
Abstract
Culture microenvironment plays a critical role in the propagation and differentiation of human embryonic stem cells (hESCs) and their differentiated progenies. Although high efficiency of hESC differentiation to keratinocytes (hESC-Kert) has been achieved, little is known regarding the effects of early culture microenvironment and pertinent extracellular matrix (ECM) interactions during epidermal commitment on subsequent proliferative capacity of hESC-Kert. The aim of this study is to evaluate the effects of the different ECM microenvironments during hESC differentiation on subsequent replicative life span of hESC-Kert. In doing so, H1-hESCs were differentiated to keratinocytes (H1-Kert) in two differentiation systems. The first system employed autologous fibroblast feeder support, in which keratinocytes (H1-KertACC) were derived by coculture of hESCs with hESC-derived fibroblasts (H1-ebFs). The second system employed a novel decellularized matrix from H1-ebFs to create a dermoepidermal junction-like (DEJ) matrix. H1-KertAFF were derived by differentiation of hESCs on the feeder-free system employing the DEJ matrix. Our study indicated that the feeder-free system with the use of DEJ matrix was more efficient in differentiation of hESCs toward epidermal progenitors. However, the feeder-free system was not sufficient to support the subsequent replicative capacity of differentiated keratinocytes. Of note, H1-KertAFF showed limited replicative capacity with reduced telomere length and early cellular senescence. We further showed that the lack of cell–cell interactions during epidermal commitment led to heightened production of TGF-β1 by hESC-Kert during extended culture, which in turn was responsible for resulting in the limited replicative life span with cellular senescence of hESC-Kert derived under the feeder-free culture system. This study highlights for the first time the importance of the culture microenvironment and cell-ECM interactions during differentiation of hESCs on subsequent replicative life span and cellular senescence of the differentiated keratinocytes, with implications for use of these cells for applications in tissue engineering and regenerative medicine.
Introduction
H
To utilize the potential of hESC-derived keratinocytes (hESC-Kert) for clinical applications, devising direct and efficient differentiation methods in the specified xeno-free system with high purity and scalability and complying with good manufacturing practice are necessary. Consequently, various strategies have been reported utilizing defined culture environments to direct the differentiation of hESCs to epithelial lineage.6,8,12 Epithelial commitment in feeder-free systems with defined culture milieu is the most promising approach to exclude or minimize the xenogeneic components. However, our prior study indicated that hESC-Kert differentiated under feeder-free condition exhibited significantly lower proliferative capacity than those differentiated under coculture feeder condition. 8 More recently, Selekman et al. reported greater expansion potential for hESC-Kert that were differentiated on synthetic extracellular matrix (ECM) in comparison with cells differentiated on Matrigel™. They concluded that the cell-ECM interactions are responsible for this discrepancy in proliferation. 12 These differences in growth ability suggest the significant influence of culture microenvironment during differentiation on the subsequent replicative capacity of the keratinocytes. The limited proliferative capacity of hESC-Kert derived in different culture systems6–8,12 is likely to pose a major obstacle for future clinical applications of these cells. Elucidating the proliferative capacity of hESC-Kert under different culture microenvironments and the underlying signaling mechanisms leading to cellular senescence would essentially pave the development of these cells for wider applications, including engineering of skin tissue grafts.
Herein, to differentiate H1-hESCs to keratinocytes in defined culture milieu, we established an autogenic feeder-free (AFF) system by applying a novel decellularized dermoepidermal junction-like (DEJ) matrix extracted from H1-hESC-derived fibroblasts (H1-ebFs) under modified culture condition. H1-ebFs offer the greatest advantage of being able to provide unlimited supply of decellularized matrix to serve as an autogenic microenvironment for epidermal differentiation and commitment of hESC-Kert.
In this study, we investigated the differentiation and replicative capacity of H1-hESC-Kert under the coculture feeder system (H1-KertACC) and feeder-free system (H1-KertAFF). We hypothesized that the culture microenvironments used for keratinocyte differentiation of hESCs might have significant downstream effects on the subsequent replicative life span and cellular senescence of the differentiated keratinocytes.
Materials and Methods
Culture of hESCs
H1-hESCs were obtained from WiCell Research Institute and cultured on mouse embryonic fibroblasts (MEFs) as described previously.
13
Briefly, hESCs were seeded on inactivated MEFs in ES medium comprising DMEM-F12 (Biowest) with 20% knockout serum replacer supplemented with 1 mM
Establishment of autogenic coculture environment and H1-KertACC differentiation
The H1-ebF feeder was prepared as described previously.14,15 For autogenic coculture (ACC) environment establishment, H1-ebFs were inactivated after eight passages before seeding at a cell density of 1.4–1.6×105 cells per well on gelatin-coated six-well plates in fibroblast growth medium comprising Dulbecco's modified Eagle's medium (DMEM) and 10% fetal bovine serum (FBS; Biowest). H1-hESCs were seeded on each well and differentiated in the presence of flavinoid adenine dinucleotide (FAD) medium comprising 3:1 DMEM:DMEM-F12 supplemented with 50 mg/mL
Establishment of AFF environment and H1-KertAFF differentiation
To establish the AFF microenvironment, the DEJ decellularized matrix extracted from H1-ebFs has been used. To prepare the decellularized matrix, 2.5×105 H1-ebF cells were seeded onto each well of human collagen I-coated six-well plates in fibroblast growth medium. Following overnight incubation, the fibroblast growth medium was changed to crowding medium. In this study, Ficoll was used as crowding agent as reported by Zeiger et al. 16 . Briefly, Ficoll cocktail comprising 37.5 mg/mL Ficoll 70 KDa and 25 mg/mL Ficoll 400 KDa (GE Healthcare Life Science) was added to DMEM medium supplemented with 0.5% FBS and 50 μg/mL AA. H1-ebFs were cultured 5–7 days under crowded condition. Cell lysis was carried out by four repeats of 15-min incubation with 0.5% sodium deoxycholate (Sigma) in 0.5× complete protease inhibitor (Roche Diagnostics GmbH) solution and two repeats of 15-min incubation on ice with 0.5% sodium deoxycholate in PBS. To remove DNA remnants, the monolayer matrix was incubated with DNAse solution comprising 10 mM Tris (pH 7.5), 2.5 mM MgCL2, 0.5 mM CaCl2, and 1 U/μL DNAse (Sigma) for 1 h at 37°C. For H1-KerAFF differentiation, H1-hESCs were seeded on ECM in the presence mTeSR™2 (STEMCELL Technologies). The hESCs were cultured for 2 days in mTeSR™2, and after 2 days the medium was changed to DKSFM supplemented with 1 μM RA and hESCs were cultured for an additional 10 days. The cells, after a period of 10 days, were transferred onto collagen IV-coated plates and expanded in only DKSFM. To investigate the effectiveness of DEJ matrix and different ECM components, culture plates were coated with 50 μg/cm2 collagen I (BD Bioscience) or 50 μg/cm2 collagen IV (BD Bioscience) or 5 μg/cm2 fibronectin (Life Technologies) or 20 μg/cm2 laminin (Sigma). hESC colonies were seeded on the DEJ matrix or respective ECM-coated plates and kept in DKSFM without supplementing RA for 10 days.
