
Abstract
As time to final coverage is the essence for better survival outcome in severely burned patients, we have continuously strived to reduce the duration for the preparation of our bilayered self-assembled skin substitutes (SASS). These SASS produced in vitro by the self-assembly approach have a structure and functionality very similar to native skin. Recently, we have shown that a decellularized dermal matrix preproduced by the self-assembly approach could be used as a template to further obtain self-assembled skin substitute using a decellularized dermal template (SASS-DM) in vitro. Thus, the production period with patient cells was then reduced to about 1 month. Herein, preclinical animal experiments have been performed to confirm the integration and evolution of such a graft and compare the maturation of SASS and SASS-DM in vivo. Both tissues, reconstructed from adult or newborn cells, were grafted on athymic mice. Green fluorescent protein-transfected keratinocytes were also used to follow grafted tissues weekly for 6 weeks using an in vivo imaging system (IVIS). Cell architecture and differentiation were studied with histological and immunofluorescence analyses at each time point. Graft integration, macroscopic evolution, histological analyses, and expression of skin differentiation markers were similar between both skin substitutes reconstructed from either newborn or adult cells, and IVIS observations confirmed the efficient engraftment of SASS-DM. In conclusion, our in vivo graft experiments on a mouse model demonstrated that the SASS-DM had equivalent macroscopic, histological, and differentiation evolution over a 6-week period, when compared with the SASS. The tissue-engineered SASS-DM could improve clinical availability and advantageously shorten the time necessary for the definitive wound coverage of severely burned patients.
Introduction
D
First introduced more than three decades ago, autologous epithelial cultures initiated the development of new tissue-engineered, structurally more complex skin substitutes in an attempt to replicate as closely as possible human skin physiology.5,6 In the realm of original new therapies, tissue engineering has allowed the development of autologous bilayered skin substitutes, composed of epithelial and dermal components, cultured in vitro from cells isolated from the patient, thereby leading to a physiological integration of the graft on the wound site.7–10 Our group proposed a bilayered self-assembled skin substitute (SASS) similar in structure and function to native human skin that is suitable for autologous grafting in human 11 and that has already been shown to allow wound closure during the treatment of human chronic wounds. 12
The self-assembly strategy entails that mesenchymal cells, cultured under adequate conditions, produce and organize their own extracellular matrix13–16 without the need for an exogenous scaffold. This method allows the production of a cutaneous substitute composed of a dermis, a functional basement membrane, and an epidermis presenting features similar to normal skin.15,17,18 Following extraction of patient skin fibroblasts and keratinocytes and their amplification in vitro, the initial standard protocol proposed for SASS production required 45–52 days of culture in vitro before grafting.12,15
We managed to reduce to three and half weeks the time frame needed for SASS production with patient cells using a new in vitro preparation strategy. 19 This new method takes advantage of previously prepared self-assembled decellularized dermal matrix as a template to seed autologous fibroblasts and keratinocytes. The resulting skin substitute, referred to as self-assembled skin substitute using a decellularized dermal template (SASS-DM), was shown to be similar to standard SASS when comparing their histology, cellular differentiation, and mechanical behavior, while maintaining a continuous basement membrane. 19 Compared to decellularized allogeneic dermis, one advantage of this strategy is the possibility to select a human fibroblast population based on its biological qualities and adequate biosafety. Thereafter, the self-assembled dermal matrix produced from such a cell bank can be homogenous in terms of thickness and elastic properties, which is not always the case with human allogeneic dermis;20,21 thus, the manufacturing method can be standardized. Furthermore, after the addition of autologous keratinocytes and fibroblasts, the resulting skin substitute can achieve a final wound coverage in a one-step surgical procedure without resorting to the use of a skin donor site for harvesting autografts.7,22–24
Guidance for industry and translational medicine recommends preclinical assessment of investigational cell therapy products. The ultimate success of the permanent integration of a skin substitute lays in the preservation of its autologous epithelial stem cells that do ensure the continuous regeneration of the epithelium. An appropriate animal model approach to show the long-term regeneration of a human skin substitute is its transplantation onto athymic mouse. These mice present cellular immunodeficiency and thus do not acutely reject human cells present in the grafted skin construct.
Our aim was to evaluate the long-term in vivo maturation of SASS-DM for future clinical use. SASS-DM was grafted on athymic mice and compared to the standard SASS. Furthermore, they were seeded with adult or newborn cells to demonstrate the applicability of the method with pediatric and adult cell populations. The evolutive histology, cellular differentiation, graft take, and healing were evaluated up to 6 weeks after grafting. Furthermore, graft integration and persistence were followed in real time using keratinocytes transduced to express the green fluorescent protein (GFP) visualized with an in vivo imaging system (IVIS). The in vivo evolution of cellular differentiation and three dimensional structure of the SASS-DM were shown to be equivalent to those of the SASS.
