
Abstract
The mechanical and physicochemical effects of three-dimensional (3D) printable hydrogels on cell behavior are paramount features to consider before manufacturing functional tissues. We hypothesize that besides good printability and cytocompatibility of a supporting hydrogel for the manufacture of individual tissues, it is equally essential to consider beforehand the desired tissue (bone, cartilage, fat). In light of its application, the structure and stiffness of printable hydrogel matrices influence cell geometry, which in turn impacts the differentiation fate. Embedded human mesenchymal stromal cells in printable type I collagen- and chitosan–agarose blends were induced to differentiate toward osteoblasts and adipocytes. Hydrogels' printability in air versus submerged printing in perfluorocarbon was evaluated according to the height, diameter, uniformity, and stability of 3D printed vertical cylinders. Bipotent differentiation within hydrogels was assessed histologically (morphology, cellularity), by immunohistochemistry (vimentin, smooth muscle actin), two-photon microscopy (spatial distribution), and real-time polymerase chain reaction (ALP, BGLAP, OPN, RUNX2, COL 1, aP2, PPARγ-2). Agarose and agarose blends revealed the most valid printability properties by generating uniform cylinders with an average height of 4 mm. Osteogenic differentiation was preferably achieved in anisotropic soft collagen-rich substrates, whereas adipogenic differentiation mostly occurred in isotropic stiff agarose-rich matrices. The conjugation of type I collagen to agarose with varying ratios is possibly a suitable bioink for a broad range of 3D printed mesenchymal tissues.
Introduction
T
Based on volumetric medical imaging techniques such as computed tomography and magnetic resonance imaging, individualized implants are fabricated entirely matching each patient's specific needs. 10 The rapid layer-by-layer deposition of cells and a supporting matrix, also known as bioprinting or tissue additive manufacturing, has allowed the construction of tissue mimics at the microscale.11–13 Submerged bioprinting is a strategy especially developed for complex, multilayered 3D engineered tissues, such as arteries, trachea, or cartilage.14–17 By submerged printing, cell-laden hydrogels are dispensed in a hydrophobic high-density perfluorocarbon drop-by-drop and layer-by-layer. After printing, the perfluorocarbon is replaced by medium to nourish cells with nutrients until tissue maturation and remodeling is completed. We have previously demonstrated that cells within agarose hydrogels remain viable allowing for cell proliferation and production of extracellular matrix (ECM).14,15 In the chase of an improved biological performance and bioprinting properties, hybrid hydrogels, such as collagen–agarose and chitosan–agarose, were examined and compared to an established and well-characterized 3D collagen model.
Collagen is a natural protein mostly present in connective tissue and, therefore, is an ideal candidate for supporting in vitro stromal cell growth and differentiation.18,19 Collagen I can be pepsin- or acid extracted from collagen-rich tissues and collected in solution. The initiation of linear fiber formation from collagen monomers, also known as fibrillogenesis, occurs spontaneously at physiological pH and temperature. 20 Acidic collagen hydrogel polymerizes both covalently in the presence of NaOH and physically at 37°C, and is composed of a loose meshwork of collagen fibers and fluid.18,20 After a rather passive polymerization, collagen gels tend to slightly deform by partly losing their water content that helped to maintain a certain volume. For this reason, collagen alone is not easily patterned in a desired 3D shape.
Chitosan is constituted of glucosamine and N-acetylglucosamine monomers and is a natural polysaccharide extracted by deacetylation of chitin. Chitosan hydrogels are commonly used for cartilage tissue engineering due to their similar content of glucosaminoglycans and hyaluronic acid to native tissue.21,22 Moreover, it has proved to be biocompatible and biodegradable as well as inert and noncytotoxic. Nevertheless, chitosan gel itself lacks fast gelling properties. 22 This evidence may trouble the 3D fabrication process introducing viable cells, once a relatively fast operation is desirable from the beginning of the printing until the culture of the constructs.
Agarose is a suitable material for the 3D freeform fabrication of large tissues by submerged printing. It has been qualified for 3D cell encapsulation due to its cytocompatibility and stable mechanical properties. Past in vitro experiments showed continuous cell functionality and differentiation in agarose hydrogels.23,24 Aqueous solutions of agar have a sol–gel transition in the range from 32°C to 47°C depending on the colloid concentration, 25 and are accordingly handled with ease as supporting biopaper for tissue additive manufacturing. Unlike other gels that require additional polymerizing agents (e.g., alginate), agarose polymerizes physically within seconds.24,26
In this study, we suggest essential combinations of hydrogels that enable both biological support, that is, by adding collagen and chitosan, and the possibility to be used as a printable scaffold, that is, by employing agarose, during the fabrication and subsequent incubation phases. Accordingly, human mesenchymal stromal cells (MSCs) are used in this study due to their multipotent ability to differentiate toward adipogenic, osteogenic, chondrogenic, myogenic, and neurogenic lineages.27,28 Stromal cell fate is directed by external stress, matrix mechanics, cell shape, and chemical stimuli. In this study we hypothesize that besides the printability of a supporting material to achieve a 3D spatial object, the material characteristics to guide cell fate are of main importance. Hydrogels with varying structure and mechanical properties confine cell shape differently and ultimately guide stromal cell fate.29,30 Hence, tendencies on stromal cell differentiation potential toward osteogenic and adipogenic lineages within agarose-based three-dimensionally printable hydrogels with diversified structure and properties were studied in this work for mesenchymal tissue substitutes.
Materials and Methods
Preparation of nonblended and blended hydrogels
Collagen
MSC-laden collagen hydrogels were produced according to Schneider et al. 18 Briefly, eight parts of collagen G (3 mg/mL collagen type I in 12 mM HCL) were mixed with one part 10-fold Dulbecco's minimum essential medium (4.5 g/L D-glucose DMEM, both Biochrom) and neutralized with 2 M sodium hydroxide. For the controls and MSC undergoing osteogenic induction, one part containing 1×106 MSC in suspension was added to the solution, whereas for MSC undergoing adipogenic stimulation one part containing 1.6×106 MSC in suspension was added. For each one of the culture conditions, 500 μL of the hydrogel (1×105 and 1.6×105 cells/mL, respectively, according to the different cell seeding densities used in established protocols for osteogenesis and adipogenesis) were poured in 24-well plates. MSC-laden collagen hydrogels were allowed to polymerize at 37°C for 1 h in a humidified incubator.
Agarose
Cell-free 3% (w/v) agarose hydrogels were generated by dissolving 3 g of low-gelling temperature agarose (suitable for cell culture; Sigma) in tap water. The gels were sterilized at 120°C for 2 h. For the controls and osteogenic differentiation hydrogels, one part of 3% (w/v) agarose hydrogel was added to one part containing 1×106 MSC in suspension (1×105 cells/mL), whereas one part containing 1.6×106 MSC in suspension (1.6×105 cells/mL) was added to adipogenic differentiation samples.
Agarose–collagen
MSC-laden agarose–collagen hydrogels were produced by mixing equal parts of the collagen, agarose hydrogels, and MSC in suspension in a 1:1:1 ratio. For the controls and MSC undergoing osteogenic stimulation, a final cell density of 1×105 cells/mL was used, whereas for MSC undergoing adipogenic stimulation, a final cell density of 1.6×105 cells/mL was applied.
Agarose–chitosan
One percent (w/v) chitosan pure hydrogel was produced by dissolving 1.0 g chitosan and 0.9 g NaCl in 100 mL 0.1 M HCl (all from Sigma) at room temperature for 48 h. The prepared chitosan hydrogel was centrifuged at 10,000 rpm for 5 min, warmed up to 60°C, and mixed with 1.1 g β-glycerophosphate in saline solution. MSC-laden agarose–chitosan hydrogels were generated by mixing equal parts of agarose hydrogel, 1% (w/v) chitosan, and MSC in suspension in a 1:1:1 ratio. For the controls and osteogenic differentiation gels, a final cell density of 1×105 cells/mL was used, whereas for adipogenic differentiation gels, a final cell density of 1.6×105 cells/mL was employed.
The solid contents used for each hydrogel as well as the used crosslinking mechanism are summarized in Table 1.
