
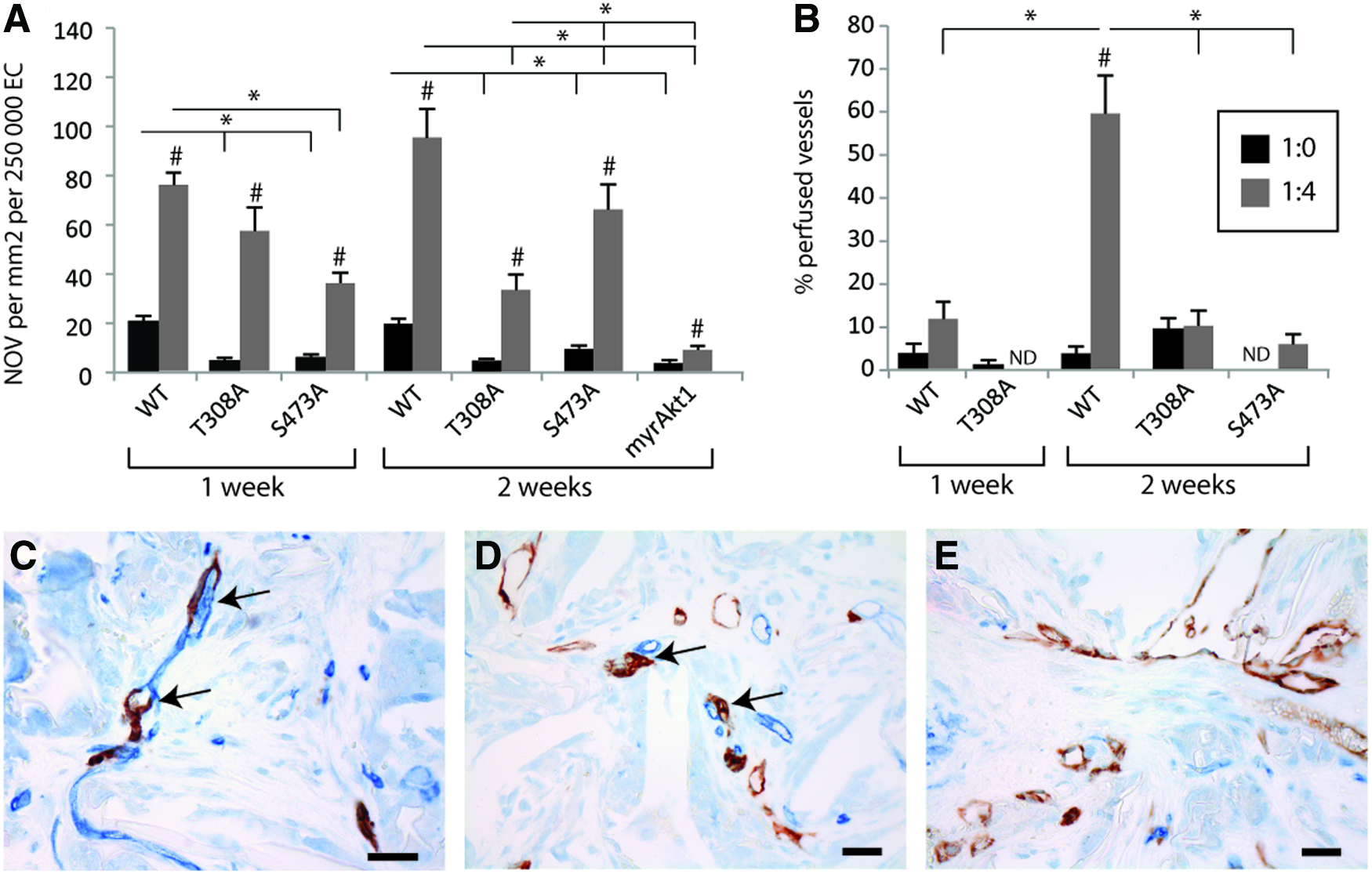
Expression of Akt1 mutant constructs in HDMVECs leads to formation of a largely nonfunctional vasculature. The microvascular density of CD31-positive human vessels in TE scaffolds was assessed. The average number of vessels formed in five fields of view from each scaffold was quantified, and represented as number of vessels/mm2/250,000 HDMVECs
The functionality of the engineered vasculature in the scaffolds was then assessed. To form functional perfused vessels, the engineered human vasculature must anastomose with the host vasculature. Thus, we expected to find chimeric vessels in the scaffolds, composed of both human and murine ECs. To assess the degree of vascular anastomosis, scaffolds from 2-week coculture experiments were double stained with anti-human CD31 and anti-mouse CD34. In scaffolds implanted with control HDMVECs (n=3) we found multiple juxtaposed small human and murine vessels (Fig. 8C) as well as scattered chimeric vessels (Fig. 8D). Among the examined scaffolds implanted with HDMVECs expressing the Akt1 mutant constructs [myrAkt1 n=3, myrAkt1(T308A) n=3, and myrAkt1(S473A) n=3], however, very few juxtaposed human and mouse vessels were observed, and no convincing anastomosed vessels were found (Fig. 8E). This indicated that there is an anastomosis defect in HDMVECs expressing the mutant Akt1 constructs.