Materials and Methods
Culture media
The fibroblast culture medium, referred to as the F medium, was composed of Dulbecco-Vogt modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS, Hyclone, Scarborough, ON, Canada), penicillin 100 UI/mL (Sigma), and gentamicin 25 μg/mL (Schering, Pointe-Claire, QC, Canada). The keratinocyte culture medium, referred to as the K medium, was composed of DMEM (Invitrogen, Burlington, ON, Canada) supplemented with HAM's F12 in a 3:1 ratio (DMEM HAM; Invitrogen, Burlington, ON, Canada) containing 5% newborn calf serum (FetalClone II, Hyclone, Logan, UT), 5 μg/mL insulin (Sigma), 10 ng/mL epidermal growth factor (Austral, San Ramon, CA), 0.4 μg/mL hydrocortisone (Calbiochem, La Jolla, CA), 0.212 μg/mL isoproterenol (Sandoz Canada, Boucherville, QC, Canada), penicillin 100 UI/mL, and gentamicin 25 μg/mL.
Cell isolation and expansion
The study protocol was approved by the Comité d'éthique de la recherche du CHU de Québec—Université Laval and cells from human biopsies were obtained after informed consent was given. Neonatal fibroblasts and keratinocytes were extracted from a single donor (4 days old) foreskin biopsy, while adult cells were extracted from two 18- and 38-year-old women, thus providing 3 distinct cell populations used for tissue reconstruction. Cells were extracted from the biopsies by the two-step thermolysin and trypsin isolation procedure, as previously described.15,25 Briefly, the epidermis and dermis were gently separated after a thermolysin (0.5 mg/mL, Sigma, St-Louis, MO) incubation process. Fibroblasts were isolated from the dermis using collagenase H (Roche Diagnostics, Laval, Qc, Canada), while keratinocytes were extracted from the epidermis using a trypsin/EDTA solution (0.05% trypsin (Intergen Company, Purchase, NY) and 0.01% EDTA/disodium salt (J.T Baker, Phillipsburg, NT). Cells were then collected by centrifugation and inoculated into culture flasks. Keratinocytes were cocultured on a feeder layer of irradiated human newborn fibroblasts in the K medium as previously described. 26 Keratinocytes were detached using trypsin [passage (P)] before becoming confluent and subcultured in P1 as described. 15 After 5–7 days, keratinocytes were cryopreserved in newborn calf serum 10% dimethyl sulfoxide as described. 15 After thawing, keratinocytes were subcultured for one passage. Therefore, they were seeded at their third passage into the skin substitute (see sections “Production of standard SASS” and “Production of SASS-DM”).
Typically, fibroblasts were cultured in the F medium until 100% confluence was attained (9–10 days) and then detached using trypsin. 15 They were cultured for one other passage (4–7 days). Fibroblasts were either seeded to produce dermal substitute or cryopreserved in FCS 10% dimethyl sulfoxide as described 15 for further utilization. Therefore, P2–P6 fibroblasts were used to inoculate flasks for the production of dermal substitutes (see below). Both cell types were maintained at 37°C in a humidified incubator containing 8% CO2 27 and culture media were changed thrice per week during the culture period.
Production of standard SASS
The SASSs were obtained using the self-assembly reconstruction method as previously described.15,19 Briefly, fibroblasts from two different adult cell populations were seeded at 4000 cells/cm2 in tissue flasks (Falcon) and cultured in the F medium containing 50 μg/mL ascorbic acid until the formation of sheets that could readily be handled, which were composed of fibroblasts embedded in their self-produced extracellular matrix. The time to obtain fibroblast-derived tissue sheets was 21–28 days depending on the intrinsic capacity of fibroblasts to secrete extracellular matrix, which varies between donors. Three of these sheets were then superimposed to form the dermal substitute using a custom-made paper frame to facilitate handling. After an additional week of maturation to allow cohesion between the tissue sheets, keratinocytes from the same donor were seeded at 1.0 × 105 cells/cm2 on the reconstructed dermis to form the epidermal component of the bilayered skin substitute. Keratinocyte expansion on the substitute was allowed through a week of immersed culture in the K medium containing 50 μg/mL of added ascorbic acid. The engineered skin construct was raised to the air–liquid interface 28 and cultured in the K medium supplemented with 50 μg/mL ascorbic acid, without epidermal growth factor for an additional 9–10 days in order for terminal keratinocyte differentiation to take place. Therefore, the total duration for the production of the SASS ranges between 45 and 52 days.
Production of a decellularized dermal template for SASS-DM culture
Decellularized dermal templates were produced as previously described. 19 Briefly, neonatal fibroblasts were seeded at 4000 cells/cm2 in a 75 cm2 flask (Falcon, BD, Franklin Lakes, NJ) and cultured for 19 days in the F medium supplemented with 50 μg/mL ascorbic acid to produce tissue sheets, further stacked three-by-three as described in the standard dermal substitute reconstruction of SASS (see previous section “Production of standard SASS”). Neonatal cells were used for their great capacity to produce quality extracellular matrix. Reconstructed dermal substitutes were cultured for a week to allow an appropriate cohesion between tissue layers. Following this culture step, dermal substitutes were decellularized using two cycles of osmotic shock, followed by a rinsing and a dehydration process, then frozen at −20°C, and stored until further use. 19
Production of SASS-DM
Frozen dermal templates previously produced and frozen (see previous section “Production of a decellularized dermal template for SASS-DM culture”) were thawed overnight at 4°C in the F medium containing 0.5 μg/mL amphotericin B (Sigma). The next day, dermal templates were immersed in the standard F medium and kept in an incubator. One newborn and two adult fibroblast cell populations were seeded at 4.2 × 104 cells/cm2 density on thawed dermal templates (n = 3 cell populations). The repopulated dermal templates were cultured in the F medium supplemented with 50 μg/mL ascorbic acid for a week; then, keratinocytes were isolated from the same donor as the fibroblasts and were seeded at 1.0 × 105 cells/cm2 density on the recellularized dermal template. Afterward, keratinocytes were cultured until differentiation, as described in previous section “Production of standard SASS”, for a total production duration with cell patients of 24 days.
