
Abstract
Surgical therapy of cardiovascular diseases frequently requires replacement of diseased tissues with prosthetic devices or grafts. Calcification is the main reason for the degeneration of implanted grafts. However, some factors reduce stenosis and attenuate calcification of implanted grafts. In this study, we used an autologous induced adipocyte cell-sheet (IACS) as a drug delivery system to determine whether its secretion ability has a beneficial effect on the remodeling process of grafts in a rat subcutaneous model. IACSs were generated from rat adipose tissue-derived cells that secreted abundant adiponectin (APN), hepatocyte growth factor, and vascular endothelial growth factor in vitro. Two types of grafts were used in the rat subcutaneous model: decellularized and IACS-wrapped decellularized porcine vascular grafts. Transplanted IACSs secreted APN into the decellularized porcine vascular graft in rats at 4 weeks. After explanting from the rat subcutaneous model at 1, 2, 4, and 8 weeks, immunofluorescence staining showed that IACS-wrapped grafts had a dominant M2 phenotype of macrophages (p < 0.001) at all time points and showed constructive remodeling and less calcification at 8 weeks. The decellularized graft showed a predominately CCR7+ cell response (M1 phenotype) (p < 0.001) and was characterized by chronic inflammation and severe calcification at 8 weeks. Furthermore, the IACS-wrapped side of the graft showed less cell infiltration compared with the other side, which may have reduced inflammation in the area. Transplantation of IACSs with a biological scaffold had a profound influence on the macrophage phenotype and downstream remodeling processes. The method might reduce inflammatory reactions during remodeling of xenogeneic scaffolds and result in less calcification.
Introduction
C
Many scientists have shown that stem cells could enhance angiogenesis, foster cell integration adhesion and growth, and reduce fibrosis by secreting various anti-inflammatory factors in a paracrine manner. 5 However, stem cell transplantation technology showed limited therapeutic efficacy in animal studies. It may be caused by insufficient paracrine effects whose figure and magnitude are largely affected by the cell type and the way they were delivered.6,7 Recently, the secretion ability of adipocytes has attracted attention. Adipocytes are a promising cell source to foster cell integration, adhesion, and growth for their ability to secrete various protein factors, including vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), and adiponectin (APN).8,9 However, their paracrine effects have not been tested in vascular engineering. To raise the functions and survival of transplanted cells, we used a cell-sheet to deliver adipose cells to decellularized porcine aorta arteries. We believe that cell-sheet treatment enhances paracrine effects compared with cell injection, resulting in better therapeutic efficacy. 10
In this study, we hypothesized that using adipocyte cell-sheet to deliver multiple anti-inflammatory factors onto implanted vascular scaffolds might attenuate an inflammatory reaction and result in an attenuated calcification of the graft.
Materials and Methods
Cell culture and seeding
Inguinal adipose tissue was excised from a Sprague-Dawley rat. Adipose tissue was digested in phosphate-buffered saline (PBS; Sigma) containing 0.1% collagenase type II (Gibco) at 37°C with vigorous shaking for 1 h, filtered through a 250 μm filter, and centrifuged at 200 g for 5 min to separate the stromal vascular fraction (SVF) cells from floating adipocytes. The supernatant mature adipocytes were discarded. SVF cells were resuspended in Dulbecco's modified Eagle's medium (Hyclone) containing 10% fetal bovine serum (Gibco) and 1% antibiotics (Gibco), and then cultured on dishes at 37°C with 5% CO2. Twenty-four hours after plating, all nonadherent cells were removed by washing. Then, the SVF cells were cultured for 3 days in the same medium. At 90% confluence, they were reseeded at 7000 cells/cm2 in an UpCell dish, coated with temperature-responsive polymers (CellSeed, Tokyo, Japan). Three days after passaging, the SVF cells were induced to differentiate into the adipogenic lineage by addition of 0.25 μM dexamethasone, 0.87 μM insulin, 500 μM isobutylmethylxanthine, and 5 μM pioglitazone (Sigma) to the original medium. Seven days after induction, the cells were detached from the culture dishes by incubation at 20°C, yielding an adipocyte cell-sheet, which we call an “induced adipocyte cell-sheet” (IACS). For direct attachment of the cell layer to the vascular surface, a membrane was used to transfer the cell sheet to the outside surface of the vascular graft, followed by incubation at 37°C for 45 min. Then, the membrane was removed gently. The seeding effect was examined by immunofluorescence and hematoxylin and eosin (H&E) staining.
Flow cytometric characterization of rat SVF cells
SVF cells (100 μL) were labeled with manufacturer-recommended concentrations of mouse anti-rat monoclonal fluorescent antibodies against CD34, ABCG2, CD29, and CD166, or the isotype control (Becton Dickinson). Cells were washed with PBS subsequently, fixed in paraformaldehyde, and analyzed by FACS Calibur, and data were analyzed with CellQuest software (BD).
Enzyme-linked immunosorbent assays
To determine the content of secreted factors, enzyme-linked immunosorbent assays (ELISA) were performed. The collected culture supernatants of SVF cell-sheets and IACSs were centrifuged to remove debris and contaminating cells. For APN, samples were diluted at 1:200 and then analyzed (SVF group, n = 6; IACS group, n = 6) by ELISA kits (CUSABIO, China) according to the manufacturer's instructions. VEGF and HGF contents in the supernatant were analyzed by ELISA kits (CUSABIO) according to the manufacturer's instruction without dilution.