To further examine vessel functionality, the engineered human vasculature was examined for intraluminal red blood cells to assess and quantify vessel perfusion. We found that ∼60% of the vessels formed by control HDMVECs coseeded with PaSMCs were perfused with intraluminal red blood cells after 2 weeks (Fig. 8B). This was significantly greater than that displayed in HDMVECs alone or in any of the myrAkt1-allele-expressing implants, where perfusion was around 10% at best. Perfusion was not detected in vessels formed by HDMVEC/myrAkt1(S473A) in monoculture or coculture after 1 week, nor in monocultures at 2 weeks. Vessels formed by cocultured HDMVEC/myrAkt1(T308A) were also not perfused after 1 week, but had around 10% perfusion both in mono- and cocultures after 2 weeks. No perfusion was detected in any vessels formed by myrAkt1-expressing cells. No microthromboses were detected in any of the sections (data not shown). These results suggested that constitutive Akt1 kinase activity in ECs effectively interferes with vessel maturation and functionality. A stronger phenotype was evident in HDMVECs expressing myrAkt1 than in cells expressing the hemiphosphorylated myrAkt1 mutants, indicating that graded Akt1 activity correlated with attenuated vessel functionality.
Discussion
In this study, we employed a tissue engineering model to assess the role of endothelial Akt1 kinase activity on vessel formation and maturation. We found that constitutive Akt1 expression attenuated PaSMC-induced vascular maturation in a kinase-activity-dependent manner. The recruitment of mural cells (pericytes/vSMCs) to the abluminal surface of nascent blood vessels is a key prerequisite in vessel maturation. Mural cells define a context comprising heterotypic cell–cell contacts, ECM deposition, and soluble factors that inhibits endothelial proliferation, maintaining capillary diameter, regulating blood flow, and providing survival signals. Failure to recruit mural cells results in abnormal vascular function, including vessel fragility, hemorrhage, and eventually vessel regression. Several diseases are associated with defective mural cell coverage, including arterio-venous malformation, diabetic retinopathy, hemangioma, and tumor vasculature.28,29 Prior to mural cell coverage, nascent vessels are susceptible to remodeling and VEGF signaling inhibitors, indicative of mural-cell-dependent changes in EC growth responsiveness. 30 Our results support a central role for Akt1 in mural-cell-dependent vessel maturation signals.
To our knowledge, the effect of endothelial-specific constitutive activation of Akt has not previously been investigated in the tissue engineering context. However, there are previous studies involving transgenic mice with endothelial-specific expression of a constitutive active Akt transgene with regulated expression. In a study of pathogenesis in cardiovascular disease, Mukai et al. found that constitutive activation of Akt in the endothelium protected the vascular wall from inflammatory and proliferative changes following vascular injury artificially induced by vessel ligation. 31 However, others have found constitutive Akt1 activity in ECs to strongly interfere with vascular homeostasis. Phung et al. found that sustained expression of myristoylated Akt1 leads to formation of an abnormal and immature vasculature that resembles the complex structural and functional abnormalities observed in tumor vasculature.14,32 This effect was found to be completely reversible when Akt activity was attenuated. These results demonstrate that dysregulated Akt1 activation is likely to contribute to pathological growth and disorganization of blood vessels. Indeed, Akt activity has also been found to be highly upregulated in both tumor cells and in tumor-associated ECs. 33
These studies analyzed overexpression of Akt in ECs in the context of extant, functional vasculature and thus attempt to model angiogenesis during the initial activation of vascular sprouting and vessel remodeling, evidenced by a dramatic change in vessel size distribution. However, in the tissue engineering setting (as illustrated in Fig. 1), ECs and PaSMCs are implanted into scaffolds as single cells, which must migrate and self-organize before vessel formation and sprouting can occur. In this sense, our model can be interpreted to resemble aspects in common with vasculogenesis. In the embryo, blood vessel growth occurs by two distinct mechanisms: angiogenesis and vasculogenesis. Vasculogenesis is defined as the de novo formation of a primitive vascular plexus by mesenchymal stem cells, 2 whereas angiogenesis is remodeling and sprouting of blood vessels from already existing vasculature. 