Production of green fluorescent protein lentiviral expression vector and transduction of keratinocytes
To induce GFP expression, 293FT keratinocytes were transfected using Lipofectamine 2000 (Invitrogen) with 2.7 μg of an equimolar mixture of PLp1, PLp2, pVSVg packaging plasmids (gracious gift from Dr. Sylvain Guerin) and plenti6.3-GFP expression vector (Invitrogen). Three days posttransfection, the lentivirus-containing supernatant was collected and cleared through a 0.45 μM filter and titrated using 293FT cells. The multiplicity of infection (MOI) was determined by flow cytometry with log amplification using a fluorescence-activated cell sorter (FACScan, BD Biosciences, Oakville, CA) for detection of GFP-expressing cells 4 days after transduction.
Adult and neonatal keratinocytes were cultured on a feeder layer of irradiated human fibroblasts until 80% confluence was reached as previously described. 15 Then, cells were trypsinized and seeded in 75 cm2 culture flasks with the K medium and incubated at 37°C, 8% CO2. After 4 h, the medium was changed by 10 mL of K medium containing 8 μg/mL Polybrene (Sigma-Aldrich Canada) and 1 MOI of lentivirus. GFP-transduced cells were isolated by flow cytometry, multiplied, and cryopreserved until used (pool of positive cells was 99% after fluorescent-activated cell sorter isolation, data not shown).
Grafting of skin substitutes on athymic mice
Adult athymic nu/nu male mice (42 days old, mean weight 29.4 g) were used as graft recipients (Charles River Laboratories, Lasalle, Quebec, Canada). Mice were maintained under sterile housing conditions and received intraperitoneal ceftazidime antibiotic (3 mg/mouse, Glaxo, Toronto, Ontario, Canada) 48 and 24 h before surgery and preoperatively to prevent infections. Mice were injected with Buprenorphine (0.05–0.1 mg/kg, Champion Alstoe, Whitby, Ont, Canada) before surgery and Carprofen (5–10 mg/kg, Pfizer, Kirkland, QC, Canada) daily for 48 h postoperatively to provide analgesia. Skin substitutes (2.5 cm2) were grafted on the mid-back of the animal as previously described. 29 After removing a circular piece of skin (2.5 cm2), a Fusenig's chamber was inserted into the opening. The poorly vascularized fascicular panniculus was removed, and the skin substitute was grafted onto the muscular bed. Fusenig's chambers were removed 21 days after surgery. Mice were inspected at regular intervals for skin necrosis, infection, and graft integration. Mice were sacrificed at 7 and 14 days, as well as 6 weeks after grafting to obtain biopsies for further analysis. Two graft series in which 3 mice were sacrificed for each biopsy time point for a total of 18 mice per condition were conducted. There were also three mice grafted with SASS-DM cultured with GFP-transduced keratinocytes and followed for 6 weeks, with weekly analyses using IVIS. Macroscopic graft take evaluation was conducted by a single plastic surgery resident at each biopsy time point.
Histological and immunofluorescence analysis
Skin samples were frozen in Tissue-Tek optimal cutting temperature compound (Bayers, Etobicoke, ON, Canada) for immunofluorescence imaging or fixed in Histochoice (Amresco, Solon, OH), embedded in paraffin, and stained with Masson's trichrome 30 for histological analysis. Indirect immunofluorescence assays were performed on 5-μm-thick cryosections of skin substitutes fixed in acetone as previously described. 31 Antibodies used were rabbit type IV collagen polyclonal (Abcam, Cambridge, MA), rat laminin monoclonal (Abcam), and rabbit loricrin polyclonal (Covance Cedarlane, Burlington, Ont, Canada). Cell nuclei were stained with Hoechst 33258 (Sigma Chemical, St-Louis, MO). Primary antibodies were replaced by PBS-BSA 1% for negative controls. Secondary antibody for type IV collagen and loricrin was chicken anti-rabbit with Alexa 594 (Life technologies, Burlington, Ont, Canada), whereas the antibody for laminin was goat anti-rat with Alexa 594 (Life technologies).
In vivo imaging analysis
Before image acquisition, mice were anesthetized with 1.5–2% Isoflurane (Abbott, Markham, Ont, Canada), and such anesthesia was maintained during scanning. Fluorescence emitted by the GFP-expressing keratinocytes used to produce SASS-DM was analyzed using an IVIS Lumina II device (Perkin Elmer, Waltham, Massachusetts). Three mice were monitored at the same time with automatic exposition times set up by the system and a binning factor of two for all animals at each time point. Images were acquired and analyzed using the Living Image software (Perkin Elmer, Waltham, Massachusetts). Strong signals emitted by the GFP-expressing keratinocytes were represented in yellow, while low signals appeared in red. Absence of fluorescent signal revealed the corresponding overlap of the black and white macroscopic picture.