Graft preparation
Aortas (10 cm in length) were harvested from 6-month-old pigs. After removing adherent tissues, the aortas were cut into 10 × 10 mm pieces for further disposal. Arterial extracellular matrices (ECMs) were decellularized as previously described by Eitan et al. 11 Briefly, after agitating five times in a 0.9% NaCl solution and double distilled water alternately for 3 min each, grafts were then washed with a fresh detergent solution (1% Triton-X-100 and 1% ammonium hydroxide, 18 h per cycle) and PBS (6 h per cycle) alternately for four consecutive cycles of 96 h at 4°C. Then, they were subjected to 3 h of enzymatic digestion (0.05% trypsin and 0.02% ethylenediaminetetraacetic acid in PBS, pH 7.4) at 37°C. After being washed in sterile saline for 24 h, the remaining ECMs were immersed in 70% ethanol for 18 h, washed completely, and lyophilized. The decellularized material was examined by H&E staining, DNA was isolated by a Wizard Genomic DNA Purification Kit (Promega, Madison, WI), and quantified by Hoechst 33258 staining and using a calibration curve with 0–500 ng/mL DNA. Values are expressed as ng DNA/mg dry sample (n = 3).
Graft transplantation
Rats were anesthetized by intraperitoneal injection of pelltobarbitalum (40 mg/kg). IACS-wrapped decellularized aortas (experimental group) and decellularized aortas (control group) were implanted under the skin of adult male Sprague-Dawley rats weighing 250–300 g (n = 24 per group). Four subcutaneous pouches were created on the back of each rat, and one scaffold (10 × 10 mm) was put in each pouch. Animals were provided with water and food ad libitum after recovery. Six rats were sacrificed at 1, 2, 4, and 8 weeks in each group. The grafts together with surrounding tissue were retrieved for histology, immunohistochemistry, and calcium analysis.
Histological analysis
After being fixed in 10% neutral buffered formalin for 24 h at room temperature, vascular scaffold samples were processed by automatic vacuum tissue processor (LEICA ASP 300S). They were sliced into 6-μm-thick sections (n = 6 sections per sample) and evaluated by H&E and von Kossa staining. Representative sections were deparaffinized for immunofluorescence staining. They were boiled in 0.01 M citrate buffer (pH 6.0; Zhongshanjinqiao, Inc., China) for 2 min to retrieve antigens. After been washed for 5 min in PBS (pH 7.4) thrice, the sections were incubated in 5% normal goat serum (Zhongshanjinqiao, Inc.) for 30 min at room temperature. Then, they were treated by the blocking solution and the diluted primary antibody, at 4°C overnight. The primary antibody used for IACS staining was a mouse anti-rat APN antibody (ab22554; Abcam). The primary antibody used for macrophages was a mouse anti-rat CD68 antibody (ab31630; Abcam) at a 1:150 dilution in PBS, rabbit anti-rat CD163 antibody (ab182422; Abcam) at a 1:100 dilution, and rabbit anti-rat CCR7 antibody (ab32527; Abcam) at a 1:150 dilution. CD163 is a surface marker of the macrophage phenotype 2 (M2), and CCR7 is surface marker of the macrophage phenotype 1 (M1). 12 After rinsing thrice in 1 × PBS for 5 min each, the specimens were incubated with a fluorochrome-conjugated secondary antibody diluted in PBS for 1 h at 37°C in the dark. The secondary antibody used was Alexa Fluor®594-Conjugated AffiniPure Goat Anti-Mouse IgG (ZF-0513, Zhongshanjinqiao, Inc) at 1:200 dilution for CD68 and APN, and fluorescein-conjugated AffiniPure Goat Anti-Rabbit IgG (ZF-0311, Zhongshanjinqiao, Inc.) for CD163 and CCR7. Rat spleen was served as the positive control tissue. Sections were coverslipped by using the Fluorescent Mounting Medium with DAPI (ZLI-9557, Zhongshanjinqiao, Inc).
Analysis
Two investigators who conducted quantitative analysis were blinded to the group of specimen and the positive antigen being identified in each group. Immunopositive cells were counted in six matched microscopic fields of each specimen at 400 × magnification. After dividing the number of CCR7+ cells by the number of CD68+ cells, we got the percentage of M1 in each field. The percentage of M2 phenotype cells was calculated by the same method. The mean value in each section was calculated by obtaining the average of the six fields, and the M1/M2 ratio was calculated. The total number of M1 plus M2 macrophages did not equal the number of CD68+ cells because not all macrophages stayed in the phase of differentiation. 13
Calcium assay
Von Kossa staining and quantitative analysis were used to determine calcium deposits of the explants. 14 The capsule was peeled of explants (n = 6 per group) and the residual graft was lyophilized, weighed, and then digested in 1 mL of 6 mol/L HCl at 95°C for 10 h. The digested samples were evaporated under a continuous stream of nitrogen gas until dry, and the residual material was dissolved in 1 mL of 0.01 mol/L HCl. The calcium concentration of dissolved sample was determined using atomic absorption spectrophotometry (Z-8000; Hitachi, Tokyo, Japan) and expressed as μg/mg dry explant.
Statistical analysis
Results are expressed as mean ± standard error of the mean. The data were analyzed using a two-tailed Student's t-test and a probability value (p) less than 0.05 was considered significant.
Results
Preparation of decellularized scaffolds
Histological analysis of the grafts showed that they were successfully decellularized by the mechanical processes, enzymatic digestion, and osmotic shock. Cells could not be detected by H&E staining (Fig. 1A), indicating that the treatments effectively removed aortic cells. DNA analysis showed less than 2%DNA content in the decellularized graft compared with the fresh aorta.(Fig. 1B).

Characterization of IACSs
Freshly isolated SVF cells showed adherence and expansion in culture. Flow cytometric analysis of SVF cells indicated expression of CD34, ABCG2, and CD29 antigens that are stem cell- and stromal cell-associated markers (Fig. 2), which was consistent with a study by March and colleagues. 15 SVF cells had a fibroblast appearance and after being inducted into differentiation for 7 days, adipocytes bearing oil droplets could be detected under microscope(Fig. 3A). IACSs were transferred onto the outside surface of vascular grafts. (Fig. 4) Each IACS was ∼25 μm thick (Fig. 3B). The result of immunohistochemistry showed that APN was secreted by adipocytes in the cytoplasm in the IACS group (Fig. 3C, D). ELISAs showed abundant APN and VEGF expression in IACSs compared with SVF cell-sheets (Fig. 3F, G). However, expression of HGF in the IACS was lower than that in SVF cell-sheets (Fig. 3E).