Three-dimensional-printability test for hydrogels
Aiming at the development of new hydrogel combinations useful for 3D submerged printing, collagen, agarose, agarose–collagen, and agarose–chitosan gels were tested. To fabricate 3D structures three main criteria were of importance: (1) the monomer solution had to be cytocompatible and dispensable, (2) gelling of single drops should happen within seconds upon a physical and/or chemical trigger, and (3) drops dispensed on top of each other had to adhere. A straightforward experiment was carried out to check these criteria. Manually using a 1-mL syringe with a 23G needle shortened to 12 mm to simulate the printing process, five to six drops of hydrogel were dispensed on top of each other to form a solid column. Using the syringe-based 3D bioprinter, columns of hydrogels were extruded submerged in perfluorocarbon. The outcome of both manual and printed scenarios was evaluated based on visual examination with respect to spreading of the printed gel as well as height, diameter, and uniformity immediately after printing, and stability of the fabricated gel column 24 h after incubation.
Isolation and expansion of MSC
MSC were isolated from femoral heads after informed consent approved by the Ethics Committee of RWTH Aachen University (EK178/08 and EK128/09), as previously described. 31 The isolation of MSC was carried out according to the minimum criteria of the International Society of Cellular Therapy. 27 Briefly, spongiosa specimens were flushed several times with MSC medium using a 22G-needle and centrifuged afterward. Cells were seeded in T75 flasks and nourished for 24 h. Hematopoietic nonadherent cells were removed by MSC medium change. Adherent cells in culture were then expanded for two to four passages in MSC medium (Mesenpan; PAN Biotech) containing 2% fetal calf serum (FCS) and 1% Penicillin/Streptomycin (both Gibco, Invitrogen).
Osteogenic and adipogenic differentiation of MSC in hydrogels
MSC were induced to differentiate toward adipocytes and osteoblasts according to protocols previously described. 18 For osteogenic and adipogenic differentiation MSC were seeded at a density of 1×105 and 1.6×105 cells/cm3, respectively. MSC were stimulated with osteogenic and adipogenic differentiation media up to 21 days. The osteogenic induction medium was composed of low glucose DMEM (PAA Laboratories), 10% FCS, 1% Penicillin/Streptomycin (both Gibco, Invitrogen), 10 mM sodium β-glycerophosphate, 100 nM dexamethasone, and 0.05 mM L-ascorbic acid 2-phosphate (all from Sigma) and was changed every 2–3 days. For adipogenic differentiation, media was changed every 3–4 days alternatively with adipogenic induction medium and adipogenic maintenance medium for 21 days. Adipogenic induction medium was composed of high glucose DMEM (Life Technologies), 10% FCS, 1% Penicillin/Streptomycin (both Gibco, Invitrogen), 0.01 mg/mL insulin, 0.2 mM indomethacin, 1 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methyl-xanthine (all from Sigma). Adipogenic maintenance medium consisted of 0.01 mg/mL insulin (Sigma), 10% FCS, and 1% Penicillin/Streptomycin (both Gibco, Invitrogen).
The study was performed with primary cultures of MSC based on up to six independent experiments from three independent donors (n=3).
Viability of MSC in hydrogels
MSC were cultured in collagen, agarose, agarose–collagen, and agarose–chitosan hydrogels for up to 21 days. The Live/Dead staining was prepared using (10 μL 5 v% PI solution [propidiumiodid 95% in Ringer's solution; Sigma] and 10 μL 5 v% FDA solution [fluorescein diacetate in acetone; Sigma] which were mixed with 600 μL Ringer's solution) and applied immediately to the samples. Cell viability of MSC in the different gels was afterward calculated based on triplicates from three independent donors.
Cytotoxicity assessment
Proliferation, apoptosis, and necrosis assays were performed to MSC-laden hydrogel specimens. Collagen, agarose, agarose–collagen, and agarose–chitosan gels were cast in 96-well plates, after polymerization MSC suspended in stromal cell media were added to the wells, and cultured at 37°C in a humidified incubator. Cells were seeded at a density of 5000 cells/well and assays were performed after 2, 4, and 6 days of culture based on sextuplicates from three independent donors for each time point. After each measurement, samples were rinsed with phosphate buffered saline (PBS) and fresh medium was added.
Proliferation
Viable cells were quantified using a proliferation assay (CellTiter-Blue; Promega). Twenty-five microliters of CellTiter-Blue were mixed with 100 μL medium and incubated at 37°C for 1 h, after which the absorbance was read at 560 nmex/590 nmem using a plate reader (Tecan Infiniti M200). MSC cultured on tissue culture polystyrene (TCPS) were used as positive controls.
Apoptosis
Cell apoptosis was measured by determination of caspase 3/7 activity using the Caspase-Glo 3/7 assay (Caspase-Glo 3/7; Promega). One hundred microliters of Caspase-Glo 3/7 reagent were added to each sample and incubation took place at 22°C for 1 h before luminescence was measured (Tecan Infiniti M200). MSC exposed to UV light (280–300 nm) were used as positive controls. MSC cultured on TCPS were used as negative controls.
Necrosis
The different hydrogels were tested on their cytotoxic effect using the ToxiLight-Assay (ToxiLight bioassay kit; Lonza). MSC incubated with 100 μL lysis buffer for 5 min were used as positive controls. Twenty microliters of each sample were transferred to white-walled 96-well plates, mixed with 100 μL adenylate kinase detection reagent and the luminescence was measured directly (Tecan Infiniti M200). MSC cultured on TCPS were used as negative controls.
Alkaline phosphatase activity in hydrogels
Alkaline phosphatase activity was measured by an Alkaline Phosphatase Colorimetric Assay Kit (ab83369; Abcam) according to the manufacturer's instructions. Briefly, 18 μL of supernatants of triplicates from three independent donors were each mixed with 30 μL assay buffer, 30 μL p-nitrophenyl phosphate and incubated for 60 min at room temperature in the dark. The reaction was terminated by adding 12 μL stop solution and the fluorescent signal was measured at 405 nm (Tecan Infiniti M200). ALP activity was empirically calculated by dividing the value of pNP (μmol) for each sample volume and time of the reaction (Eq. 1), where A is the pNP value in μmol, V is the volume of the sample added to the assay (mL), and T is the reaction time (min).
Collagen ECM production by assessment of hydroxyproline
Collagen production and tissue development was tested by measuring hydroxyproline content within cell-laden hydrogels using the method of Reddy and Enwemeka. 32 This test was based on quadruplicates from three independent donors. MSC were encapsulated in the different hydrogels at densities of 5×104 and 8×104 cells/cm3, respectively for controls and osteogenic induction samples, and for samples undergoing adipogenic differentiation. MSC-laden hydrogels were cast in 24-well plates and cultured up to 21 days. Osteogenic induction media was changed thrice a week, whereas adipogenic induction and maintenance media were changed alternately twice a week. After the incubation period, MSC-laden hydrogels were washed in PBS and transferred to cryo vials (VWR). Standards containing 0, 6.3, 12.5, 25, 50, 100, 200, and 400 μg were prepared by mixing hydroxyproline with distilled water. One hundred microliters of 2 M NaOH were added to each sample, which were afterward autoclaved at 120°C for 20 min. Fifty microliters of the hydrolyzed hydrogels were transferred to 1.5 mL reaction tubes, and 450 μL of chloramine T were added. Samples were mixed gently and afterward oxidized at room temperature for 25 min. Subsequently, 500 μL Ehrlich's reagent (Sigma) was added for 20 min at 65°C. Then, 100 μL of each sample were transferred to 96-well plates and absorbance was measured at 550 nm (Tecan Infiniti M200). The values obtained from the standard curve were used to calculate the hydroxyproline content (μg/mg) of each sample. Collagen level for each sample was determined empirically (Eq. 2).
Histomorphological and immunofluorescence analysis
For histological and immunohistological evaluation, MSC-laden hydrogels were fixed in 3.7% formaldehyde for 24 h. After fixation, samples were embedded in paraffin, cut in 2 μm thick slices, and stained with Hematoxylin and Eosin (HE) according to a routine protocol. Elastic van Gieson (EvG) staining was used to visualize ECM deposition. Briefly, samples were stained in Weigerts Resorcin-Fuchsin (Chroma) for 15 min at room temperature, and washed with tap water differentiated in acid alcohol. Then, samples were stained in Picro-Fuchsin (Chroma) for 3 min and washed once in tap water. Samples were counterstained in 0.1% Nuclear Fast Red Solution (Merck) for 5 min. Slides were dehydrated in ascending alcohol series, rinsed in xylene, and mounted in Vitro-Clud (Langenbrinck).