Results
Macroscopic appearance and histological analysis
As observed for the SASS, the SASS-DM was resistant, could be easily handled for grafting, and presented a uniform whitish macroscopic aspect (Fig. 1A). The SASS-DM microscopic structure was similar to standard SASS as well as native normal human skin (Fig. 1B). Histological analyses of SASS-DM reconstructed from both adult and newborn cells before grafting showed a fully differentiated epidermis, including a stratum corneum, above a dense and fully repopulated dermal substitute (Fig. 1B).

Sometimes, histological analysis of SASS and SASS-DM cultured in vitro revealed ovoid structures, hereinafter referred to as epithelial cell inclusions, in the dermal component of some SASS and SASS-DM when adult fibroblasts were used. Some of the structures were in contact with the epidermis forming an invagination of the dermoepidermal junction. The occurrence of epithelial cell inclusions was much more frequent in SASS-DM compared to SASS made with corresponding adult cell population of fibroblasts and keratinocytes. Over culture time, innermost cell layers of the epithelial inclusions resembled anucleate squames detaching in the center (Fig. 1C, right panel), probably as a result of cell proliferation and differentiation. Epithelial cell inclusions were not observed in SASS or in SASS-DM when the dermal component was produced from neonatal fibroblasts.
To demonstrate adequate engraftment and safety, SASS-DM was transplanted on athymic mice and compared with SASS. The macroscopic graft evolution was similar between SASS-DM and SASS at 14 days after grafting (Fig. 2). Complete healing, graft take, and integration were observed in every mouse up to 6 weeks after grafting as indicated by fully adherent, well-colored, and uniform grafts (Fig. 2). The appearance of SASS-DM produced with adult and newborn cells was also similar. No cases of infection or graft lysis were observed in any of the conditions studied.
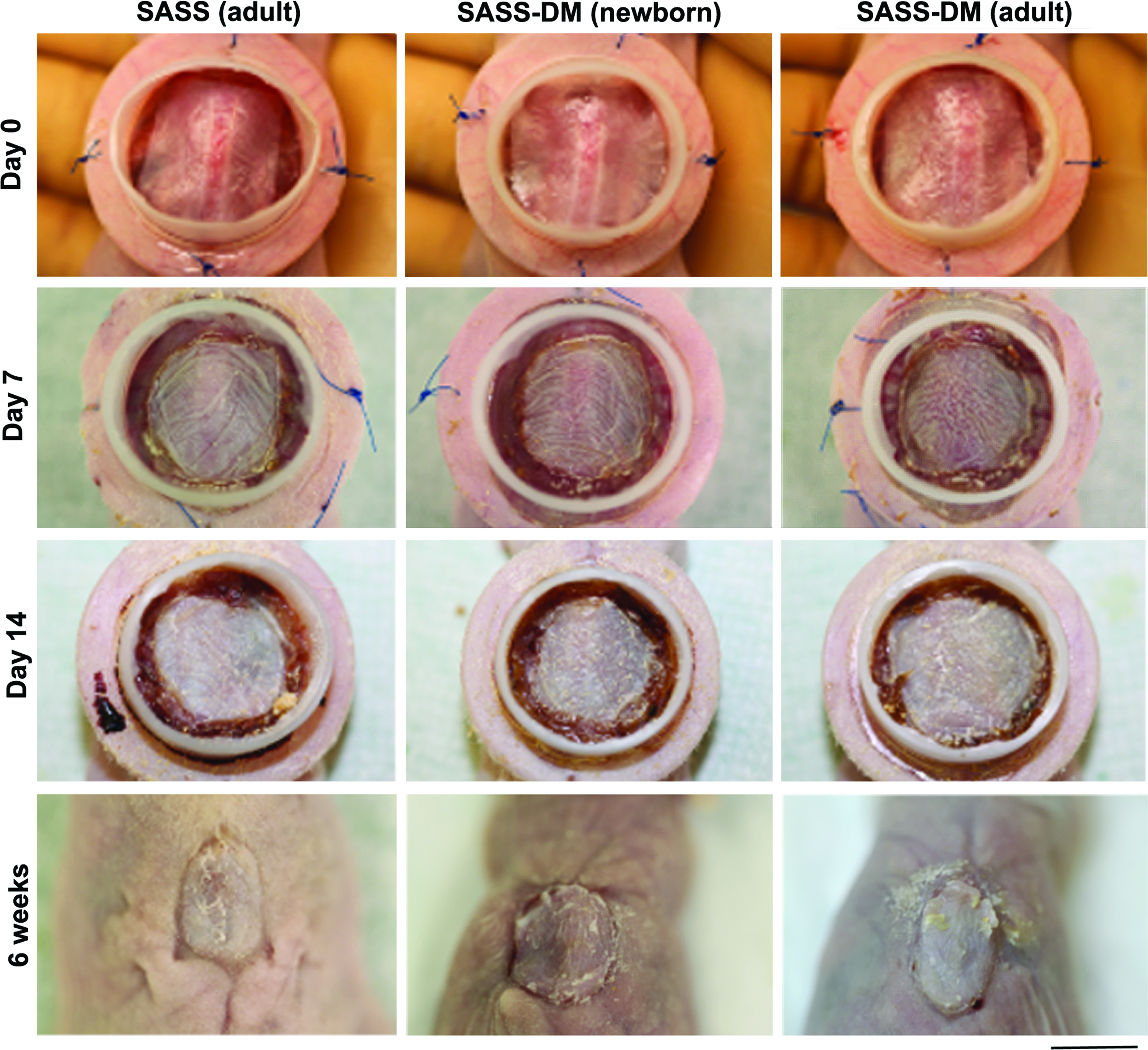