Flow cytometry histogram of adipose-derived cells. The flow cytometry histograms for selected stem cell and stromal cell markers are displayed at the SVF cells. The percentage of cells staining positive is indicated in the upper right corner of each panel. The blue line indicates the positive staining cells, whereas the red line indicates the isotype-matched monoclonal antibody control. Phase contrast micrographs of plated SVF cell demonstrate that cells are adherent. Color images available online at www.liebertpub.com/tea
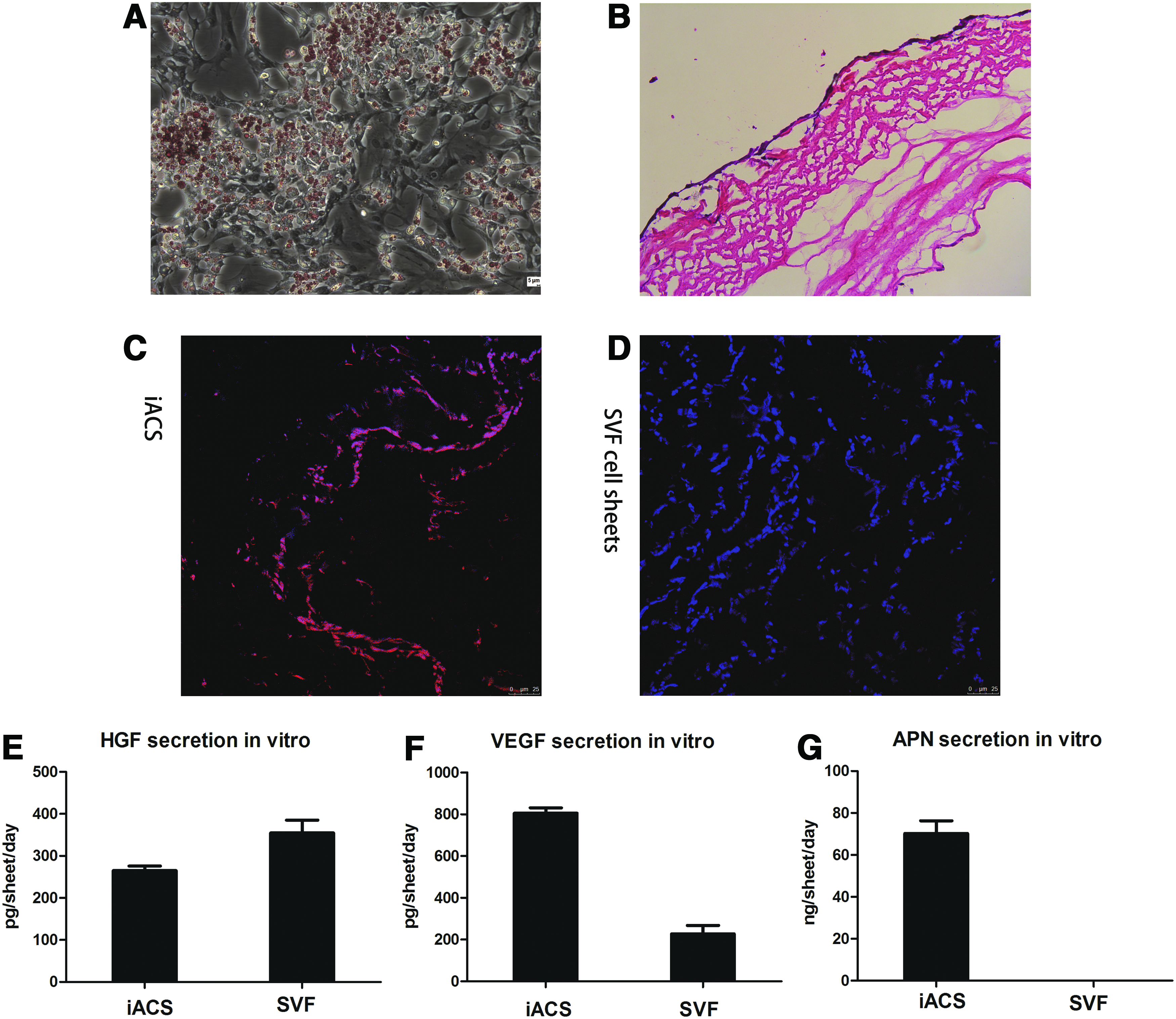
Characterization of induced adipocyte cell-sheet (IACS) in vitro and in vivo.