Replicative capacity
To determine possible difference in replicative capacity of hESC-Kert cultured in ACC and AFF systems, 1×105 cells per well were replated onto collagen IV-coated six-well plates and cumulative population doubling (PD) was calculated every 10 days as log2 (number of counted cells after 10 days/number of initial seeded cells). As there are differences in keratinocyte differentiation kinetics and efficiency in the two culture systems (ACC and AFF), hESC-Kert cultured in the two systems were matched based on the p63+ population to be more than 85% before replating onto collagen IV-coated plates in DKSFM for measurement of the replicative capacity.
Immunofluorescence staining
For immunofluorescence (IF) staining of monolayer culture, the cells were fixed in 4% paraformaldehyde (PFA) at room temperature for 15 min, followed by 10 min of permeabilization with 0.4% Triton X-100. Blocking was performed by 2% bovine serum albumin (BSA; Sigma) for 60 min at 4°C. Then, cells were incubated by primary antibodies at 4°C overnight. Cells were washed with 0.05% Tween-20 and incubated for 45 min with fluorochrome-conjugated secondary antibodies and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) before examination by an IX70 inverted fluorescence microscope (Olympus). For IF staining of DEJ matrix, under either crowded or uncrowded conditions, the similar protocol of IF staining was performed and images were acquired by the fluorescence microscope. The primary antibodies, suppliers, and working dilutions and their corresponding applications are listed in Table 1. Fluorescent secondary antibodies, Alexa Fluor 488 goat anti-mouse, Alexa Fluor 488 goat anti-rabbit, and Alexa Fluor 594 goat anti-mouse (all from Life Technologies), were used at 1:100 dilution.
Colony-forming assay
To determine the colony-forming ability of hESC-Kert at day 50, mitotically inactivated H1-ebFs were plated at 2.5×105 cells per well on gelatin-coated six-well plates in triplicate experiments and incubated overnight. The following day, 1000 viable cells in FAD medium were plated on H1-ebFs. After 8 days, the monolayer cultures were fixed with 4% PFA and washed thrice with PBS. The cultures were incubated with 1% rhodamine B (Sigma) for 20 min before acquiring images.
Flow cytometry analysis
Flow cytometry analysis was performed for p63, K14, K10, p16INK4a, and p21CIP1 protein expression. Briefly, after trypsinization, the same procedure of fixation, permeabilization, and blocking, as explained in the IF staining process, was carried out. Cell suspension was incubated with p63, K14, K10, and p16INK4a primary antibodies (Table 1) for 60 min at room temperature, followed by washing with 0.05% Tween in PBS. Subsequently, cells were incubated with 1:300 Alexa Fluor 488 goat anti-mouse or goat anti-rabbit for 30 min. For p21CIP1 staining, cells were incubated with FITC-conjugated p21CIP1 antibody for 60 min, then washed, and transferred to FACS buffer (Miltenyi Biotec). Samples were analyzed by DAKO cytomation CYAN LX using Dako summit v 4.3 software.
SA-β-Gal activity analysis
To measure the SA-β-Gal activity, the ImaGene Green™ C12FDG lacZ Gene expression kit (Molecular Probes) was used following the manufacturer's instructions. The monolayer culture was washed with PBS twice and images were captured by inverted fluorescence microscopy or cells were trypsinized and transferred to FACS buffer solution for flow cytometry analysis.
Telomere length analysis
Genomic DNA was extracted from H1-Kert at every 10-day interval until growth arrest, using the DNA extraction kit (Roche) following the manufacturer's instructions. Mean telomere length was measured by terminal restriction fragment (TRF) analysis using the TeloTAGGG telomere length assay kit (Roche) following the manufacturer's instructions. The telomere length was quantified according to standard molecular weight signals by ImageJ software v 1.47.
Enzyme-linked immunosorbent assay for TGF-β1
To measure the level of TGF-β1 in the culture supernatant, 1×105 cells were seeded in each well of a six-well plate. The expended medium was collected at every 5-day interval. Total and active TGF-β1 concentration was measured in triplicate samples using the TGF-β1 DuoSet ELISA development kit (R&D systems) according to the manufacturer's instructions.
Statistical analysis
All data were calculated and represented as mean±standard error (SE). Statistical significance between each group at different time points was evaluated by Student's t-test and one-way ANOVA analysis with p<0.05 considered as significant.
Results
Differentiation of hESCs to keratinocytes in ACC system
Successful differentiation of hESCs to epithelial lineage can be achieved through direct contact with an inducing lineage like fibroblasts and/or in the presence of the specific growth factor combination. 17 In this study, H1-hESCs were differentiated to H1-KertACC by coculturing hESCs on the H1-ebF feeder support. Each step was termed according to expression of p63 and K14 (Fig. 1A). Analysis by flow cytometry showed that by 10 days of differentiation, 25.0%±10.2% were p63+, but a very low percentage (1.1%±0.5%) of cells were K14+. By day 20, at the induction step, 85.3%±4.3% of cells were p63+ and 4.3%±1.5% of cells were K14+. With further differentiation, there was a robust increase in K14+ cells (4.3%±1.5% to 80.2%±5.2%) (p<0.05) between days 20 and 30 termed as the maturation step. However, the level of p63+ remained relatively consistent, with 82.4%±3.7% of cells remaining p63+ by day 30 of differentiation. With further culture at the expansion step, there was a decreasing trend in the number of p63+ cells (56.5%±7.7%), with an increasing trend of K14+ cells (87.2%±8.5%) by day 50 of differentiation. With extended culture, upon reaching the cellular senescence phase at day 120, there was a drastic decrease in number of p63+ cells (4.2%±2.4%), although majority of the cells remained K14+ (95.0%±2.2%; Fig. 1B). Consistently, IF staining showed high expression of K14 in H1-KertACC colonies at days 30 and 120 (Fig. 1C).