Representative macroscopic results of SASS produced with adult cells (left panels) and SASS-DM produced with newborn and adult cells (center and right panels, respectively) at 0 (first line), 7 (second line), and 14 (third line) days and 6 weeks (fourth line) after grafting. Note that the SASS as well as SASS-DM appeared uniform without any sign of lysis throughout evolution, up to complete healing. Scale bar: 1 cm. Color images available online at www.liebertpub.com/tea
To characterize the microscopic evolution of grafted SASS and SASS-DM, full-thickness skin biopsies were harvested at 1, 2, and 6 weeks after grafting. Histological analysis of these biopsies revealed that the evolution of SASS and SASS-DM was similar and was characterized by the persistence of a dense extracellular matrix whose thickness increased with time (Fig. 3). SASS- and SASS-DM-respective epithelium showed typical features of human epidermis and were prone to a normal desquamation phenomenon (Fig. 3). These observations suggested the presence of a cell population able to renew, differentiate, and self-regulate the epithelium within SASS and SASS-DM. No difference was observed between SASS-DM cultured from adult and newborn cells. As expected in successful graft take, adherence of skin substitutes to the wound bed was also observed throughout tissue manipulation and histological preparation as none of the grafted SASS or SASS-DM separated from the deeper subcutaneous tissue (Fig. 3).
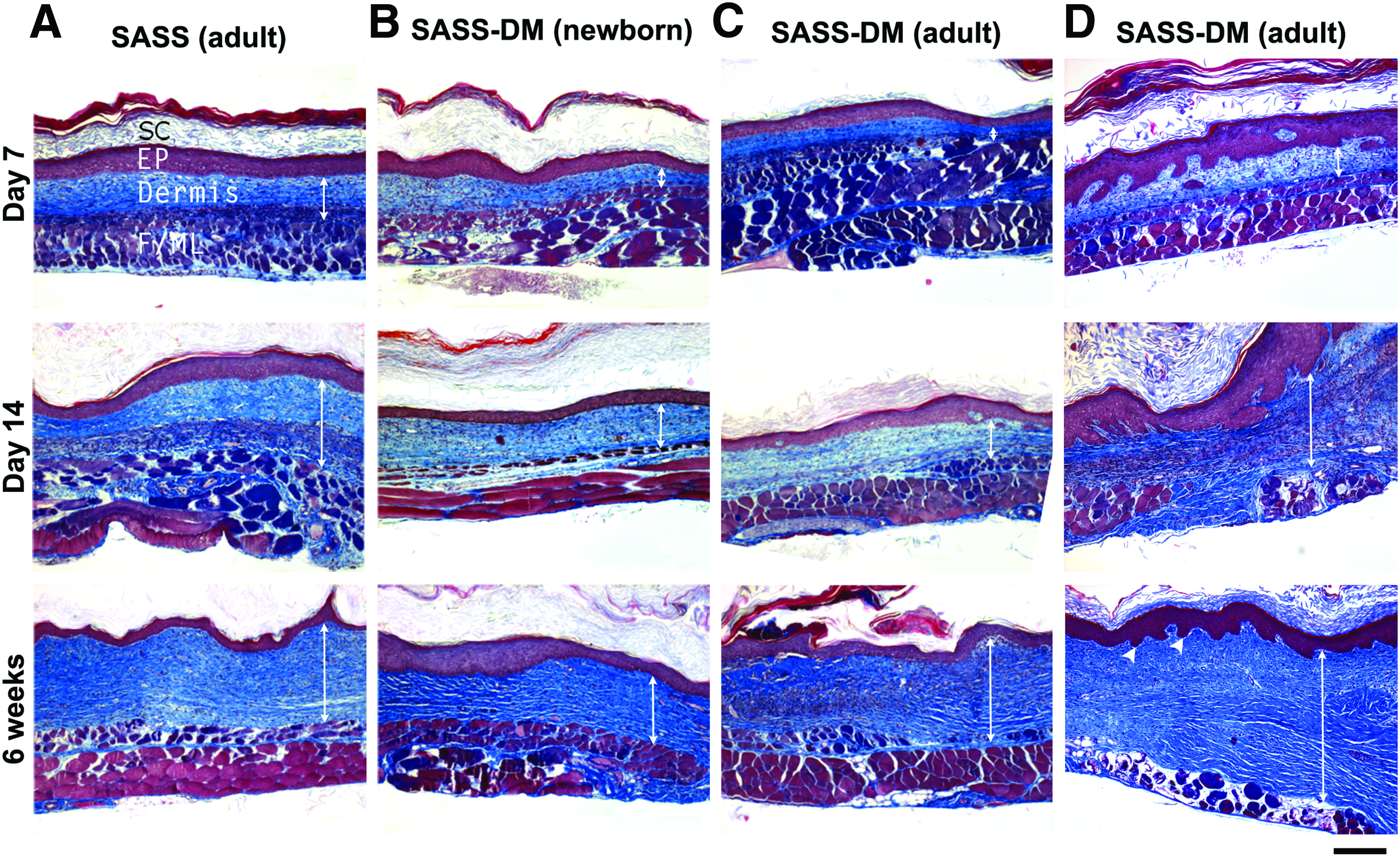
Histological analysis (Masson's trichrome staining) of SASS produced with adult cells
The epithelial cell inclusions observed in vitro resorbed with time after grafting. Some ovoid structures were present 7 days after grafting. However, none was observed in any grafted skin substitutes 6 weeks after grafting (Fig. 3). At this time point, only small invaginations of the dermoepidermal junction resembling rete ridges were observed; all epithelial cells were in continuity with the epidermis.