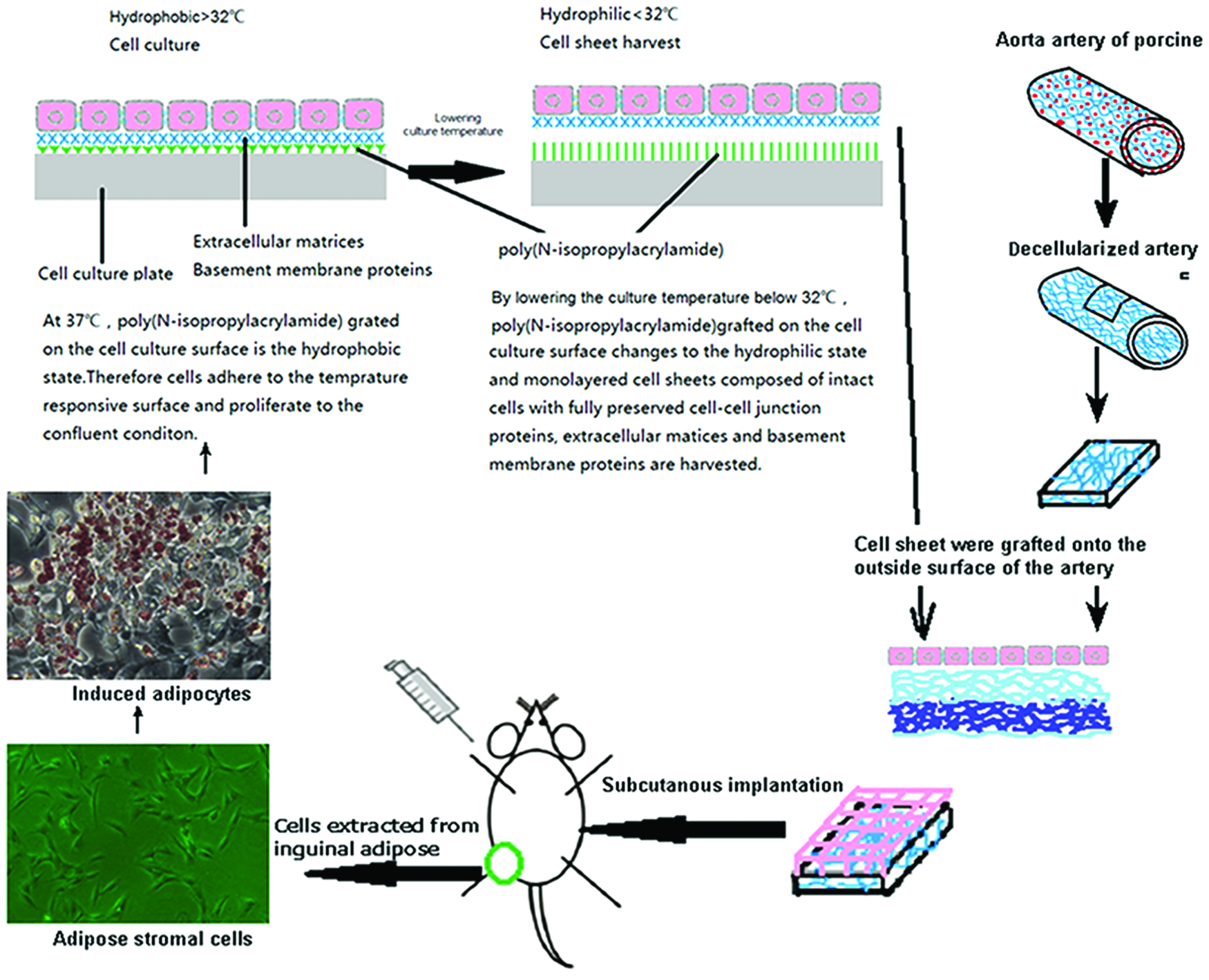
Overview of the cell differentiation steps and sheet creation. Color images available online at www.liebertpub.com/tea
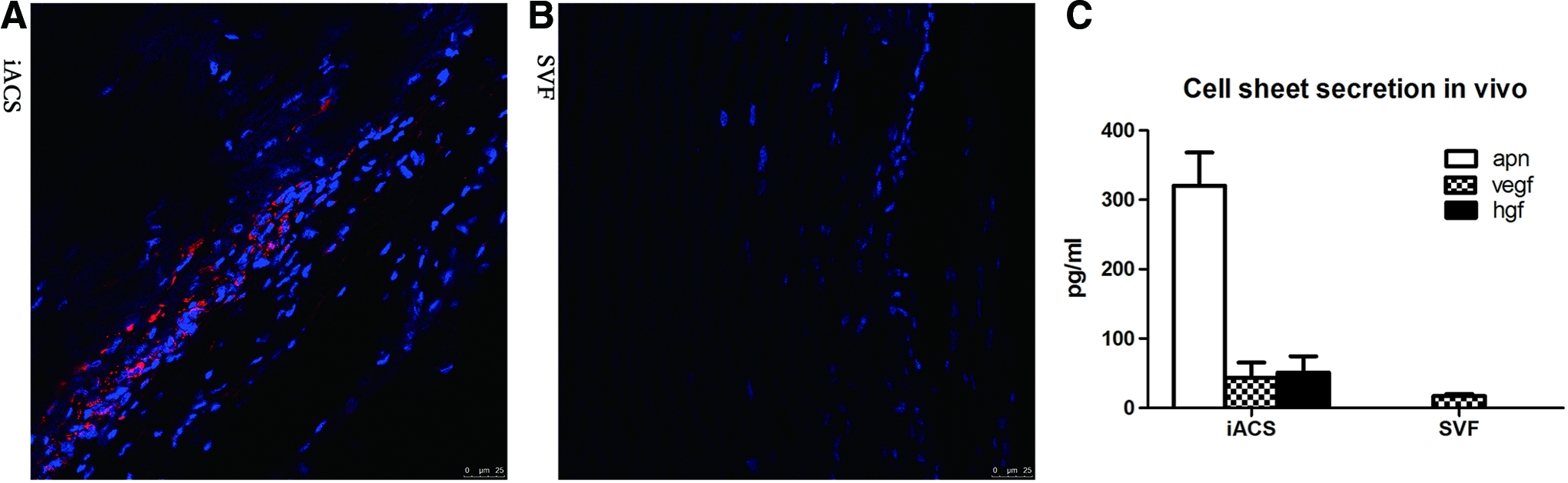
Transplanted IACSs supply APN to decellularized grafts
IACSs were transplanted onto the outside surface of the vascular graft to evaluate the influence of the IACS on the transplanted graft. The IACS had survived 4 weeks after transplantation and was tightly integrated with the grafts, which was ∼25 μm thick. APN could be detected in the scattered surviving cells (Fig. 5).
IACS implantation reduces inflammatory responses
Macroscopically, grafts were covered by a thin capsule and substantially kept their original shape. Substantial host cells infiltrated into the porcine aorta scaffold, exhibiting some differences between decellularized and IACS-wrapped scaffolds. Most infiltrated cells were CD68+ macrophages and all explants contained CD68+ mononuclear cells when they have been harvested at 1, 2, 4, and 8 weeks. The decellularized group was characterized by a large number of CCR7+ cells (M1 phenotype mononuclear cells) compared with CD163+ cells at 4 and 8 weeks (p < 0.001), and multinucleate giant cells were present at 8 weeks associating with fibrosis of the scaffold (Fig. 7). However, the IACS group was predominantly dominated by M2 phenotype mononuclear cells (CD163+) at all time points (p < 0.001) (Fig. 7). At the second week, M1 cells infiltrated on the outside surface of the grafts in the IACS group were much less than those in the decellularized group. Multinucleate giant cells were present in the decellularized group then (Fig. 6).