34 Rearrangement and expansion of the primitive vascular plexus by sprouting, intussusceptions, and bridging leads to a transformation of the fairly uniform plexus into a complex vascular network consisting of fewer large vessels progressively increasing in number as the vessels branch and become smaller. 35 A similar process is also likely to occur in the tissue engineering scaffolds. ECs seeded into scaffolds as single cells are likely to proliferate and migrate, forming a homogenous vascular network consisting of mainly large vessels (Fig. 6A). Thereafter, vessel remodeling results in a heterogeneous network consisting of fewer large vessels branching into progressively more numerous smaller vessels (Fig. 6B). Coculturing HDMVECs with PaSMCs was found to accelerate this process, allowing the intrascaffold vasculature to reach maturation faster (Fig. 6B), likely by facilitating mural cell recruitment to the nascent vessels. However, partial upregulation of Akt1 kinase activity led to a dramatic change in the vascular size distribution, and the hierarchical pattern of a normal vasculature was lost. Already after 1 week, a high percentage of small-diameter vessels (<20 μm) were observed. After 2 weeks, a higher percentage of larger vessels were also apparent, an opposite pattern of what we observed with control cells. This suggests that constant remodeling of the vasculature occurs by vessel fusion as well as sprouting. Overexpression of Akt1 kinase activity led to a similar result (Fig. 6G), with formation of a fairly homogenous vascular network comprising a high percentage of large vessels. Importantly, there were only few and inconsistent significant differences in the vascular size distribution of mono- and cocultures in HDMVECs expressing mutant Akt1 constructs (Fig. 6C–G), suggesting that constitutive Akt1 activity interferes with the EC–mural cell crosstalk. Indeed, we found that vasculature formed by HDMVECs expressing the mutated Akt1 constructs colocalized to a lesser extent with mural cells than vasculature formed by control HDMVECs expressing wild-type Akt1 (Fig. 7A, B). Recruitment of mural cells to the vessel wall is essential during angiogenesis because mural cells suppress EC proliferation and migration, thereby stabilizing the newly formed vasculature. Mural cell recruitment is activated directly by ECs in the immature vessel wall through secretion of several paracrine factors, including platelet-derived growth factor-B (PDGF-B), which binds to the PDGF receptor-β on mural cells in adjacent vessels and stimulates their migration toward the immature vessel wall.36,37 In a recent study by Uebelhoer et al., it was shown that mutations in the EC tyrosine kinase receptor TIE2, associated with inherited sporadic venous malformations, are caused by a deficit in endothelial PDGF-B production. This defect, in turn, was found to be caused by a chronic ligand-independent activation of Akt by the mutant receptor. 38 In support of these findings, our results also suggest that constitutive Akt1 activation in ECs in our TE model interferes with endothelial PDGF-B secretion. This suggests that a downregulation of Akt1 signaling in ECs is necessary for PDGF-B secretion and subsequent recruitment of mural cells to the vessel wall.
Compared with vasculature formed by control cells, vessels formed by HDMVECs expressing all Akt1 mutants show very little evidence of perfusion (Fig. 8B). Vessel perfusion is achieved through anastomosis of the engineered vasculature with the host vasculature, likely through a “wrapping-and-tapping”-like anastomosis, as previously observed in engineered blood vessels by Cheng et al. 39 This generates chimeric blood vessels comprising both implanted human ECs as well as mouse ECs, as was observed in control scaffolds in this study (Fig. 8C). The formation of a largely nonfunctional vasculature by HDMVECs expressing mutant Akt1 constructs could indicate a direct failure of anastomosis in these cells. Indeed, we did not observe any chimeric vessels formed by HDMVECs expressing mutant Akt1 constructs (Fig. 8E). However, around 10% perfusion was found in vessels expressing the myrAkt1(T308A) and myrAkt1(S473A) constructs (but not the myrAkt1 construct) (Fig. 8B), indicating that some anastomosis occurred in these vessels. The role of endothelial Akt1 in vessel anastomosis is to our knowledge not yet elucidated, and further studies are needed to examine the mechanism of vessel anastomosis and the role of Akt1 in this process.