Immunofluorescence analysis
The presence of collagen type IV as well as laminin, two important components of the basement membrane, was assessed by immunofluorescence assays. Type IV collagen and laminin formed a continuous line at the dermoepidermal junction up to 6 weeks after grafting for each condition (Fig. 4A–F, arrows). As expected, loricrin, a major protein of the cornified cell envelope found in terminally differentiated keratinocytes of the epidermis, was present in the upper layers of SASS and SASS-DM 6 weeks after grafting (Fig. 4G–I).
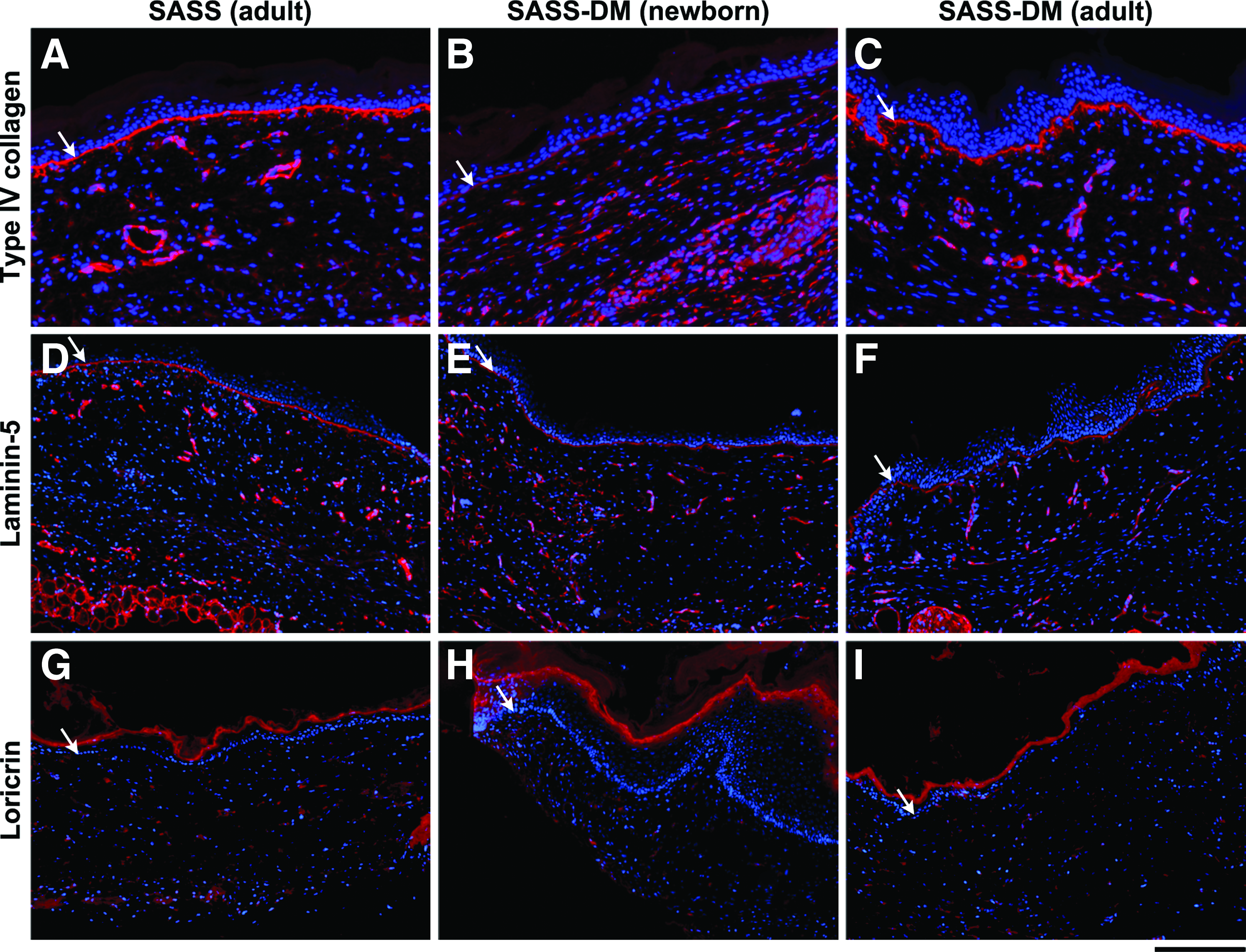
Immunofluorescence detection of type IV collagen
In vivo imaging
To demonstrate that SASS-DM integration and healing were achieved from the human cells forming the tissue and not due to mouse cell migration or wound contraction, newborn and adult human keratinocytes were transduced with GFP, allowing the follow-up of these cells within SASS-DM through time after grafting using IVIS. Results showed the persistence of human GFP-transduced newborn (data not shown) and adult keratinocytes within SASS-DM (Fig. 5) and SASS (data not shown) up to 6 weeks after grafting, demonstrating permanent engraftment of human SASS-DM to the host. Following the removal of Fusenig's chambers after 21 days, mild wound contraction was observed at the graft periphery, as expected since mouse skin tends to heal with more contraction compared to human skin, but the presence of human cells was demonstrated by the GFP labeling.