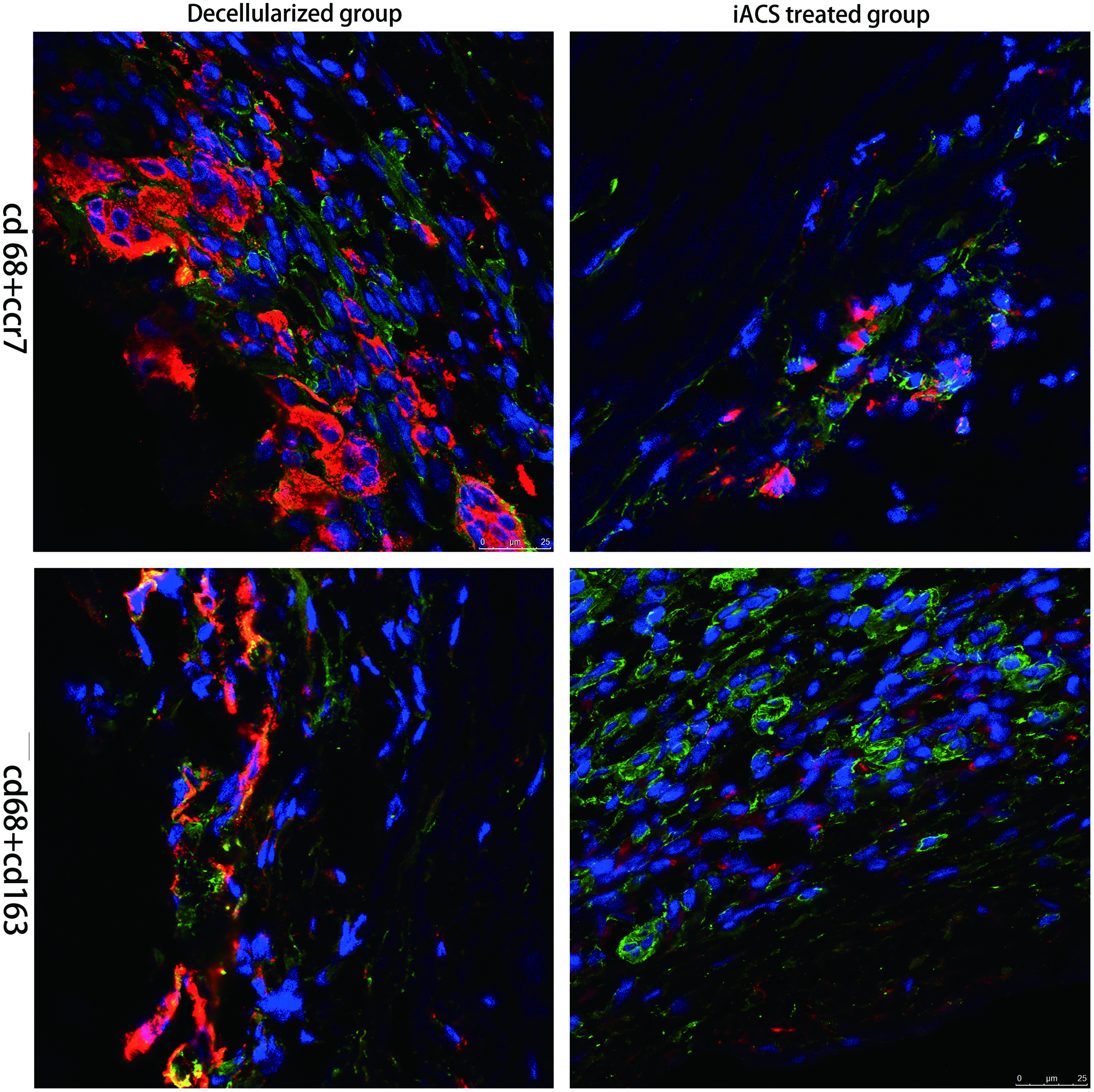
Host cell infiltration at the outside surface of the graft at the second week. The cells infiltrated at the outer side of the graft were much less in the IACS-treated group than in the decellularized group. CCR7 cells were present in the decellularized grafts and in much smaller numbers in the IACS-treated graft (scale bar = 25 μm, red indicates CD68 and green indicates CCR7 and CD163). Color images available online at www.liebertpub.com/tea
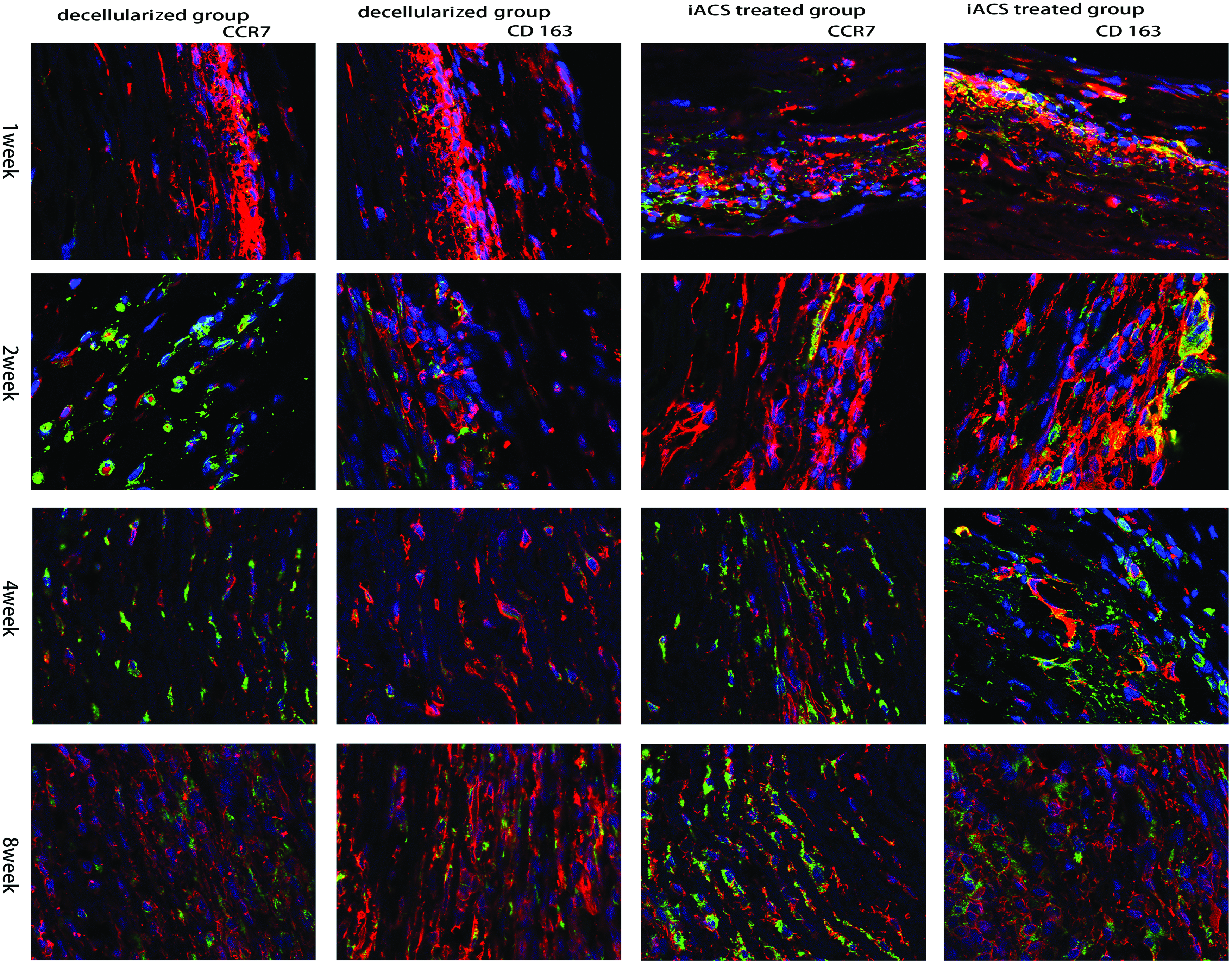
Macrophage cells infiltrated at the implanted grafts at 1-, 2-, 4-, and 8-week time points. The macrophage phenotype within each section was identified using immunofluorescence staining for CD68 (pan macrophages), CCR7 (M1 macrophages), and CD163 (M2 macrophages). CD68+ cells were present in all grafts. CD163+ cells were present in large numbers in the IACS-treated graft and in much smaller numbers in the decellularized grafts at all time points. CCR7 cells were present in the decellularized grafts and in much smaller numbers in the IACS-treated graft (Scale bar = 25 μm). Color images available online at www.liebertpub.com/tea