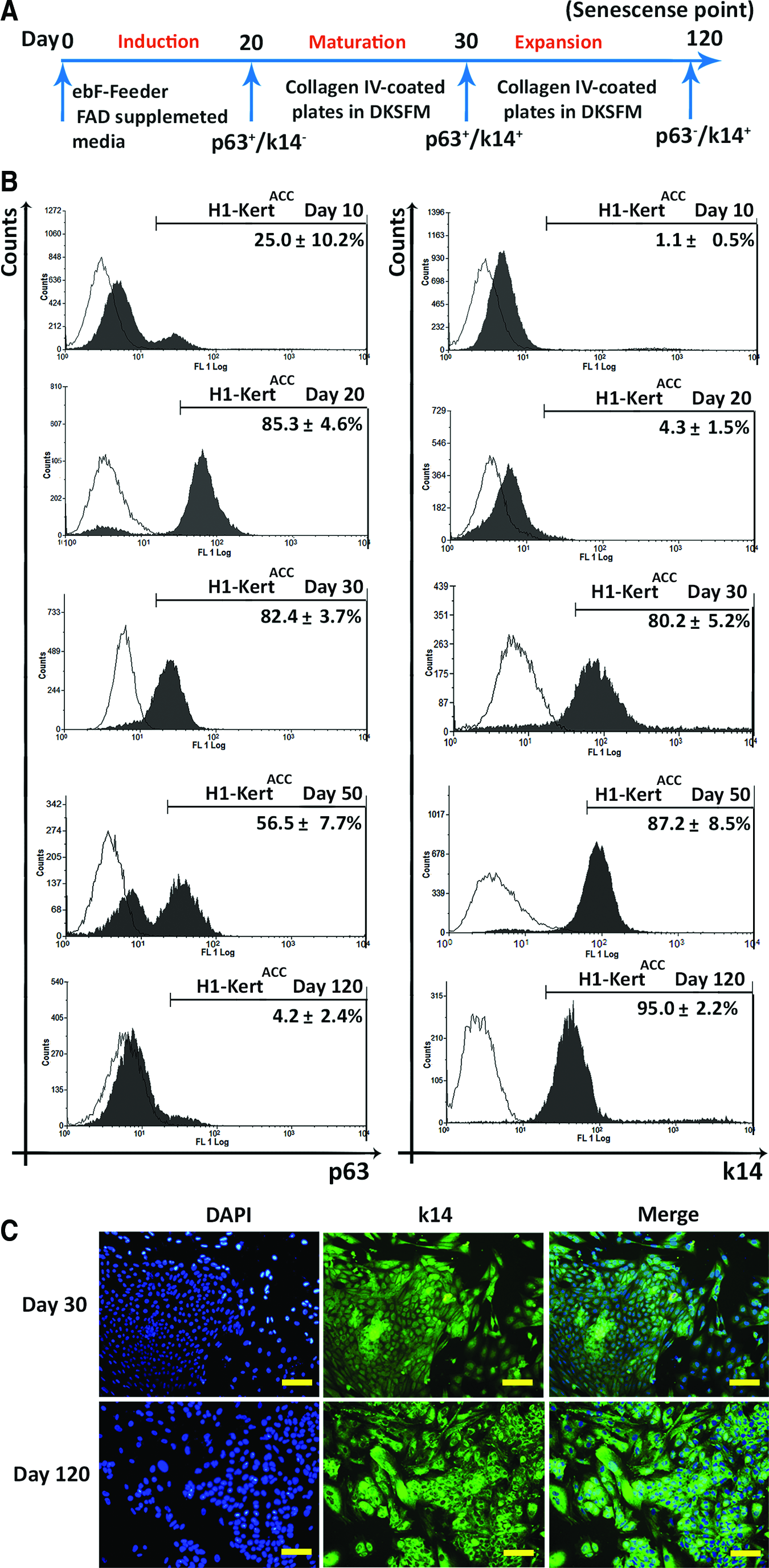
Differentiation of H1-hESCs to epithelial cells in the autogenic coculture (ACC) system.
Establishment of DEJ matrix and evaluation of matrix composition
To minimize the presence of xenogeneic components, the feeder-free system that employed the DEJ matrix from H1-ebFs was applied during differentiation of hESCs. To induce ECM deposition, we enhanced the medium crowding degree with the aid of Ficoll 70/400 cocktail. We observed that the H1-ebFs exhibited marked increase in deposition of cross-linked fibers of both dermal ECM components, collagen I and fibronectin, and basement membrane matrix proteins, including collagen IV, collagen VII, and laminin, after 6–7 days of culture in the crowded medium (Fig. 2A). To examine the effect of DEJ matrix to induce hESCs to epidermal progenitors, hESC colonies were cultured on DEJ matrix or different nonxenogeneic ECM-coated plates in DKSFM for 10 days. A significantly higher p63+ population was observed in cultured cells on DEJ matrix (p<0.05, compared with all groups) (Fig. 2B).

Establishment of basement membrane (dermoepidermal junction-like [DEJ]) matrix for autogenic feeder-free (AFF) system under Ficoll macromolecular crowded condition.