To visualize MSC differentiating into osteoblasts within the different hydrogels Alizarin Red staining (Sigma) was performed. After 21 days of incubation, samples were fixed in 3.7% formaldehyde for 24 h, embedded in paraffin and cut in slices. After contact with a descendent alcohol series, slices were rinsed in distilled water and stained for 20 min at room temperature with Alizarin Red solution (40 mM pH 4.1). Samples were washed three times with PBS and imaged using light microscopy. To evaluate the presence of uni- or multivacuolated cells derived from MSC undergoing adipogenic induction, H&E was applied. Cellularity of all histological samples was estimated with Equation 3. The area of 10 high power fields (HPF) is equivalent to 2.8 mm2.
To screen for cellularity and phenotypic changes of MSC within hydrogels, immunohistochemistry was applied. Primary antibodies were used and diluted as follows: Ki67 (1:1000, M7240; DAKO), vimentin (1:4000, M0725; DAKO), SMA (α-smooth muscle actin, 1:3000, A2547; Sigma). The deparaffination was performed according to manufacturer's instructions. Slide preparations were stained by an autostainer for immunohistochemistry (DAKO cytomation). Primary antibodies were applied for 25 min and secondary antibodies (rabbit/mouse; DAKO) were added. Counterstaining of slides was achieved by Hematoxylin staining, after which slides were dehydrated and mounted.
Real-time polymerase chain reaction
Collagen and agarose–collagen hydrogels were selected for analyzing MSC adipogenic and osteogenic gene expression. The hydrogel blend consisting of agarose and chitosan was not further analyzed due to its less satisfactory printability properties. Samples were stored in 350 μL RLT buffer (RNeasy Mini Kit; Qiagen) and stored at −80°C. Papain digestion of samples was performed according to Kim et al. 33 Frozen samples were thawed and incubated at 60°C for 24 h in sterile 1.5-mL reaction tubes with 50 μL of papain (0.2 μL filter sterilized) concentrated digestion buffer (1 M potassium phosphate buffer, 50 mM cysteine, 50 mM EDTA, and 1.25 mg/mL papain [all from Sigma]). Thereafter, MSC-laden hydrogel samples were heated to 70°C for 10 min to stop the enzymatic reaction, and vortexed. Total RNA was isolated using the RNeasy Mini-Kit (Qiagen). For reverse transcription, 500 ng of RNA were used to initiate the cDNA synthesis (Fermentas First Strand cDNA Synthesis Kit; Thermo Scientific). Thereafter, cDNA was diluted 1:10 with RNA-free water. Semi-quantitative polymerase chain reactions were performed with SYBR Green (Mastermix, Diagenode). For each sample, 1 μL of cDNA was added as a template for polymerase chain reaction (PCR). Gene expression of all samples was normalized against the housekeeping gene ACTB, and relative expressions were analyzed with the 2−ΔΔCt method. The nucleotide sequences of the studied genes are listed in Table 2.
Two-photon laser scanning microscopy
After 21 days of incubation, MSC-laden hydrogels were fluorescently labeled with wheat germ agglutinin-Alexa Fluor 594 (WGA 594), Hoechst 33342, and CellLight Actin-GFP BacMam 2.0 (all from Molecular Probes, Invitrogen). Cells were labeled and incubated with actin-GFP 12 h before imaging. The staining concentration was adjusted to 10 particles per cell. Shortly before imaging, cell nuclei were stained with Hoechst 33342 (2 μg/mL in PBS) for 20 min and cell membranes were labeled with WGA 594 (1 μg/mL in PBS) for 5 min both at room temperature. Subsequently, specimens were rinsed once in PBS and imaged by two-photon laser scanning microscopy (TPLSM, Olympus FV1000MPE; Olympus) with a 25×NA1.05 water dipping objective. Excitation wavelength was mode-locked to 800 nm using a pulsed Ti:Sapphire laser (Mai Tai DeepSee; Spectra Physics). Single images were recorded in the X-Y axis and collected at different 1.0 μm Z heights. Imaging processing and 3D reconstruction of the samples were carried out using the Imaris 7.4 (Bitplane).
Scanning electron microscopy
The microstructure of hydrogels was evaluated by scanning electron microscopy. Specimens were fixed in 2% glutaraldehyde and 0.1 M Sorenson's buffer (pH 7.4) for 24 h. Subsequently, samples were dehydrated in acetone and critical point dried (E-300 Critical Point Dryer; Polaron Equipment). Then, hydrogels were coated with gold and imaged using a scanning electron microscope (ESEM XL 30 FEG, FEI; Philips) in a high vacuum environment.
Statistical analyses
Data are presented as means±SD. Data analyses were performed using one-way analysis of variance followed by Bonferroni's post-hoc test. Statistical significance was defined as *p<0.05, **p<0.01, and ***p<0.001.
Results
Printability of hydrogels
The feasibility of manufacturing 3D individual and blended hydrogel constructs was evaluated at first manually using a syringe with a needle, and thereafter using a 3D bioprinter. In general, collagen hydrogels showed a tendency to spread onto the surface where they were dispensed, contrarily to agarose and agarose hybrid hydrogels, which kept a vertical structure (Fig. 1). Collagen hydrogels alone, that is, without additional additives such as stronger polymerizing agents or in combination with other gels are highly disable to form a 3D structure. For this type of printing technique, it is essential to work with polymers which covalently or physically polymerize within a few seconds to freeform 3D constructs. Due to the elongated polymerizing period of collagen hydrogels (up to 1 h, according to the manufacturer), while a drop was placed on top of another drop of the same gel, it leaked and flowed downward to the lower part of the construct forming one big drop. Agarose and agarose blends, however, polymerize very fast and by placing a drop of these gels immediately after the latter one it was possible to build a solid vertical column.
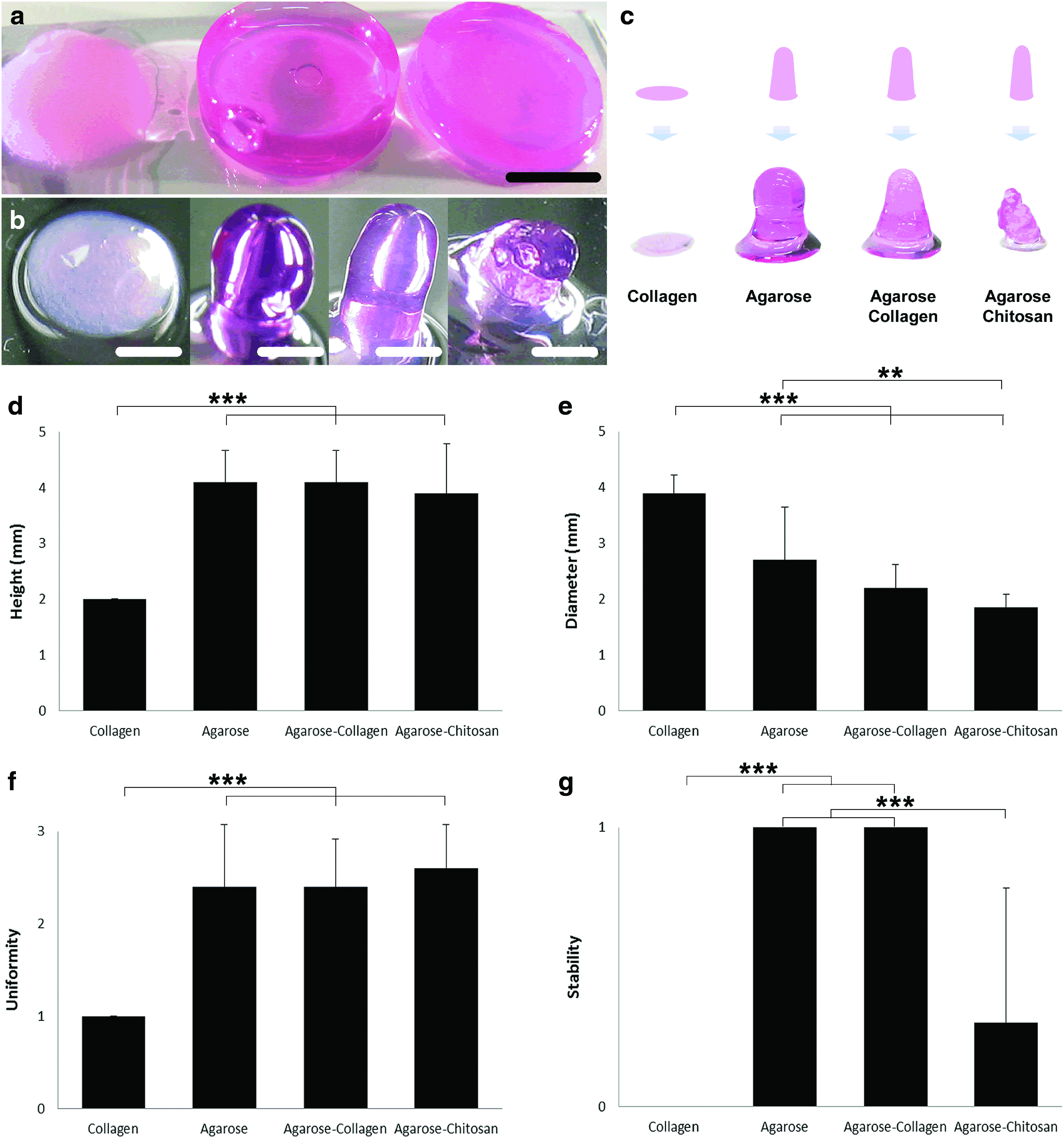
Manual test for hydrogel printability.