Evolution of SASS-DM produced using adult fibroblasts paired with GFP-transfected keratinocytes, from 1 to 6 weeks after grafting. Mice were observed with IVIS, allowing to follow the persistence and integration of grafted tissue with fluorescence emission. Since athymic mouse cells are not transfected, the detection of fluorescence at each time point (up to 6 weeks after grafting) confirms that healing and regeneration of the SASS-DM epithelium is due to the persistence of human epithelial cells within the graft and not due to mouse cell migration. Mice 1 through 3 are presented in the same order for each panel. Subtle wound contraction following Fusenig's chamber removal at day 21 is notable on the mouse 1 (left). Also worth mentioning, mouse 1 scratched her back following chamber removal, causing the loss of center and right inferior quadrants of the graft during follow-up. However, GFP-transduced human keratinocytes were still detected in the graft at 6 weeks with probable mouse intrinsic healing contribution in the center of the graft. GFP, green fluorescent protein; IVIS, in vivo imaging system. Color images available online at www.liebertpub.com/tea
Discussion
In the last few years, our group has continuously endeavored to optimize our production of skin substitutes to mimic nature as close as possible. Our self-assembly approach has enabled us to provide skin substitutes that are very similar to the human skin, allowing to replace skin when lacking. However, this technique is very time-consuming and in the context of severely wounded burn patients, this step can be problematic as survival improves dramatically with prompt complete definitive coverage.
Despite this, SASS produced according to the previously reported method15,32 is currently used in clinical trials for burns and was used on chronic wounds.12,15 With the self-assembly approach, the extracellular matrix composing the dermal portion of the SASS is entirely produced from fibroblasts. Furthermore, SASS allows the preservation of epithelial stem cells localized in the basal layer of the fully differentiated epidermis.18,29 However, as previously explained, the time necessary for SASS production is quite long 15 and needs to be shortened to be more quickly used to treat severely burned patients.
Recently, we described an optimized method allowing to produce a skin substitute faster, SASS-DM, which presented histological and mechanical characteristics equivalent to those of SASS. 19 SASS-DM takes advantage of a self-assembled dermal matrix made from allogenic cells, which is further devitalized and used as a template to seed autologous fibroblasts and keratinocytes. Herein, we showed that the in vivo maturation level of the SASS-DM when compared to SASS was equivalent. Moreover, we confirmed that SASS-DM integrated and was stable overtime after grafting on a mouse model, as no difference between skin substitutes produced with both methods was observed as pertains to their architecture and evolution following surgery.
A shorter production delay of any engineered tissue represents a huge advantage by helping with optimal surgical planning and brings to the bedside an enhanced patient quality of care. As allografts currently used for temporary coverage of severely burned patients with insufficient donor sites need to be replaced every 2–4 weeks due to immunologic reaction, 33 the prompt availability of a permanent autologous skin substitute could lower the number of surgeries needed, in turn, reducing the time before full permanent coverage as well as the total length of stay in the intensive care unit. 8 Considering that presently, about 13–17 days are required to obtain enough cells to reconstruct autologous SASS-DM from a 3 cm2 skin biopsy, in addition of a SASS-DM production time of 24 days, the overall production delay is about 1 month and a half. We consider this production time to be acceptable in a clinical setting.
SASS-DM possesses interesting characteristics when compared to other available bilayered skin substitutes. Among the autologous bilayered skin substitutes developed, some use fibrin as a dermal extracellular matrix equivalent8,34 with hydrogels, 35 hyaluronic acid, 36 or a human donor acellular dermal layer combined to cultured epithelial autograft (CEA)37,38 or a thin split-thickness autograft.39,40 These substitutes, available between 2 and 5 weeks, need temporary allograft application for wound bed preparation, harvesting skin donor sites, or application of CEA.