Differentiation of hESCs to keratinocytes in AFF system
The DEJ matrix, obtained under crowded condition, was utilized as an autogenic microenvironment to induce differentiation of H1-hESCs toward epidermal lineage (Fig. 3A). Comparing with the ACC system that yielded 85.3%±4.3% p63+ cells and 4.3%±1.5% K14+ cells after 20 days, the AFF system was able to induce a more rapid and efficient differentiation of hESCs to keratinocytes with higher percentages of p63+ (95.8%±3.3%) and K14+ (7.5%±2.4%) cells following only 10 days of induction. After 30 days of differentiation, 74.4%±5.0% of cells were p63+ and 90.4%±3.6% were K14+ at the end of the maturation step. Upon reaching cellular senescence at day 50, only low percentage of p63+ cells (8.5%±1.3%) could be detected, while majority of cells (91.6%±4.5%) remained positive for K14 (Fig. 3B). Furthermore, IF staining analysis at days 30 and 50 showed a high expression of K14 in H1-KertAFF colonies with enlarged and flattened morphology at the senescence phase (Fig. 3C).
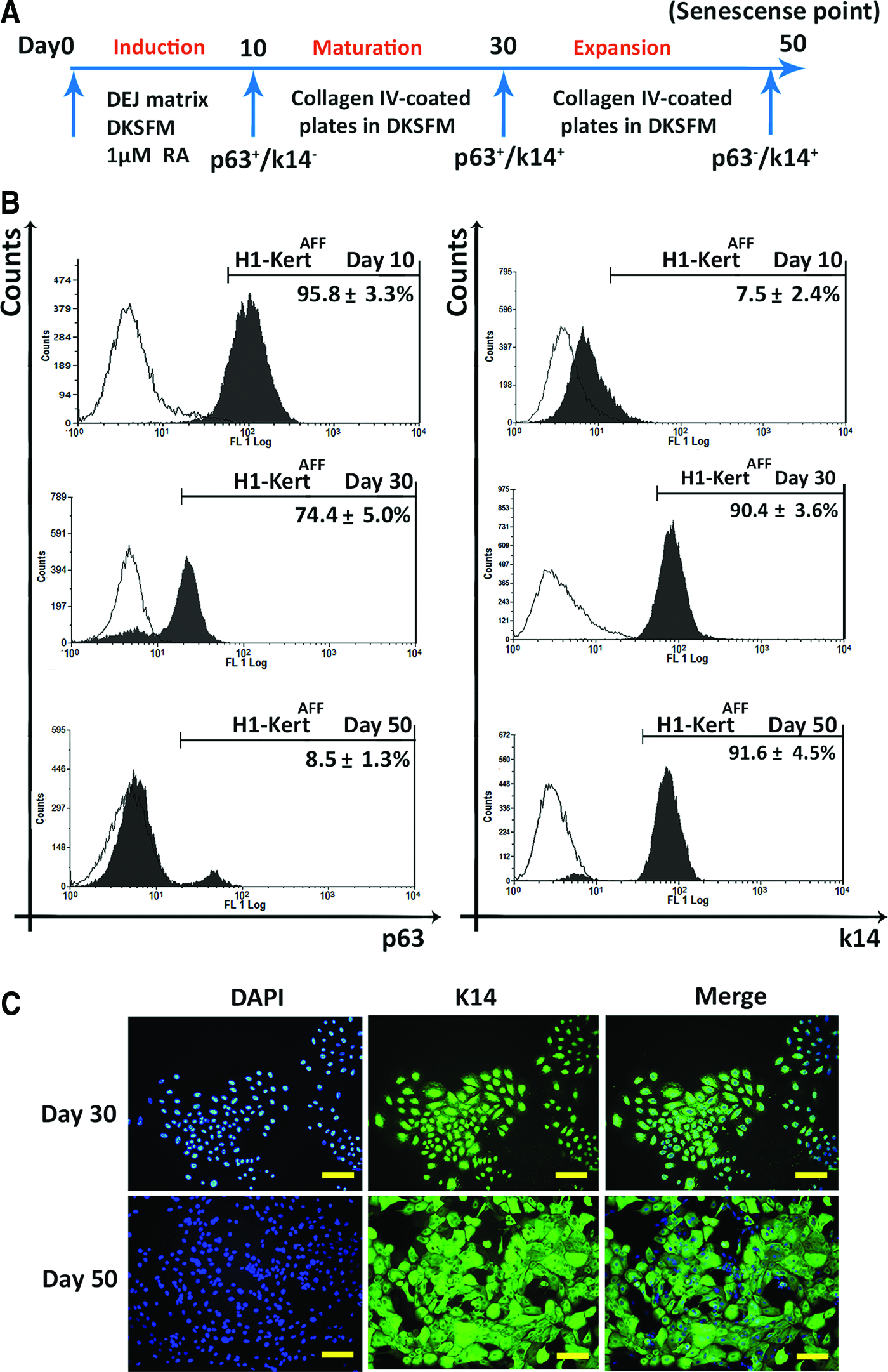
Differentiation of H1-hESCs to epithelial cells in the AFF system.
Evaluation of replicative capacity of hESC-Kert
To resolve the issues of differential keratinocyte differentiation kinetics and efficiency in the two systems (ACC and AFF), hESC-Kert differentiated under the two systems were matched based on expression of early keratinocyte marker, p63, to be more than 85% before replating onto collagen IV-coated plates for measurement of the replicative capacity. This will reduce the likelihood of having undifferentiated and other contaminant cell types affecting the measurements. Therefore, H1-KertACC were differentiated till day 20 of differentiation to yield 85.3%±4.3% p63+ cells, and H1-KertAFF were differentiated till day 10 of differentiation to yield 95.8%±3.3% p63+ cells, before replating onto collagen IV-coated plates and propagating under the same medium conditions. Extending the induction step until day 20 in the AFF system resulted in pronounced decrease in the population of epidermal progenitors, implying an unfavorable effect of overinduction in the AFF system (Supplementary Fig. S1A).
Following replating, H1-KertAFF were not able to be propagated by more than 8 PD over 50 days, while H1-KertACC were able to undergo 18 PD for up to 120 days (Fig. 4A). Furthermore, H1-KertACC besides retaining a significant p63+ population at day 50, demonstrated a much superior colony-forming ability compared with H1-KertAFF (Fig. 4B). To exclude the effect of the medium on limited replicative life span of H1-KertAFF, AFF differentiation medium was supplemented with the same concentration of AA and serum as that present in the ACC differentiation system. In a separate experiment, ACC differentiation medium was applied during epidermal commitment in the AFF system for 20 days. We were not able to obtain a representative population of p63+ and K14+ cells (Supplementary Fig. S1B).