Using a 1-mL syringe with a 23G needle shortened to 12 mm to simulate the printing process, it was conceivable to dispense hydrogel drops of ∼1.5 to 2.5 mm of diameter, depending on the viscosity and density of the gel. An average diameter of 3.9 mm of collagen specimens was measured after the simulating 3D printing process, which validates the difficulty to build up 3D freeform constructs. In terms of uniformity of the 3D vertical columns, agarose and blends of agarose appeared to have similar rates ranging from 2.4 to 2.6 uniformity units. After 24 h of incubation, however, agarose and agarose–collagen gels were the most stable ones by keeping their original structure (Fig. 1g). Even though the combination of agarose and chitosan worked very satisfactorily immediately after the printing process, the gel lost its form after incubation with cell medium (Fig. 1b, c, g).
Using a custom-made drop-on-demand bioprinter, columns of hydrogels were extruded submerged in perfluorocarbon (Fig. 2a). The printer head comprises a heatable metal housing (Fig. 2b) that helps to keep a constant temperature around 38°C, avoiding premature gelation. This experiment was performed only using agarose–collagen hydrogels, since the combination agarose–chitosan was shown less successful as soon as the samples were removed out of the perfluorocarbon. For this test, tubes of agarose–collagen hydrogels loaded with MSC were printed (n=10). It was possible to notice the buoyant influence of the perfluorocarbon during printing, both on the achieved height (Fig. 2c) and diameter (Fig. 2d) of the constructs. The uniformity and stability of the printed specimens appeared to have a similar performance whether the manufacturing steps were done in the air or submerged in perfluorocarbon (Fig. 2e, f).

Custom-made bioprinter tests for hydrogel printability.
Microstructure of nonblended and blended hydrogels
Besides the printability of hydrogels for 3D bioprinting, the microstructure after polymerization is an important parameter. Using electron microscopy, a different microstructure between collagen and agarose-based hydrogels was observed (Fig. 3). Collagen hydrogels were rather fibrous, whereas agarose and agarose hybrid hydrogels revealed a more porous microstructure. Interestingly, the combination of agarose with collagen resulted in an intermediate yet more agarose-like structure of both hydrogels. Agarose hydrogel alone showed the formation of small spheres around and within the gel. When in combination with collagen, these spheres became increased in size and were homogeneously distributed throughout the hydrogel. Moreover, agarose–collagen hydrogels revealed larger pores compared with agarose gels. When agarose was combined with chitosan, an increased porous structure was observed similar to agarose–collagen hydrogels. All gels appeared to have an attractive topology for cell growth and proliferation. The presence of such spheres similar to nodes in combination with the porosity of agarose–collagen hydrogels may be of interest for cell adhesion and migration.

Microstructure of cell-free
Cytotoxicity and cell behavior in 3D cultures
Cytotoxicity was accessed for all hydrogels by measuring the number of viable cells (Live/Dead staining and proliferation assay), necrotic cells (bioassay), and apoptotic cells (caspase-3/7 activity assay; data not shown). Live/dead staining showed 99.1%±0.7%, 98.8%±2.1%, 98.4%±2.6%, and 96.6%±3.4% viable cells within collagen, agarose, agarose–collagen, and agarose–chitosan, respectively, after an incubation period of 21 days (Fig. 4a–e). Human MSC showed a four- to five-fold higher proliferation within collagen hydrogels compared with agarose and agarose–chitosan hydrogels (Fig. 4f; data from three independent donors). Nevertheless, proliferation potential was comparable for cells embedded in collagen and those embedded in the agarose–collagen blend. There was a significant increase on cell proliferation when the incubation occurred up to 6 days in agarose hydrogels alone or in the combination agarose–collagen. Hence, the addition of collagen to agarose polymer revealed a three-fold increase in cell growth. Additionally, it did not negatively influence cell necrosis (Fig. 4g) or apoptosis (data not shown).
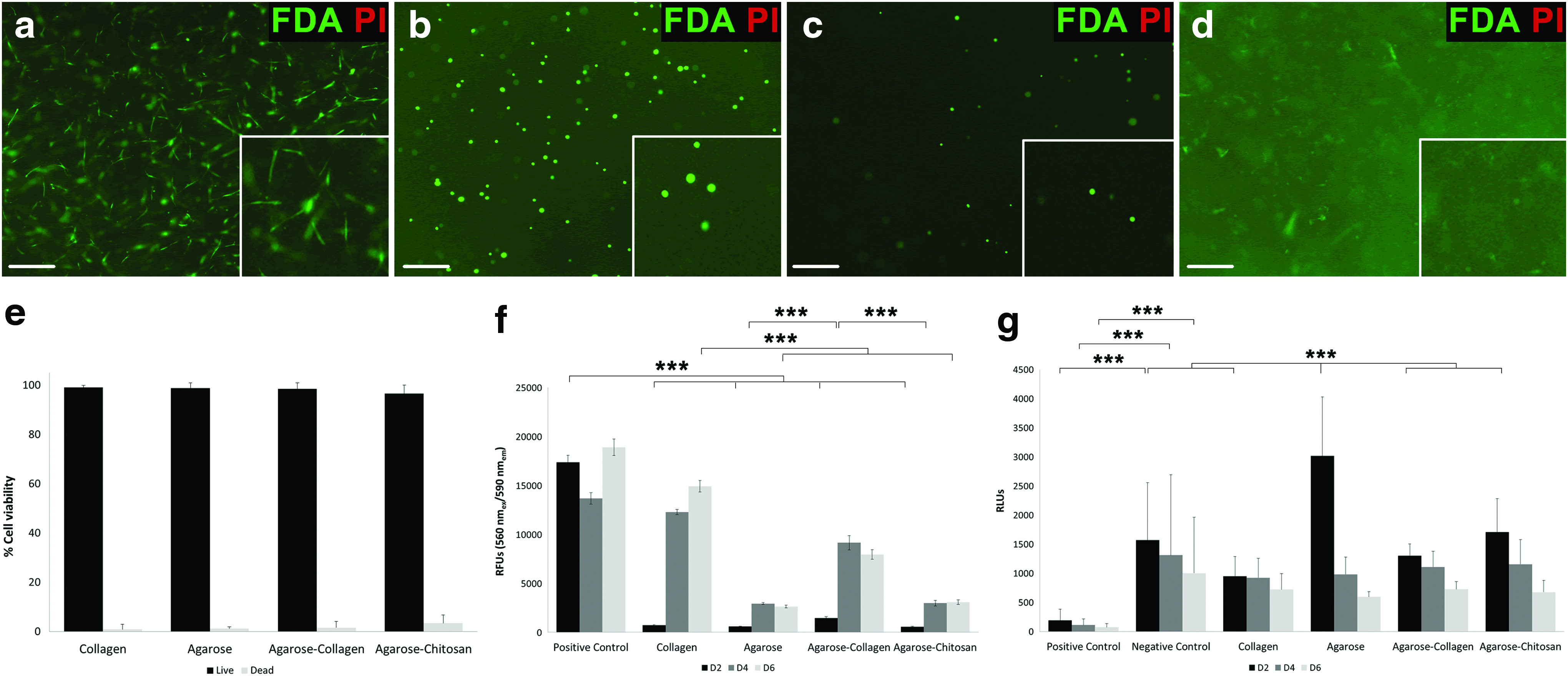
Cytocompatibility of cell-laden hydrogels. Live/dead staining of MSC-laden
Spatial cell morphology and distribution
Using TPLSM it was possible to reconstruct the 3D distribution of MSC inside the hydrogels (Figs. 5a–d and 6). MSC within collagen gels showed the elongation of filopodia and were in general bigger compared to cells in agarose and agarose hybrid matrices (Fig. 5f). Interestingly, the biggest cells were found in collagen independently of the culture conditions, followed by cells encapsulated in agarose and agarose–collagen gels upon adipogenic induction. We observed a homogeneous cell distribution inside all the hydrogels. Nevertheless, specimens subject to adipogenesis appeared to have lower cellularity compared to the ones undergoing osteogenesis and controls.