Bar graphs of the cells' phenotype for each explant at each time point are shown in Figure 8A and B. The M1:M2 ratio for the two groups at all time points is shown in Figure 8C. In the IACS-wrapped group, M2 showed a higher proportion. However, the bare decellularized group showed a dominant M1 response at all time points.

The macrophage polarization percentages for
Quantitative analysis of calcium
IACS-wrapped grafts did not show significant calcification up to 8 weeks (Fig. 10A). However, in the decellularized group, calcification was severe and increased progressively. (Fig. 10B) Calcification of grafts was evaluated at three different time points (Fig. 9). Quantitative analysis on grafts of the IACS group exhibited minor calcium levels at 2 weeks (0.90 ± 0.22 μg calcium/mg dry explant), 4 weeks (1.43 ± 0.13 μg calcium/mg dry explant), and 8 weeks (10.61 ± 1.10 μg calcium/mg dry explant), while calcification tendency of the decellularized grafts was more pronounced after subdermal implantation at 2 weeks (6.98 ± 0.23 μg calcium/mg dry explant), 4 weeks (38.54 ± 2.03 μg calcium/mg dry explant), and 8 weeks (27.91 ± 1.75 μg calcium/mg dry explant. Pairwise comparison showed that the differences between the two groups at 2, 4, and 8 weeks were statistically significant (p < 0.05).