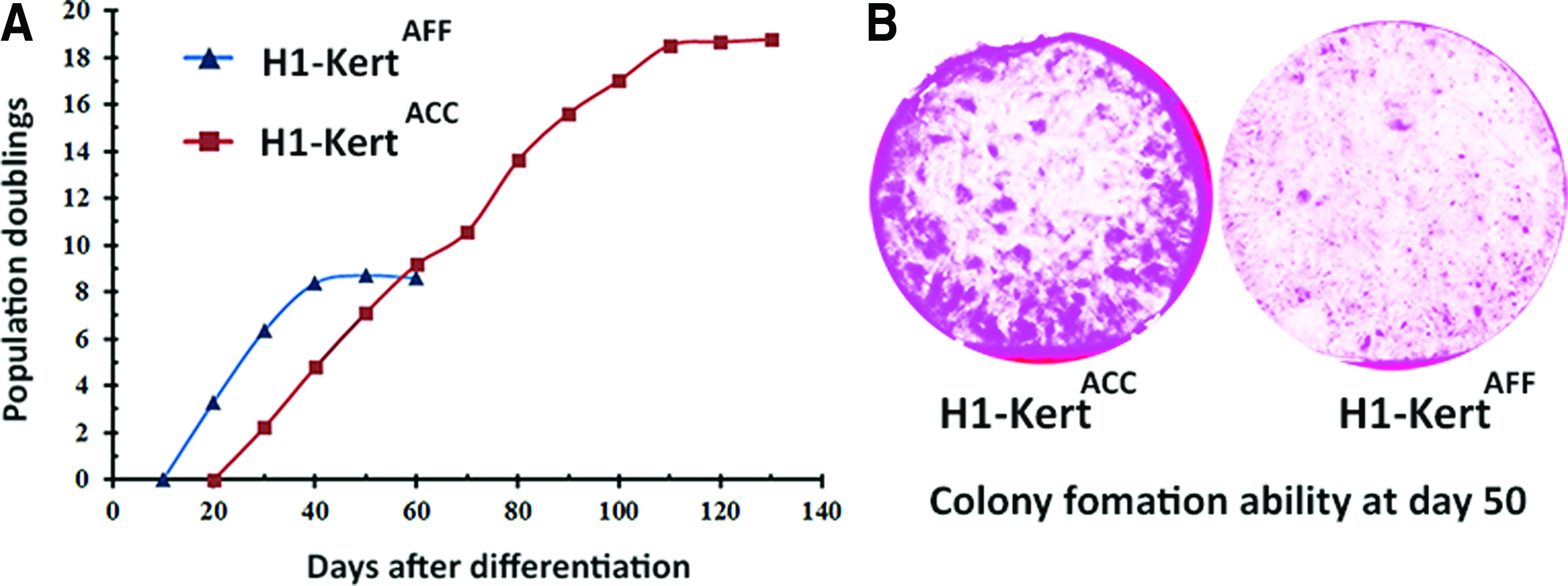
Evaluation of replicative capacity of hESC-Kert.
Cellular senescence of hESC-Kert and potential underlying mechanisms
The difference in replicative capacity of hESC-Kert derived under ACC and AFF systems led us to examine the role of extracellular microenvironments during epidermal commitment in the modulation of cellular senescence of hESC-Kert and the potential underlying mechanisms. It is well established that primary human keratinocytes will eventually enter the senescence phase with telomere loss during progressive subculturing.18,19
Mean telomere length analysis by Southern blot revealed a substantial decrease in telomere length from 10.14 to 8.55 Kbp (difference of 1.59 Kbp) by the H1-KertAFF over a period of 30 days. Conversely, in the H1-KertACC, there was only a gradual decrease in telomere length from 10.34 to 9.27 Kbp (difference of 1.07 Kbp) over a longer period of 50 days (Fig. 5A).
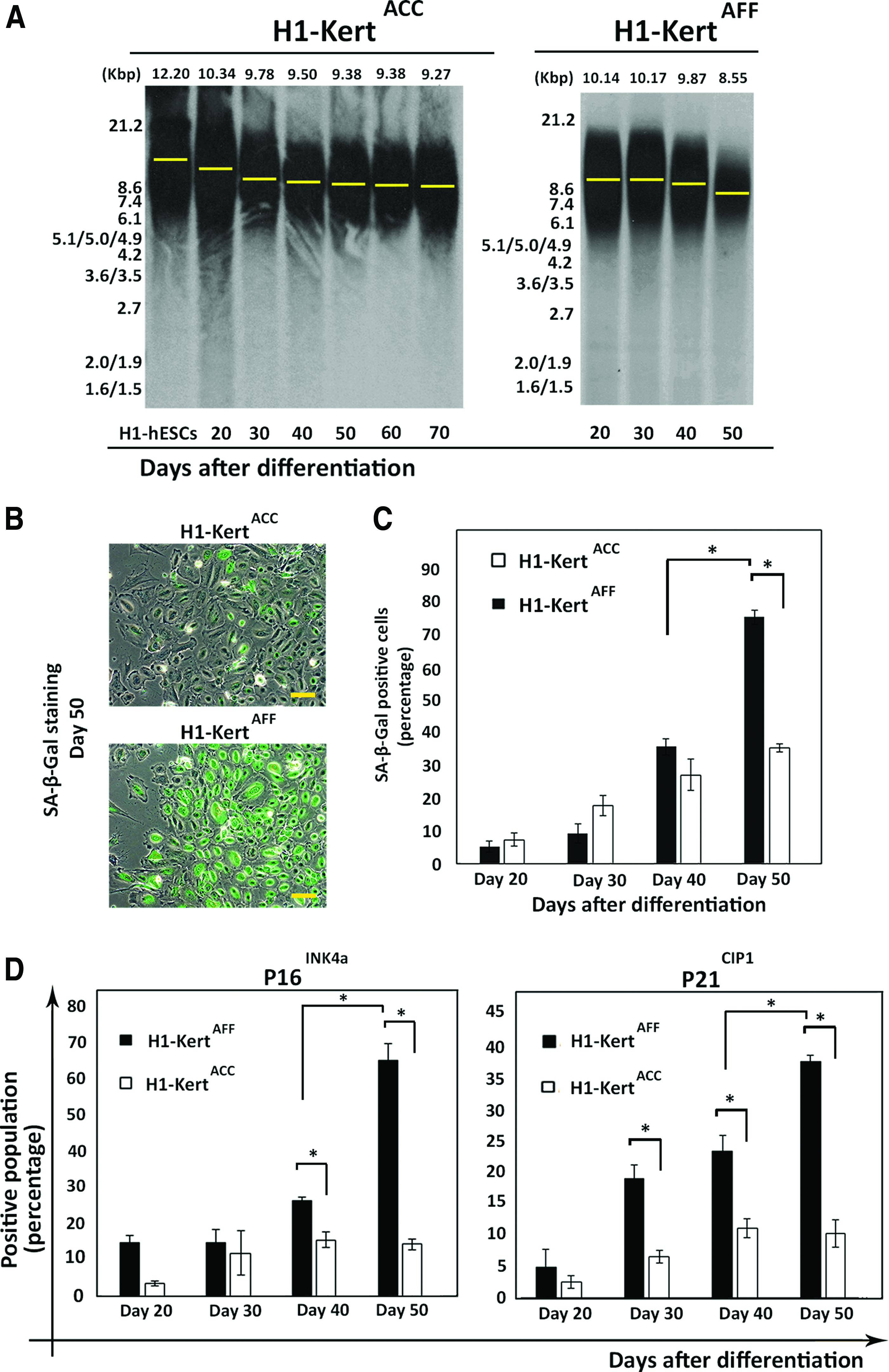
Cellular senescence of H1-Kert differentiated in ACC (H1-KertACC) and AFF (H1-KertAFF) systems and potential underlying mechanisms.
SA-β-Gal, the biomarker of cellular senescence, can be detected in replicative growth-arrested cells in vitro and in vivo. 20 Concomitant with growth arrest at day 50 of differentiation, SA-β-Gal cytochemical evaluation revealed high expression in H1-KertAFF, but not in H1-KertACC (Fig. 5B). To accurately measure SA-β-Gal activity, the SA-β-Gal+ population was quantified by flow cytometry analysis. Our results revealed a time-dependent increase in cells positive for SA-β-Gal by both H1-KertACC and H1-KertAFF. Notably, there was a significant 2.5-fold increase of SA-β-Gal+ population in H1-KertAFF from day 40 (31.6%±2.5%) to day 50 (74.6%±5.8%) (p<0.05). In contrast, there was only a slow and gradual increase in the SA-β-Gal level in H1-KertACC from day 20 (6.33%±1.8%) to day 50 (31.4%±1.2%). As a result, there was a significant 2.5-fold difference in percentage of SA-β-Gal+ cells between H1-KertACC and H1-KertAFF by day 50 of differentiation (p<0.05) (Fig. 5C).