Evaluation of 3D cell morphology, volume, spatial distribution, and expression of GFP actin by two-photon laser scanning microscopy (TPLSM). TPLSM photographs of MSC-laden

Assessment of cell spreading and distribution by TPLSM. MSC-laden collagen
To investigate the relation between migration and differentiation potentials, cells were stained with GFP-actin and the intensity levels expressed in each sample were calculated (Fig. 5e). In theory, undifferentiated stromal cells show more migration capability than differentiated cells. Collagen showed, very often, the lowest actin expression among the others, independently of the culture conditions. Focal adhesion formations were mostly found in collagen samples. Both cell-laden agarose and agarose–collagen hydrogels revealed actin downregulation when cultured in differentiation media compared with the controls. Contrarily, collagen specimens cultured in differentiation media showed actin upregulation. Exceptionally during adipogenesis, MSC-laden agarose–chitosan showed lower actin expression compared with the controls.
MSC differentiation potential toward osteoblasts in hydrogels
The potential of MSC to differentiate toward the osteogenic lineage was accessed qualitatively by macroscopic and microscopic histological evaluation as well as quantitatively by measuring alkaline phosphatase activity, collagen production, and immunoexpression of α-smooth muscle actin. Gene expression toward the osteogenic fate was assessed by semiquantitative qPCR. Once cell viability was assured after 3D printing (Fig. 4), cell-laden hydrogel discs were cast in 24-well plates and cultured in osteogenic differentiation medium for up to 21 days. Alkaline phosphatase activity increased significantly in all cell-laden hydrogels subject to osteogenic differentiation compared with the controls (Fig. 7a). Additionally, alkaline phosphatase was expressed in a comparable amount in all cell-laden hydrogels incubated in adipogenic induction medium for 21 days.
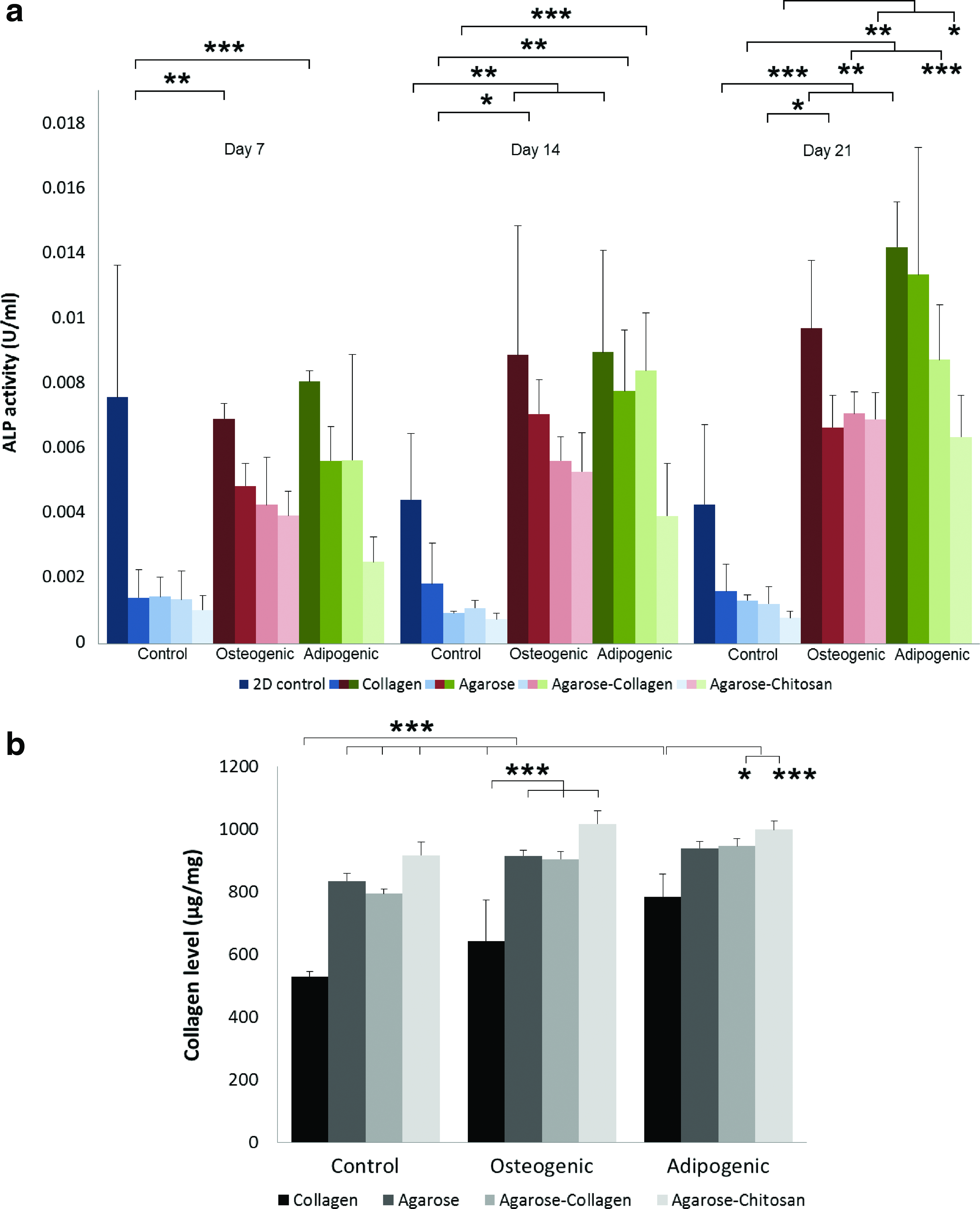
Measurement of alkaline phosphatase and collagen levels after incubation in differentiation media.
Hydroxyproline content was measured for all samples after an incubation of 21 days (Fig. 7b). Hydroxyproline is a main component of collagen, where it serves to stabilize the helical structure. The measurement of hydroxyproline levels can be used as an indicator of collagen content. Elevated hydroxyproline levels are indicators for increased collagen turnover. In this study, osteogenically differentiated MSC in hydrogels expressed higher collagen levels compared with the controls. Interestingly, cell-laden agarose and agarose hybrid hydrogels revealed significantly higher extracellular collagen production compared to MSC encapsulated in collagen gels.
The expression of cytoplasmic actin was evaluated as an indicator of cell migration (Fig. 5e). Undifferentiated MSC in agarose and agarose–collagen hydrogels showed higher actin expression compared with osteogenically differentiated samples (Fig. 5e). Contrarily, MSC-laden collagen and agarose–chitosan gels (controls) revealed lower actin expression compared with samples undergoing osteogenic differentiation. Human MSC has a decreased migration capability when incubated in agarose and agarose–collagen hydrogels for up to 21 days in osteogenic differentiation medium. Yet, osteoblasts encapsulated in collagen and agarose–collagen hydrogels had comparable actin expression. Additionally, cell migration within collagen hydrogels was confirmed by the measurement of cell volume (Fig. 5f). MSCs encapsulated in collagen gels showed, in general, a higher cell volume. There was, however, no significant difference between cell volumes of undifferentiated MSC within collagen gels compared to osteoblasts after 21 days of incubation. Nevertheless, osteogenic MSC-laden agarose samples showed higher cell volumes compared with the controls.