An alternative explanation to the loss of functionality in vessels formed by HDMVECs expressing Akt1 mutant constructs could be a defect in the regulation of EC cytoskeletal dynamics. Endothelial migration during angiogenesis is dependent on a tightly regulated process of constant remodeling of the actin cytoskeleton. 40 Morales-Ruiz et al. have previously shown that constitutive activation of Akt through myristoylation in ECs leads to a profound reorganization of the actin cytoskeleton as well as increased spontaneous endothelial migration independent of VEGF migratory stimuli. 41 Similarly, we found that HDMVECs expressing Akt1 mutant constructs, most notably myrAkt1-expressing cells, had a strongly reorganized actin cytoskeleton compared with control cells (Fig. 3). These results indicate that constitutive activation of Akt1 in HDMVECs induces a constant migratory signal in these cells, which would inhibit maturation and subsequent perfusion of the engineered vessels. This notion is further supported by the apparent reorganization of the vessels into a more homogenous vascular network observed at 2 weeks in all Akt1-mutant-expressing HDMVECs (Fig. 6C–G), which suggests a constant remodeling of the engineered vasculature through vessel fusion as well as sprouting.
In a previous study, Sun et al. investigated the role of Akt in vascular development and remodeling in the retina and skin of neonate transgenic mice by the use of an inducible endothelial-specific driver of myrAkt. They found that constitutive activation of Akt during vessel development led to a formation of vascular malformations, edema, and resultant embryonic lethality. Consistent with our findings, they found that this phenotype was due to a loss of normal vascular patterning and a disturbance of the normal vessel hierarchy. 42 Vascular patterning is regulated by a combination of environmental factors (such as hemodynamics and tissue demand for oxygen) and genetic factors. 43 Thus, loss of functionality of vessels formed by HDMVECs expressing Akt1 mutant constructs may in part explain the loss of normal vascular patterning. Further, we have also shown that vascular remodeling is regulated by endothelial–mural cell interactions (Fig. 6A, B). 16 Our results confirm that constitutive activation of Akt1 in ECs interferes with this exogenous regulation of EC proliferation and migration. Hence, the activity of Akt1 in ECs appears to be tightly regulated by contextual cues, including heterogeneous mural cell–EC signaling.
To our surprise, we found that myrAkt1-expressing cells were challenging to grow. They had an altered morphology and decreased proliferation rate due to increased spontaneous apoptosis in these cells (Fig. 4). This has not been reported by others using myrAkt1 in ECs in vivo. On the contrary, Akt is a well-known antiapoptotic protein, 26 and overexpression of Akt has been shown to protect ECs from apoptosis due to vascular injury, 31 hyperoxia, 42 or anoikis. 44 However, Miyauchi et al. found that constitutive Akt activity in cultured primary ECs induced a senescence-like growth arrest and decreased the lifespan of these cells. 45 The authors suggest that this mechanism might be important in vascular pathologies, such as in diabetes, where hyperinsulinemia could cause constitutive Akt activity in ECs. Importantly, ECs in these studies displayed similar morphological alterations as we found in our studies, indicating that a similar mechanism could also be the cause of the decreased growth rate and apoptosis in myrAkt1-expressing HDMVECs.
Increased apoptosis was not observed in HDMVECs expressing hemiphosphorylated Akt1 constructs. Indeed, no significant differences were observed in the apoptotic and dead cell count of cells expressing myrAkt1(T308A) and myrAkt1(S473A) constructs compared with control cells. However, both flow cytometric (Fig. 2A, B) and western blot analyses (Fig. 2C, D) showed that whereas expression of hemiphosphorylated Akt1 constructs led to an increased Akt1 phosphorylation 10–15 times that of control cells, expression of the myrAkt1 construct increased Akt1 phosphorylation 30–40 times. This is a dramatic increase in kinase activity, and increased apoptosis therefore appears to be directly related to the grade of Akt1 kinase activation. It has been shown that overexpression of oncogenes under some circumstances can induce cellular senescence and/or apoptosis, as a mechanism to protect cells against malignant transformations.46,47 Oncogene-induced apoptosis could also explain why we see increased apoptosis in myrAkt1-expressing HDMVECs, although more studies are necessary to elucidate this mechanism.
In this study, we investigated intracellular regulation of blood vessel formation in the tissue engineering setting. Our results emphasize the importance of Akt1 signal transduction in productive mural cell recruitment, vessel stability, and maturation. We demonstrate that coimplantation of HDMVECs with PaSMCs in tissue engineering scaffolds accelerates vessel maturation. Gradual increase of Akt1 kinase activity in HDMVECs was found to interfere with PaSMC-induced vessel maturation in a grade-dependent manner, suggesting that a temporal and spatial fine tuning of Akt1 kinase activity is essential in the regulation of normal vascular development and growth.
Footnotes
Acknowledgments
The authors thank Gerd Lillian Hallseth, Bendik Nordanger, Paula Ruurs, Marianne Enger, and Sissel Vik Berge for expert technical assistance.
Disclosure Statement
No competing financial interests exist.