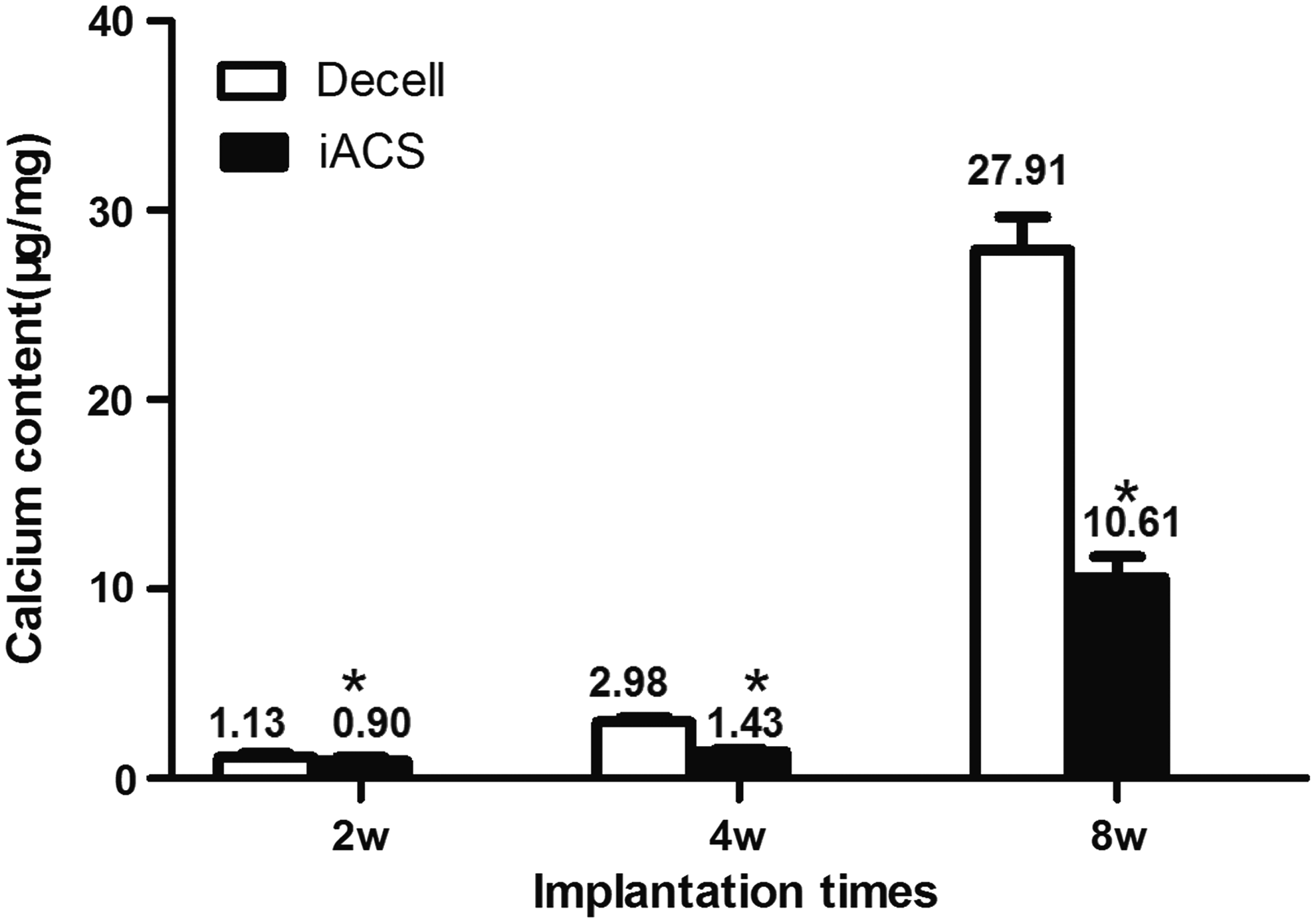
Calcium content of scaffolds at 2, 4, and 8 weeks in a subdermal model. Decell (decellularized grafts, controls), IACS (IACS-treated group) (*p < 0.001 versus control, Student's t-test; n = 6).
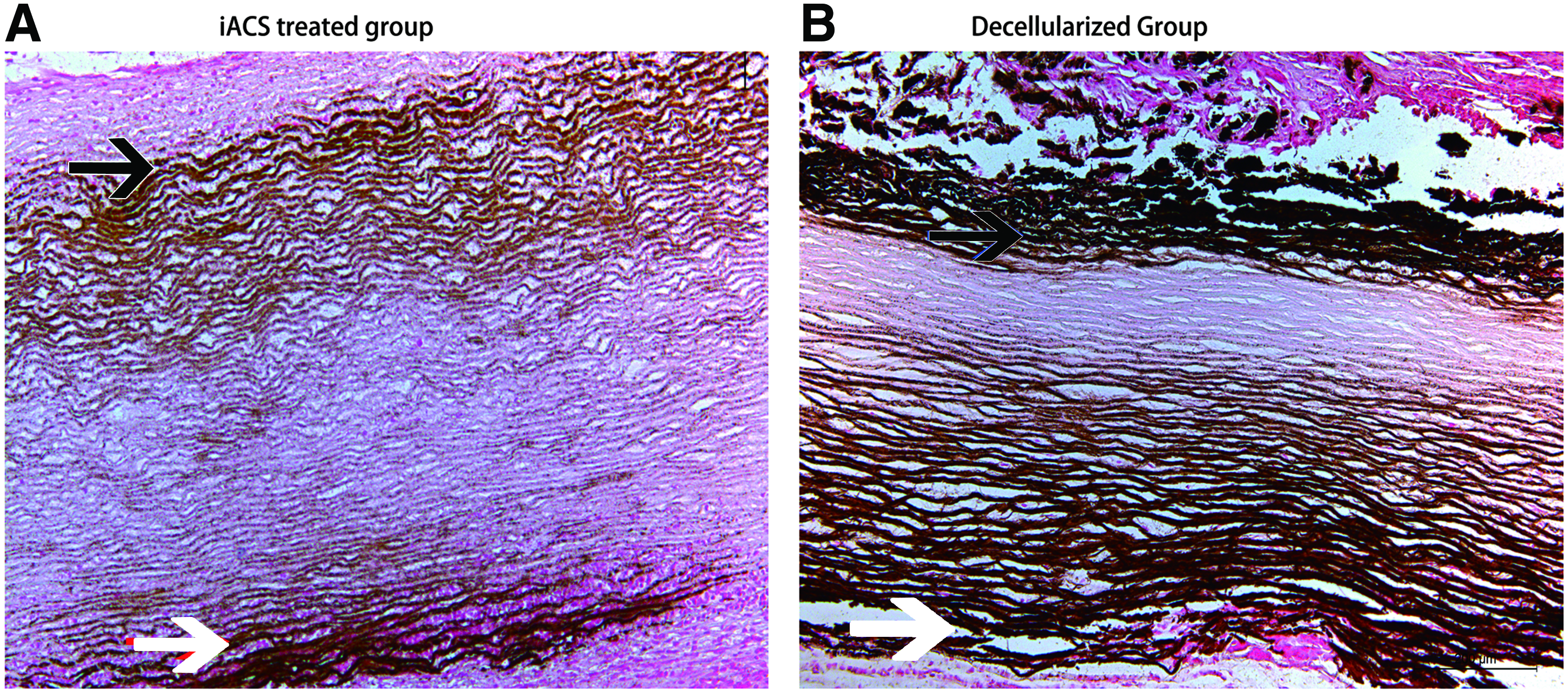
Extracellular matrix calcification of scaffolds at 8 weeks in rat subdermal model between the two groups. Arrows show calcification of the porcine graft tagged by von Kossa staining. (Scale bar = 200 μm). Color images available online at www.liebertpub.com/tea
Discussion
We placed IACSs on the surfaces of decellularized vascular grafts. These sheets, which were composed of induced adipocyte cells, secreted multiple anti-inflammatory and growth factors in vitro, including VEGF, HGF, and APN. Our result confirmed previous findings that SVF cells express the surface markers ABCG2, CD34, and CD29, 16 which were also expressed on progenitor and hematopoietic stem cells. SVF cells could be induced into adipocytes that secreted APN, in addition to VEGF and HGF. Furthermore, secretion of these factors from the IACSs into the grafts lasts at least 1 month. The IACS-wrapped grafts were infiltrated prevailingly by M2 cells at each time point, showing a constructive remodeling process. Furthermore, calcification of the IACS-wrapped grafts was significantly attenuated at the late stage. However, the bare decellularized grafts were occupied by M1 cells and have much severe calcification of the graft tissue.