Histologically, we observed a lower cellularity in agarose and agarose blends, both with and without osteogenic induction (Fig. 8f). There was a significant increase of the cell number in osteogenic MSC-laden collagen gels compared to the control. The presence of elastic fibers in osteogenic collagen samples was verified by EvG staining (Fig. 8a). In addition, calcium accumulations (marked with arrows) were detected in osteogenic collagen and agarose–chitosan samples after Alizarin Red staining (see inserts in Fig. 8a, c). Cells from osteogenic differentiation samples were α-smooth muscle actin positive (Fig. 8b), which is an indicator of a myofibroblastic phenotype, capable of cell migration and matrix remodeling. Osteogenic differentiation of MSC maintained positive immunohistochemical reaction for the intermediate filament vimentin (Fig. 8c), which is a marker of mesenchymal derived cells.
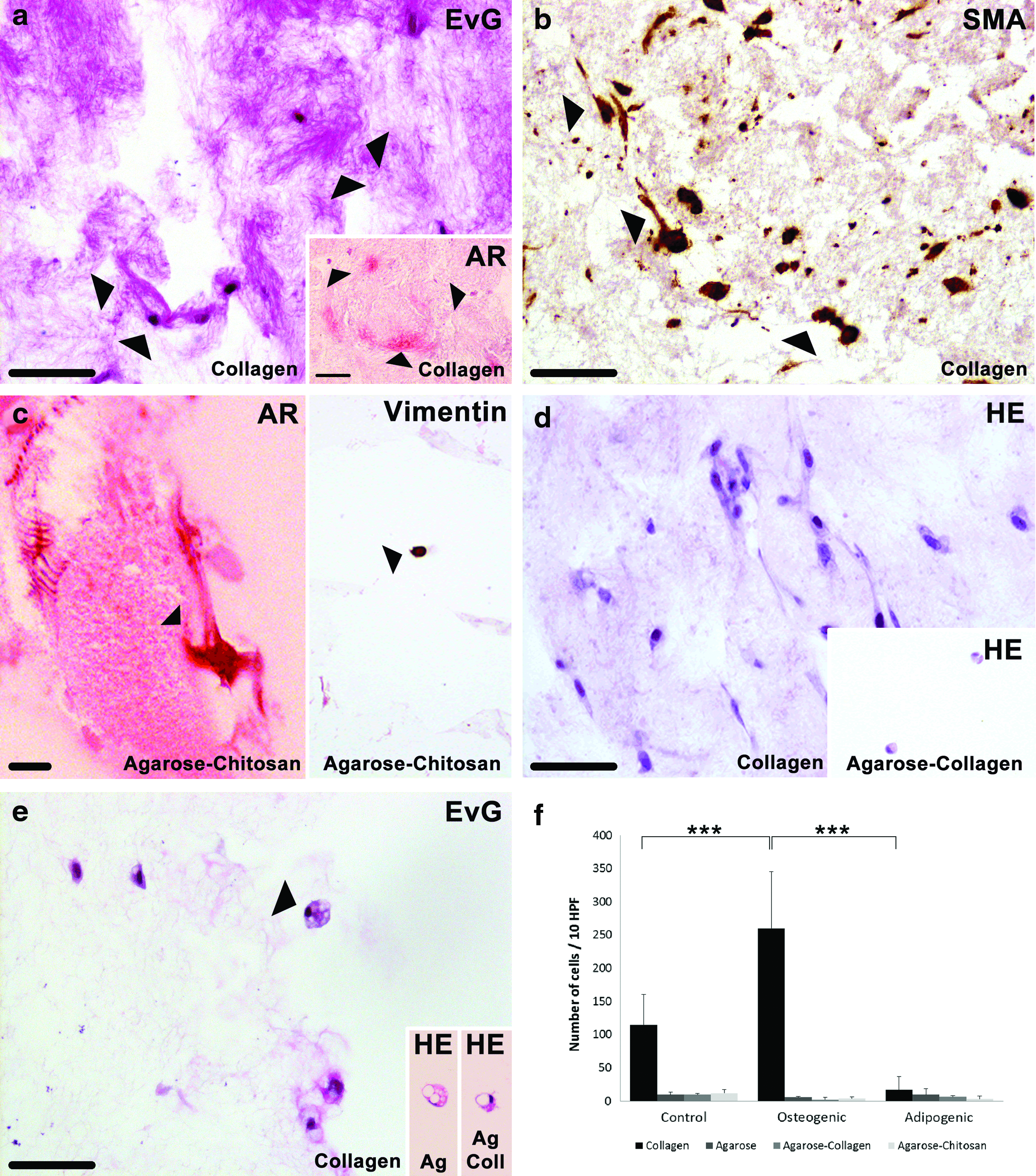
Histological and immunohistochemical analysis.
Real-time PCR conducted for the osteogenic differentiation markers alkaline phosphatase and osteocalcin revealed similar gene upregulation patterns both for MSC cultured in collagen and agarose–collagen hydrogels (Fig. 9a, d). After 21 days in culture, the levels of osteopontin were mostly expressed in adipogenic MSC encapsulated in collagen (Fig. 9e), and were statistically higher than in osteogenic and adipogenic MSC in agarose–collagen gels. The early osteogenic marker RUNX2 was significantly more expressed in osteogenic MSC in collagen hydrogels compared with both adipogenic and osteogenic MSC in agarose–collagen blends. The expression of certain osteogenic markers, such as osteocalcin, alkaline phosphatase, and osteopontin was comparable in osteogenic and adipogenic MSC independently of the type of matrix.
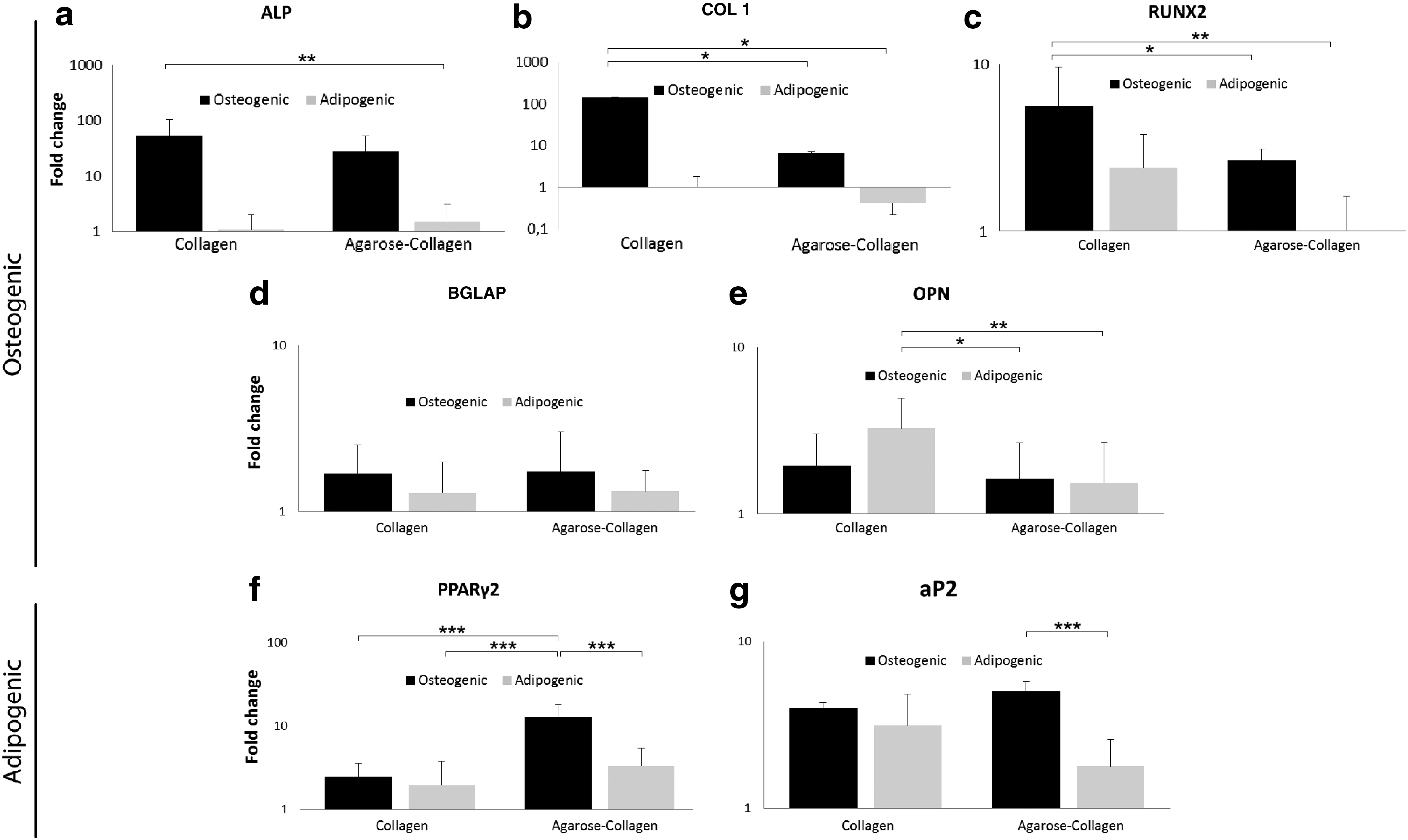
Gene expression in agarose and agarose–collagen gels after an incubation of 21 days. The osteogenic genes
MSC differentiation potential toward adipocytes in hydrogels
MSC differentiation toward adipocytes was evaluated both qualitatively and quantitatively, similarly as performed with the osteogenic differentiation assays. Macroscopically, cell-laden hydrogels in 24-well plates which underwent adipogenic differentiation for up to 21 days appeared more yielding and pulpy compared with both controls and osteogenic samples (data not shown). Alkaline phosphatase activity increased significantly in all cell-laden hydrogels subject to adipogenic differentiation for 21 days compared with the controls (Fig. 7a). Additionally, alkaline phosphatase was expressed in a comparable amount in collagen, agarose, and agarose–collagen cell-laden hydrogels incubated in adipogenic induction media for 21 days. The activity of alkaline phosphatase was, however, significantly lower in adipogenic agarose–chitosan samples compared with both adipogenic collagen and agarose specimens.