We found that H1-KertAFF with higher propensity for cellular senescence consistently expressed higher levels of p16INK4a and p21CIP1 compared with that of H1-KertACC throughout the experiment course. Of note, at day 50 of differentiation, there was higher accumulation of p16INK4a and p21CIP1 proteins by H1-KertAFF, with 65.1%±4.7% and 38.6%±1.1% of cells expressing p16INK4a and p21CIP1, respectively. By contrast, only 14.51%±1.4% and 9.1%±3.6% of H1-KertACC expressed p16INK4a and p21CIP1, respectively (p<0.05) (Fig. 5D). Immunofluorescent staining of p16INK4a and p21CIP1 in H1-KertAFF further confirmed the high accumulation of these proteins at day 50 (Supplementary Fig. S2A). The rapid increase in cell population expressing p16INK4a and p21CIP1 was concomitant with the onset of cellular senescence at day 50, where no K10-positive cells could be detected (Supplementary Fig. S2B), indicating that the cells did not undergo terminal differentiation upon growth arrest.
Role of TGF-β1 in cellular senescence of hESC-Kert
TGF-β1 protein is an isoform of the TGF-β superfamily. This protein has essential roles in stem cell differentiation, proliferation, and migration. 21 Due to the essential role of TGF-β1 in controlling keratinocyte proliferation, we further investigated whether TGF-β1 was responsible for the lower replicative capacity of H1-KertAFF. Enzyme-linked immunosorbent assay (ELISA) was performed to measure the concentration of released TGF-β1 in the culture medium at every 5-day interval. The levels of total and active TGF-β1 production were consistently higher in H1-KertAFF throughout the course of differentiation (p<0.05) (Fig. 6A). The concentration of active TGF-β1 in the H1-KertAFF system remained high at ∼120 pg/mL for up to day 50 of culture when the cells entered the senescence phase. In contrast, the level of TGF-β1 produced by H1-KertACC remained constantly low at ∼30 pg/mL and remained at this concentration till the end of the extended culture at day 70 of differentiation (data not shown).