Exogenous proteins like VEGF and APN that could reduce stenosis and attenuate calcification of implants have been demonstrated by previous experimental and clinical studies.15,17,18 However, naked proteins could be inactivated quickly, which resulted in the lack of long-term efficacy. 19 Vascular remodeling is a progressive process influenced by the material of the implanted graft and subsequent inflammation. A constitutive and balanced supply of anti-inflammatory factors rather than using single naked protein maybe an effective way to inhibit chronic inflammation. 20 Our results showed that the secretion of APN and VEGF was increased abundantly when SVF cells were induced into adipocytes. Furthermore, the IACS had survived and was tightly integrated with the grafts at 1 month after transplantation, which is the acute inflammation period. Therefore, IACSs could constitutively deliver multiple anti-inflammatory factors during the acute inflammation period after graft implantation, leading to vascular remodeling, a reduction in fibrosis, and cell integration. Therefore, using cell-sheets might be an effective method to deliver therapeutic proteins to biological scaffolds.
Adipose tissue is an important endocrine organ participating in autocrine regulation. 21 It includes 50% adipocytes and 50% other cell types, including leucocytes, vascular, neural and immune cells, and preadipocytes. Anti-inflammatory molecules could be synthesized and stored by adipocytes at normal conditions; however, at some circumstance, inflammatory factors such as monocyte chemoattractant protein-1 and interleukin (IL) 6 could also be produced. Cells derived from primary adipose tissues staying at different develop stages could not be controlled as preadipose cell lines. Therefore, adipocytes differentiated from SVF are a promising cell source to use in vascular tissue engineering. In this study, we found that the IACSs significantly changed the phenotype of phagocytes during the remodeling of implanted grafts. We believe that the adipocytes promoted polarization of anti-inflammatory M2 macrophages, which is consistent with a study by Jenke et al. 22 Their results indicated that APN could promote polarization of M2 macrophages and reduce the number and activation of natural killer cells, and finally attenuated acute antiviral immune responses in AVB3 myocarditis. An in vitro experiment showed that APN could increase Beclin1 expression and LC3-I processing, as well as downregulate p62, which indicates autophagy of primary macrophages. 23 According to these findings, APN secreted by adipocytes regulate the polarization and autophagy of macrophages, which may be the reason M1 cells occupied a bigger amount in the IACS group. However more research is needed to clarify the underlying mechanism.
Macrophages play a critical role in regulating inflammatory response that may be responsible for calcification of the implants.24,25 They could acquire different polarization phenotypes by reprogramming after being exposed to various cytokine signals in an internal environment. 26 M1 could accelerate monocyte recruitment and lead to tissue damage by producing proinflammatory cytokines (IL-12, IL-6, and tumor necrosis factor-α) and reactive oxygen species. However, M2 secretes anti-inflammatory cytokines (transforming growth factor-β and IL-10) and promotes tissue repair and scavenging of apoptotic cells. In our study, higher M2 cell polarization was associated with less calcification of the explanted graft, which could be partly explained by the anti-inflammatory cytokines secreted by M2 cells. Also, Li et al.'s result showed that APN promotes M1 cells to secrete more proinflammatory cytokines (IL-12, TNF-α, and IL-6), and induces M2 cells to produce an anti-inflammatory cytokine (IL-10). Thus, APN may affect polarization of macrophages and promote the secretion of anti-inflammatory cytokines at the same time, and both effects reduce the inflammatory reactions at the acute stage of graft remodeling and calcification in the long run.
A potential limitation of this study is that the identification of M1 versus M2 phenotype was based on a limited choice of cell surface markers, for most of those were not suitable for rat tissue detection. CD68 has been used to identify monocyte–macrophage lineage cells, including activated macrophages, resting-tissue macrophages, and activated monocytes; CD163 was used to identify M2-activated cells and CCR7 shows high expression in M1-activated macrophages. 27 Although the staining affinities of these two markers used in our research were distinct, more criteria need to be used to indentify cells' phenotype. Flow cytometric analysis of cells inside grafts would be a more objective method. However, because only a small number of macrophages could be recovered from the graft, we were unable to conduct the flow cytometric analyses of macrophage phenotypes. Also, more research need to be done at cytokine tissue levels to explain the relationship between cytokines secreted by adipocytes and polarization of macrophages.
In summary, an IACS may be a powerful drug delivery system for cytokines, including VEGF, HGF, and APN. Implantation of an adipocyte cell-sheet onto tissue-engineered vascular grafts reduces acute inflammation and attenuates long-term calcification. This method could be applied as a drug delivery system during the construction of tissue-engineered vascular grafts.
Footnotes
Acknowledgment
Funding for this study was provided by a grant from the Natural Science Foundation of Beijing Municipality (7122149).
Disclosure Statement
The authors have no professional or commercial associations that might create a conflict of interests in connection with this article.