MSC-laden hydrogels undergoing adipogenic differentiation expressed higher collagen levels compared with the controls after 21 days of incubation (Fig. 7b). Cell-laden agarose–collagen and agarose–chitosan hydrogels revealed significantly higher extracellular collagen production compared with MSC encapsulated in collagen gels.
The expression of cytoplasmic actin in adipogenic differentiation cell-laden collagen hydrogels was higher compared with the control (Fig. 5e). Contrarily, adipogenic MSC embedded in agarose and agarose–chitosan gels revealed lower actin expression compared with undifferentiated MSC. Nevertheless, adipocytes within collagen and agarose gels expressed actin in comparable amount. Relatively to cell volume within hydrogels, the values were equivalent independently in which hydrogel cells were embedded (Fig. 5f). Adipocytes in collagen hydrogels were slightly bigger than in other gels, yet there was no significant cell volume difference.
Histological evaluation revealed, in general, a lower cellularity in all the samples with adipogenic induction (Fig. 8f). There was a higher cell number within adipogenic induced MSC in agarose hydrogels, yet not statistically significant. Human MSC induced to differentiate toward the adipogenic lineage maintained positive immunohistochemical reaction for the intermediate filament vimentin (data not shown). The formation of multivacuolated cells was detected in collagen, agarose, and agarose–collagen hydrogels cultured in adipogenic media for up to 21 days.
The expression of fatty acid binding protein (aP2) was found the highest in MSC incubated in agarose–collagen hydrogels after osteogenic induction for 21 days (Fig. 9g). This result is statistically higher than in adipogenic MSC embedded in the same type of hydrogel. Also, the adipogenesis marker PPARγ-2 is expressed the highest in osteogenic MSC agarose–collagen matrices. The latter result in osteogenic MSC in agarose–collagen is significantly higher than adipogenic MSC embedded both in collagen and agarose–collagen, as well as significantly higher than osteogenic MSC in collagen gels.
Discussion
Agarose and agarose–collagen hydrogels as printable materials for mesenchymal tissue substitutes
New concepts for regenerative medicine such as drug and tumor model testing ought to be, in the coming years, one of the most useful matters in which 3D bioprinting is employed. Even more encouraging, yet challenging, is the hope to deliver fabricated body organs on request. Hence, the generation of living tissue-like structures that mimic the natural environment of human organs is a key issue. Not only the mechanical stability of the supporting biomaterial matrix (i.e., hydrogels) is important for such applications, but also the biological performance of the containing cells. In this study, we focused on physical and biochemical effects on MSC interaction with nonblended and blended supporting materials for 3D biofabrication.
Three-dimensional viable tissue-like objects can be manufactured based on CT or MRI volumetric medical images. 10 Subsequently, processing algorithms are used to create a special model that is exported to a 3D bioprinter. The printing resolution is a fundamental concern in this technology. Printers or bioprinting technologies that fail to deliver accurate tissue-like specimens may be breaking down the success of specialized tissue manufacturing for individual use. Besides good cytocompatibility and dispensability, cell-laden hydrogels as bioinks must have satisfactory printability characteristics. According to this, we demonstrated an enhanced uniformity of nonblended and blended agarose hydrogels compared to the biologically well-established individual collagen gels. Previously, other research groups used collagen hydrogels applied to 3D additive manufacturing for rather small or planar constructs which did not require a stable shape.34–36 However, nonblended collagen and chitosan hydrogels lack fast gelling properties, which ultimately slows or disables printability.19,22 In our experiments both collagen and chitosan gels alone often leaked before polymerization was completed. Nevertheless, this problem seemed to be overcome when both materials were conjugated with agarose.
Moreover, agarose and agarose–collagen hydrogels revealed excellent stabilities after standard incubation times. Agarose–chitosan blends showed, however, worse results in respect to stability compared to agarose and agarose–collagen. This may be explained by the porosity of chitosan blends, which allows for the migration of inner nonconnected molecules and eluted degradation products. 37 For this reason, agarose–chitosan was not considered printable and excluded from further experiments, albeit, it might potentially be an osteogenic and adipogenic differentiation promoting material. Also printing submerged in perfluorocarbon influences the diameter and height of 3D printed agarose–collagen samples, as shown in previous studies.15,16 This may be advantageous when scaling up size and complexity compared with other techniques. 38
Cytocompatibility of hydrogels for 3D bioprinting
Novel hybrid hydrogels for biofabrication must be cytocompatible. Collagen and agarose blends tested positive for cytocompatibility, whereas chitosan specimens were slightly below the trend. Nonetheless, no statistical differences were found for the different hydrogels and consequently there are no major concerns that could yield to an exclusion of these for tissue engineering. The higher proliferation rate in collagen and agarose–collagen hydrogels is remarkable compared with individual agarose and agarose–chitosan. One possible explanation for the increased proliferation in collagen-containing matrices is related to the presence of RGD-peptide ligands, which contribute to an improved cellular function, including proliferation, migration, ECM synthesis, and tissue remodeling. It is a result of more and better adhesion points of cells to the biomaterial. This is in accordance with previous observations that have shown differences on cellular performance whether cells were embedded in peptide ligand-based hydrogels or in the matrices alone. 39 For this reason, we endorse the sense of employing agarose–collagen blends for 3D human mesenchymal tissue manufacturing.
Hydrogel properties influence MSC differentiation fate
In the literature three main features are described which may influence stromal cell fate and hence a controlled tissue morphogenesis in 3D cultures: (1) matrix stiffness, (2) cell shape (spindle-like vs. polygonal), and (3) chemical stimulation (growth factors).30,40 In this work biomaterials with varying mechanical stiffness (3.9–7.4×104 Pa40,41) and microstructures were applied, as well as different chemical stimulators. The conjugation of agarose to type I collagen formed a fine web matrix that is interspersed among the collagen meshwork. According to Lake et al., agarose does not seem to change the formation on structural topology of collagen fibers, nor does the self-assembly of the latter appear to disrupt agarose gelation. 42 The three component mixture of agarose, collagen, and water showed macroscopic signs of a higher resistance to compression than the two-component system collagen–water, which may be an interesting characteristic for hard- as well as soft-tissue engineering.