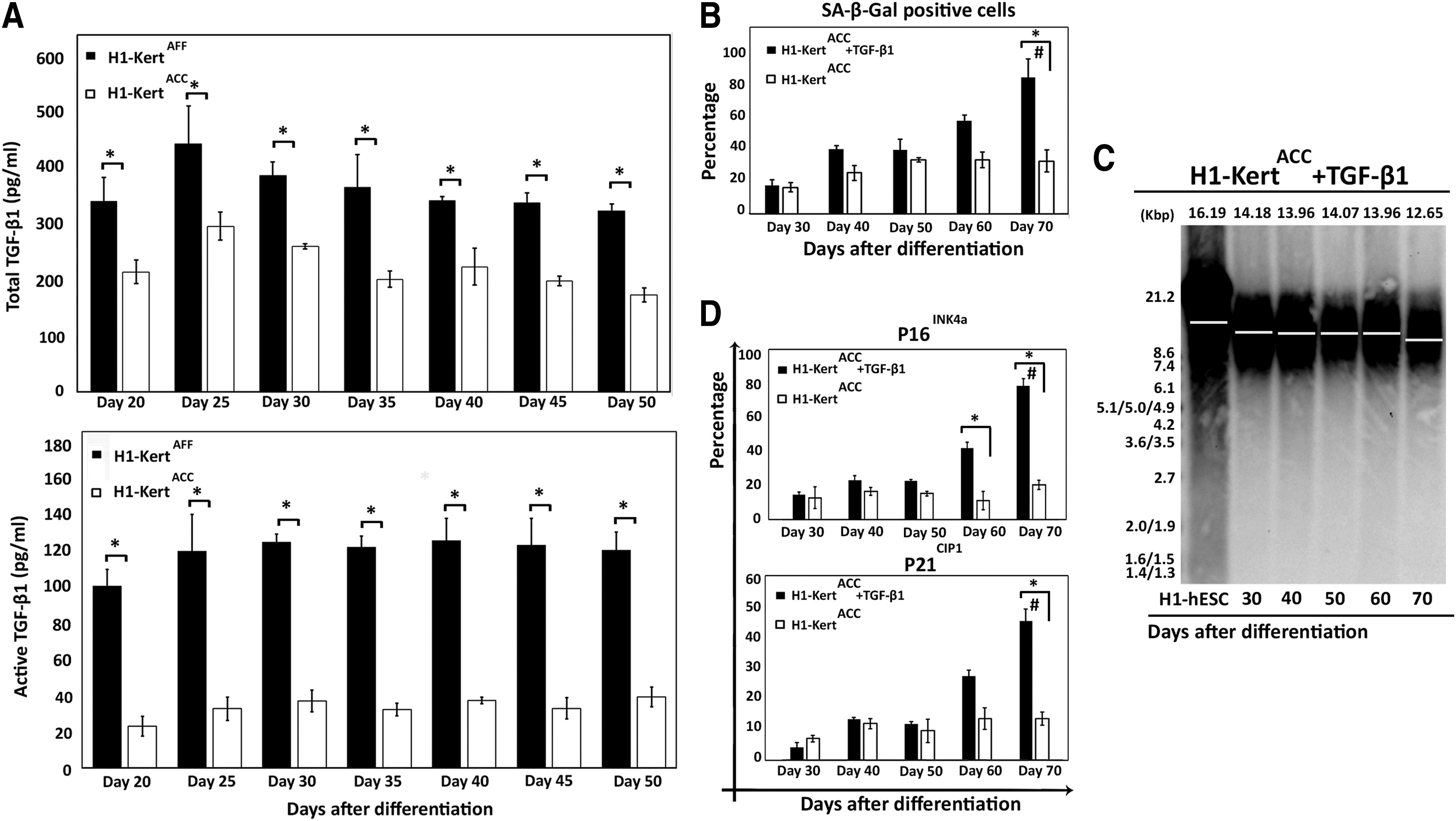
Role of TGF-β1 in cellular senescence of H1-Kert.
To investigate whether the higher concentration of TGF-β1 in H1-KertAFF is responsible for the limited propagation ability with increased propensity for cellular senescence, we hypothesized that H1-KertACC under the same physiological concentration of TGF-β1, as measured in H1-KertAFF, will exhibit limited propagation with the early onset of the senescence phase. To achieve active TGF-β1 concentration of 120 pg/mL, 90 pg/mL of exogenous active TGF-β1 was applied to H1-KertACC cultures after 20 days of differentiation when the cells were transferred onto collagen IV-coated plates. As a result of prolonged TGF-β1 treatment, H1-KertACC entered earlier into the senescence phase at day 70 of culture, with a high percentage of cells (>80%) positive for SA-β-Gal (p<0.05) (Fig. 6B). Analysis of mean telomere length by Southern blot further revealed a substantial decrease in telomere length from 14.18 to 12.65 Kbp (difference of 1.53 Kbp) by the H1-KertACC over a period of 40 days as a result of TGF-β1 treatment (Fig. 6C).
Prolonged TGF-β1 treatment over a period of 50 days also resulted in increased accumulation of p16INK4a and p21CIP1 proteins in the H1-KertACC. Analysis of p16INK4a+ and p21CIP1+ populations by flow cytometry showed high accumulation of p16INK4a and p21CIP1 proteins (p<0.05) in H1-KertACC after 50 days of culture in the presence of 120 pg/mL TGF-β1 (Fig. 6D). Furthermore, only a low number of the K10+ population (<6%) could be detected at the growth arrest point (Supplementary Fig. S3).
Discussion
Despite the extensive studies on the importance of culture condition for keratinocyte growth and differentiation, the effects of extracellular microenvironment on differentiation of hESCs to keratinocyte lineage and subsequent replicative capacity of hESC-Kert is still not well understood. Additionally, it has been shown that keratinocytes derived from hESCs under various differentiation systems exhibited different proliferation ability 17 and posed lower replicative capacity in comparison with postnatal keratinocytes. 22 In this study, for the first time, we have investigated the effect of culture microenvironment during differentiation and the possible mechanism underlying the limited proliferative capacity of hESC-Kert differentiated under feeder-free condition. We demonstrated that the DEJ matrix creates a robust and stable extracellular microenvironment to provide a pure population of epidermal precursors in a defined culture milieu. However, the lack of feeder support during epithelial commitment was responsible for the higher expression of TGF-β1 by hESC-derived keratinocytes, resulting in elevated expression of senescent markers and subsequent limited replicative potential. These observations provide relevant insights toward future directions in establishing effective human pluripotent stem cell differentiation systems for engineering of clinically relevant skin tissue constructs.
TGF-β1 signaling pathways are involved in several aspects of keratinocyte migration, proliferation, and differentiation, as well as ECM deposition and remodeling.23,24 It is well known that TGF-β1 acts as a reversible antiproliferative agent for human keratinocytes. 25 Application of this growth factor suppresses expression of p63 26 and triggers expression of senescence markers. 27 Furthermore, culturing human keratinocytes without feeder support resulted in substantial decrease in number of PDs 28 and upregulated expression of TGF-β-associated genes. 19 These reports suggested a strong correlation between the presence of feeder support and TGF-β1 production for the keratinocyte replicative life span and cellular senescence.