An autologous bilayered skin substitute model based on an esterified hyaluronic acid scaffold with autologous fibroblasts, keratinocytes, and melanocytes was applied in patients for permanent coverage purposes. 9 However, the loss of the epithelial portion was observed in 10 of 11 treated patients, highlighting the importance of preserving epithelial stem cells and the basement membrane within skin substitutes for their permanent integration. As shown previously, SASS and SASS-DM both present a continuous basement membrane at the dermoepithelial junction. 19 The presence of a well-structured basement membrane is a key element for the reappearance of a niche allowing stem cell preservation at this expected location in a skin substitute.18,41,42 Its absence results in the loss of stem cells in collagen gel skin equivalents. 31 We previously demonstrated that SASS provide a microenvironment supporting the maintenance of keratinocytes presenting stemness features such as quiescent state, K19 expression, contact with basement membrane, and long-term regeneration of the SASS epithelium after grafting. 18 In this study, we demonstrate that epithelial stem cells are maintained in SASS-DM since its epithelium renews after grafting, as confirmed by the preservation of the GFP signal from transduced human keratinocytes throughout in vivo evolution.
The observation of epithelial cell inclusions within the dermal component was reported to occur in other models of skin substitutes production and, in all instances, were shown to disappear following skin graft integration and maturation.8,43 As observed by Compton et al., a complete disappearance of epithelial cell inclusions observed within skin substitute in vitro occurred within 6 weeks after grafting. 43 The number of epithelial inclusions, referred to as keratin peals, observed in skin substitutes was positively correlated with the keratinocyte seeding density. 44 However, a minimal seeding density of at least 50,000 cells/cm2 was deemed necessary to obtain a continuous epidermis and prevent epidermal degeneration. 44 Herein, a keratinocyte seeding density of 100,000 cells/cm2 was used, allowing a consistent and well-developed epidermal layer. Since all epithelial cell inclusions observed in SASS and SASS-DM resorbed overtime in vivo, their initial quantity before grafting was not considered to be clinically significant.
This study showed no difference in graft take, regeneration of the epithelium, and macroscopic or microscopic evolution for both SASS and SASS-DM, when grafted onto athymic mice. Histological studies demonstrated the ability of the grafts to offer an environment stimulating extracellular matrix production by fibroblasts even after grafting, yielding thicker dermis through time. Results of SASS-DM produced from both adult and newborn were similar, thus indicating that the optimized protocol could also be used in a pediatric population. However, even if the athymic mouse model is a useful tool to evaluate the in vivo evolution of a human skin graft, it does not assess the risk of graft rejection caused by residual heterologous cellular material within the extracellular matrix following decellularization. Therefore, our next step will be to evaluate if the decellularized self-assembled matrix that we developed elicits adverse host responses after grafting onto an immunocompetent in vivo model. While residual cellular material sufficient to induce a negative remodeling response may vary depending upon the model, the residual amount of double-stranded foreign DNA within the extracellular matrix substitute after the decellularization process should not exceed 50 ng/mg dry weight. 45
Another investigator, Dr. Steven T Boyce, created a cultured skin substitute (CSS) by combining human cells with a sponge-like biomaterial. 46 The fabrication of CSS consists in seeding autologous fibroblasts onto an acellular collagen and glycosaminoglycan bovine matrix, on which were added autologous keratinocytes allowing the formation of a fully differentiated epidermis. Excluding the time required to isolate and amplify autologous skin cells, the CSS production requires 3 weeks. The CSS was used for the treatment of severe burns in a clinical study conducted on 40 severely burned patients. 7 Excellent results were obtained for CSS integration (graft take of 81.5%) and long-term regeneration of the epithelium.
Conclusions
We conclude that SASS-DM is a very promising skin substitute for the permanent coverage of skin wounds since it displays an excellent graft take, its histological architecture is similar to native skin, and its epithelium continually renews after transplantation. The shorter production time required compared to SASS should certainly raise the laboratory unit production capacity in acute necessity. Such a reduction in the production step with patient cells represents a huge clinical gain, thus optimizing burn care and improving patient prognosis by reducing the time before definitive total skin coverage.
Footnotes
Acknowledgments
The authors would like to thank Anne-Marie Moisan, Todd Galbraith, and Sebastien Larochelle for technical assistance, Sylvain Guerin for gracious gift of plasmids, and Dr. Jamil Sawaya for kindly reviewing the article. This work was supported by the Fondation des Pompiers du Québec pour les Grands Brûlés (FPQGB), Canadian Institutes of Health Research (CIHR), MOP-14364 (F.A.A.) and MOP-12087 (L.G.), and the Cell and Tissue Therapy Network (ThéCell) from the Fonds de Recherche du Québec en Santé (FRQS). L.G. is the holder of a Canadian research chair (Tier 1) on stem cells and tissue engineering from CIHR. MOP-115093 (F.A.A. and V.J.M.) and FND 143213 (L.G.).
Author Contribution Statement
C.B.C. and R.G.1 conceived the experiment and together with C.P., carried it out; B.G. produced GFP-expressing cells; C.B.C. and D.L. cowrote the article; V.J.M., L.G., R.G.3, and F.A.A. cosupervised experiments and revised the article.
Disclosure Statement
No competing financial interests exist.