When it comes to the choice of a supporting hydrogel for a specific application, such as fat, cartilage, or bone tissue, its effects featuring cell shape and matrix mechanics are of main impact. The surrounding environment of a stromal cell niche is essential for the ultimate direction of their differentiation fate. Hence, the mechanical and physicochemical effects of biomaterials on cell fate are crucial parameters for 3D tissue engineering and especially for 3D bioprinting. Stiffer materials seem to influence MSC toward the osteogenic path, whereas on the contrary softer matrices trigger adipogenesis.43,44 In alginate hydrogels MSC differentiation is dictated by matrix stiffness irrespective of cell morphology, once cells remain rounded independently of stiffness. 29 Hence, fate is additionally regulated by mechanical properties which are consequently dependent on the type of hydrogel. Moreover, the hydrogel's degradation influences cell traction, allowing for the assembling of focal adhesion and cytoskeletal structures. 20 Undifferentiated stromal cells show higher migration ability in two-dimensional (2D), whereas osteogenic differentiation decreases cell motility. 45 In this study, in the presence of differentiation media, MSC embedded in hydrogels showed lower actin stress fiber expression, which corresponds to a lower motility. This was a first positive indicator of cell differentiation toward osteogenesis or adipogenesis.
On the one hand, scaffolds which promote cell spreading such as a meshwork of collagen linear fibers are more utilized for musculoskeletal or neural applications, 46 because they force a physical and subsequent phenotypic cell change to a rather spindle-like morphology, typical for osteoblasts. On the other hand, a mesh of porous and quasi-rigid fibers typical for polysaccharide polymers are more suitable for hosting large polygonal cells such as multivacuolated adipocytes. 30 This evidence was presented by Lee et al. where restricted spreading enhanced signaling associated to adipogenesis. Matrices with similar stiffness such as collagen and fibronectin promote neurogenesis and adipogenesis, respectively. 30
Differentiation tendency toward osteogenic and adipogenic lineages in 3D printable hydrogels
Our study shows that embedded MSC in soft collagen hydrogels which were chemically forced to differentiate toward the osteogenic fate showed higher upregulation of ALP and collagen I genes, in comparison to the ones with adipogenic induction. Interestingly, the presence of osteogenic growth factors in culture, such as dexamethasone, was less effective compared to the influence of cell shape in osteoblast differentiation, as shown by qPCR. MSCs in large fiber soft collagen matrices could better elongate and hence adapt an osteogenic phenotype than when embedded in stiffer agarose hydrogels. This evidence is in accordance with Kumar et al. who proved MSC osteogenesis in fibrous matrices in the absence of osteogenic supplements and, therefore, indicating the role of cell shape in the differentiation fate.47,48 COL 1 and RUNX2 gene expression levels were mostly expressed in osteogenic MSC in collagen gels possibly due to a combination of an elongated cell geometry and chemical induction. Subsequently, the stiffness of the supporting material also seemed to influence MSC phenotype, but less effectively in osteogenic induced samples. This was proved by the significantly higher expression of osteopontin after 21 days in culture of adipogenic MSC in collagen (softer) in contrast to cells in agarose–collagen (stiffer). The loose meshwork of collagen gels partially allowed for cell spreading and phenotype change toward the osteogenic lineage even when adipogenic growth factors were applied. This was shown before in 2D cultures by Köllmer et al. who described an overlapping osteoblast- and adipocyte-associated gene expression. 49
The differentiation from preadipocytes to adipocytes is triggered by changes in cell shape, the expression of adipogenic specific genes, and the evidence of lipid accumulation. 50 Cells hosted in agarose hydrogels hold a polygonal rounded shape which is also characteristic for adipocytes. The change to a rather round shape seemed more likely to promote the expression of aP2 and PPARγ-2 in MSC in agarose–collagen blends than in individual collagen scaffolds, even when the incubation conditions were chemically defined to enhance osteogenesis. Nevertheless, softer supporting networks could possibly be more suitable for adipogenesis once osteogenic MSC in agarose–collagen and in collagen express equivalent levels of the adipogenic gene aP2, whether they were adipogenically or osteogenically induced. Beyond the changes in cell shape and the expression of adipogenic genes, we detected the formation of multivacuolated adipocytes in collagen, agarose, and agarose–collagen blends as proved by histology, suggesting that any of these matrices could be accepted for the fabrication of 3D bioprinted fat tissue.
In this work we have shown that on-demand fabrication of novel tissues depends on specific features such as hydrogel printability, matrix mechanics, and chemical factors from the beginning until the end of the process line. To make use of a 3D bioprinter for the creation of tissue-like objects it is essential to achieve a successful cell-supporting material printability, allowing for the freeform fabrication of individual structures. By drop-on-demand bioprinting, only agarose and agarose–collagen hydrogels have shown satisfactory results.
Moreover, not all printable inks are suitable for a desired application. Up to now, housing stromal cells in such scaffolds is limited to certain applications such as fabrication of bone, cartilage, fat, and capillaries. Osteogenesis seemed to be preferably achieved in anisotropic collagen-rich substrates, whereas adipogenesis trended to be mostly expressed in isotropic agarose-rich matrices. These results reveal that also cell morphology seems to have an effect guiding the differentiation fate in 3D mesenchymal cultures, as do matrix mechanics, and chemical induction.
Table 3 shows osteogenic and adipogenic differentiation tendencies of MSC within each of the studied hydrogels. By pointing out agarose and agarose–collagen as the most promising matrices in terms of printability, it is possible to distinguish an improvement on both osteogenic and adipogenic differentiation potential in the combination of type I collagen with agarose compared to agarose alone. The addition of type I collagen fibers to agarose hydrogel seems to increase the expression of genes related to osteogenesis, which were phenotypically visible through a slight change in cell shape. Additionally, the decrease in mechanical stiffness as well as the maintenance of a polygonal cell shape as a result of the latter combination is partly contributing to an enhanced MSC adipogenesis.
Each parameter was assigned as follows: good/positive (++ ), fair/neutral (+), and poor/negative (−), with the exception of microstructure that was attributed as fibrous (F) or porous (P) organization.
Future experiments for this work will include the development of new printable hydrogels. For example, hydrogels consisting of a higher content of collagen conjugated with a lower content of agarose and calcium phosphate particles for promoting osteogenesis. Also the conjugation of a higher content of agarose with fibronectin or laminin will be investigated for enhancing adipogenic differentiation. By scaling up the size of 3D printed objects using submerged conditions cell hypoxia may deserve more attention, because it additionally affects MSC differentiation. 51 Therefore, branched endothelial- and MSCs-derived pericyte-rich printed capillaries are to be combined in a more complex 3D coculture.
Conclusions
The use of 3D bioprinted objects for medical research is currently an increasing demand. Human mimic tissues with personalized 3D shape and appropriate functionality are to be requested as disease testing models or replacement organs in patients. Therefore, cell-laden hydrogels used for 3D printing must (1) be accurately and reproducibly printable (individuality) and (2) allow for cell differentiation guidance according to the desired application (functionality). In this study, agarose hydrogel showed to be essential for the fabrication of such 3D objects that allowed adipogenesis by cell shape confinement and osteogenesis by the conjugation with type I collagen. In conclusion, osteogenesis seemed to be preferably achieved in anisotropic soft collagen-rich substrates, whereas adipogenesis trended to be mostly expressed in isotropic stiff agarose-rich matrices. The conjugation of type I collagen to agarose with varying ratios might, therefore, be suitable for a broad range of 3D printed mesenchyme-derived tissue objects. This is of importance as biomanufactured tissues will be used as functional models for disease modeling, replacement of animal testing, and ultimately in the future even implanted in patients to replace failed tissues or organs.
Footnotes
Acknowledgments
This work was supported by the two-photon imaging facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University. The authors thank Roswitha Davtalab and Jose Gerardo-Nava (Dental Materials and Biomaterials Research, RWTH Aachen Universtity Hospital) and Norina Labude (Institue of Pathology, RWTH Aachen University Hospital) for the kind technical assistance in the cell culture lab.
Disclosure Statement
No competing financial interests exist.