In this study, we comparatively evaluated the replicative life span of hESC-Kert differentiated in the ACC system with the H1-ebF feeder support and in the AFF system, established by use of decellularized DEJ matrix extracted from H1-ebFs. Our approach has the unique advantage of deriving keratinocytes and fibroblasts for supporting keratinocyte differentiation from single-sourced hESCs, which overcomes the problem of interdonor variability. It has been shown that mesenchymal decellularized matrix can induce differentiation of mouse embryonic stem cells to keratinocytes. 29 Similarly, Fu et al. showed that ECM derived from H9-ebFs under dextran sulfate crowded condition can maintain hESCs in the pluripotency state for up to 20 passages. 14 In our study, to induce ECM deposition, we enhanced medium crowding degree with the aid of Ficoll 70/400 cocktail. Ficoll served as a neutral macromolecular crowding agent 16 to induce deposition of dermal ECM proteins, such as collagen I and fibronectin, as well as major basement membrane molecules, including collagen IV, collagen VII, and laminin.14,30 It is well established that basement membrane and dermal ECM play a crucial role for epidermal cell differentiation and proliferation.31,32 Comparatively, the AFF system with the DEJ matrix was able to induce a more rapid and efficient differentiation of hESCs to keratinocytes with higher percentages of p63+ (∼96%) and K14+ (8%) cells following only 10 days of differentiation. Enhanced physisorption of extruded ECM components from the cells onto the tissue culture surface is the hallmark of crowded culture environment. 16 However, in contrast to our previous protocol, which employed dextran sulfate macromolecular crowding, 14 H1-ebFs deposited higher levels of collagen I and collagen IV under Ficoll-induced crowding condition.
Previous studies have reported a strong correlation between the PDs that the cells can undergo and the telomere length status.33,34 Similarly, in our study, TRF analysis showed a dramatic reduction in telomere length in H1-KertAFF after 50 days, concomitant with rapid increase in SA-β-Gal activity. By contrast, there was no significant change in H1-KertACC in either telomere length or SA-β-Gal activity level after day 20 up to day 70. When the telomeres are shortened and/or the protection cap is abrogated, the chromosome ends are sensed as double-stranded breaks, which trigger DNA damage responses and permanent cell cycle arrest through the p21CIP1 signaling pathway. 35 Furthermore, other in vitro studies have shown that culturing human keratinocytes in the absence of a feeder layer can substantially affect proliferative potential in a telomere-independent manner. 34 This limited replicative life span that correlated with upregulation of p16INK4a is likely to be attributed to cellular stress caused as a result of inadequate in vitro culture conditions.19,35
Our observations confirmed that the lack of cell–cell interactions in the feeder-free system of hESC differentiation is responsible for the simultaneous upregulation of cell cycle inhibitor proteins, p16INK4a and p21CIP1, in differentiated keratinocytes. Higher accumulation of both p16INK4a and p21CIP1 implicates the involvement of both telomere-dependent and independent pathways in impaired replicative potential of hESC-Kert, which is in agreement with other studies performed using mouse 36 and human keratinocytes.34,37 Darbro et al. investigated the effect of dermal feeder on expression of p16INK4a by human keratinocytes and concluded that the higher level of p16INK4a expression in keratinocytes cultured without the feeder contributed to elevated migratory response and TGF-β-associated genes expression due to lack of dermal–epidermal contacts. 19 Furthermore, Vijayachandra et al. reported that TGF-β1 treatment resulted in p16INK4a production in mouse keratinocytes. 38 Similarly, p21CIP1 plays a central role in keratinocyte growth arrest and terminal differentiation. p21CIP1 can be induced by DNA damage signals 35 and upon exposure of keratinocytes to TGF-β1-supplemented medium. 39 In our study, there were significantly higher productions of TGF-β1 in H1-KertAFF compared with H1-KertACC, implicating the role of cell–cell interactions between the fibroblasts and keratinocytes during epidermal commitment in suppressing TGF-β1 production and preventing/slowing the progression of hESC-Kert to cellular senescence. However, the effect of higher TGF-β1 production was only evident after long-term expansion of the cells, which can be attributed to low concentrations of TGF-β1 production. Strikingly, the level of TGF-β1 (∼120 pg/mL) that resulted in impaired replicative potential of hESC-Kert is in agreement with the previous report on human precursor keratinocytes, where TGF-β1 of concentration higher than 100 pg/mL inhibited cell growth and only promoted cell proliferation at a lower concentration of 10–30 pg/mL. 40 The pivotal role of TGF-β1 in causing cellular senescence was further confirmed when H1-KertACC also exhibited limited propagation ability with the same expression of senescence markers upon prolonged exposure to TGF-β1 at 120 pg/mL concentration by the addition of exogenous TGF-β1. Importantly, our study indicates that the lack of cell–cell interactions during epidermal commitment led to increased production of TGF-β1 by hESC-Kert during extended culture, which in turn is responsible for resulting in the limited replicative life span with cellular senescence of hESC-Kert derived under the feeder-free culture system. To shed light on the effect of cell–cell and cell-ECM interactions, further studies should investigate the presence of markers such as cadherins for cell–cell interactions and different integrins for cell-ECM interactions.
To the best of our knowledge, no previous study investigated the role of culture microenvironment during epidermal differentiation of stem cells and its associated effects on subsequent replicative life span of differentiated keratinocytes. We have shown in our study that the culture microenvironment not only affects the kinetics and efficiency of keratinocyte differentiation of hESCs but also the subsequent replicative life span of the differentiated keratinocytes. Future efforts would therefore focus on differentiation of human pluripotent stem cells, including human iPSCs and ESCs, into functional keratinocytes with enhanced proliferation ability and reduced cellular senescence for applications in tissue engineering and regenerative medicine.
Footnotes
Acknowledgments
The authors acknowledge the expert assistance of Dr. Paul Hutchinson from Flow Cytometry Laboratory in the Life Science Institute, Immunology Program at National University of Singapore. This work was partially funded by the National University Health System Research grant R221-000-053-515 and the Singapore Ministry of Education Academic Research grants R221-000-023-112, R223-000-024-133, R221-000-050-133, and R221-000-067-133.
Disclosure Statement
The authors indicate no potential conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.