
Abstract
Adipose-derived stem cells (ADSCs) can differentiate into various cell types and thus have great potential for regenerative medicine. Herein, rat ADSCs were isolated; transduced with lentiviruses expressing Osterix (Osx), a transcriptional factor essential for osteogenesis. Osx overexpression upregulated key osteogenesis-related genes, such as special AT-rich binding protein 2, alkaline phosphatase, osteocalcin, and osteopontin, at both mRNA and protein levels. In addition, mineral nodule formation and alkaline phosphatase activity were enhanced in Osx-overexpressing ADSCs. The expression of dickkopf-related protein 1, a potent Wnt signaling pathway inhibitor, was also increased, whereas that of β-catenin, an intracellular signal transducer in the Wnt pathway, was decreased. β-catenin expression was partially recovered by treatment with lithium chloride, a canonical Wnt pathway activator. The Osx-expressing ADSCs were then combined with 3D gelatin-coated porous poly(ɛ-caprolactone) scaffolds with a unique release prolife of entrapped recombinant human vascular endothelial growth factor (VEGF). The controlled release of VEGF promoted osteogenic differentiation capacity in vitro. When the scaffold-ADSC complexes were transplanted into rat calvarial critical-sized defects, more bone formed on the gelatin/VEGF-coated scaffolds than on other scaffold types. Taken together, the results indicate that, Osx-overexpression promotes ADSCs' osteogenesis both in vitro and in vivo, which could be enhanced by release of VEGF.
Introduction
B
Osterix (Osx), a zinc finger-containing transcription factor, is essential for bone formation and osteoblast differentiation. In Osx-null mice, no bone formation occurs and preosteoblasts cannot differentiate into fully functional osteoblasts. 9 Acting downstream of Runx2, Osx enhances proliferation and osteogenic lineage commitment in mesenchymal stem cells.10,11 Osx also downregulates the canonical Wnt/β-catenin signaling pathway by activating dickkopf-related protein 1 (Dkk1) in osteoblasts.12–14 However, to date, the osteogenic effects of Osx on ADSCs and the associated molecular mechanisms have not been fully elucidated.
Several synthetic polymer scaffolds have been explored as seed-cell carriers for tissue engineering applications.15–18 With its excellent biocompatibility, favorable mechanical properties, and low degradability, poly(ɛ-caprolactone) (PCL) is one of the few biomaterials approved by the Food and Drug Administration for such purposes. 19 The modification of polymer scaffolds with biochemical cues can enhance their osteoconductivity. Among all the cytokines, VEGF, especially the VEGF165 isoform, has been shown to have the highest capacity to promote both osteogenic and angiogenic differentiation.20,21 However, the use of high doses of VEGF can cause severe inflammation and deformities. 22 As a result, numerous studies have been performed to optimize the VEGF release profile.23–25 A 3D porous PCL scaffold immobilized with recombinant human bone morphogenetic protein-2 exhibited enhanced osteoconductivity and favorable physical and mechanical abilities. 26 Notably, gelatin-coated PCL scaffolds displayed an optimized biphasic release profile composed of a transient burst release followed by a sustained release. Such a profile meets the intrinsic needs of cell differentiation and bone formation processes. 26 These findings raise the questions of whether such PCL scaffolds could achieve controlled release of VEGF and whether they could exhibit osteoconductive effects in bone-tissue engineering applications.
In this study, we investigated the influence of Osx on the osteogenic differentiation of ADSCs. Then, gelatin-coated/VEGF PCL scaffolds containing Osx-overexpressing ADSCs were constructed and transplanted into critical-sized calvarial defects in rats. Through the in vitro and in vivo studies, we try to demonstrate the effect of the complex of porous scaffold entrapped with VEGF and Osx-modified ADSCs on bone formation, which may advance our understanding of ADSC fate and facilitate the clinical translation of ADSC-mediated bone therapies.
Materials and Methods
Isolation and culture of rat ADSCs
Four- to six-week-old female Sprague Dawley rats were obtained from the Ninth People's Hospital Animal Center (Shanghai, China). All procedures were approved by the Animal Research Committee of the Shanghai Ninth People's Hospital. Briefly, inguinal fat pads were separated, sliced, and digested with 0.1% collagenase type I (Sigma). After centrifugation, cell pellets were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin (all from Invitrogen) at 37°C in 5% CO2 for 5–7 days. ADSCs were detached with a 0.25% (w/v) trypsin/1 mM EDTA solution (1:1, v/v) after reaching 80–90% confluency, and 1.0 × 106 cells were seeded onto 10 cm dishes in 10 mL medium.
ADSC identification
ADSC proliferation ability was quantified for 8 days using a cell counting kit-8 (Sigma) according to the manufacturer's instructions. In a typical flow cytometry experiment, 1.0 × 106 cells were collected in PBS and incubated with a CD45-PE, CD11b-PE, and CD90-FITC solution (Invitrogen) for 30 min at 37°C in the dark. Analysis was performed using a FACS Calibur flow cytometer (BD Biosciences). For cell cycle detection, the cells were harvested and fixed with 75% ice-cold ethanol at 4°C for 30 min in the dark. Then, DNA content was measured using the flow cytometer, and cell cycle fractions were determined. To perform a colony-forming assay, 1.0 × 103 cells were seeded onto 35 mm Petri dishes, grown for 10 days, fixed, and stained with crystal violet (Sunshine Biotechnology). Colonies were visualized under an inverted microscope and photographed. The multi-lineage differentiation potential of the ADSCs was confirmed by incubation of the cells in osteogenic media and adipogenic media (Cyagen). To induce osteogenic differentiation, the cells were cultured in osteogenic medium consisting of DMEM, 10% FBS, 50 μM ascorbic acid (Sigma), 10 mM β-glycerophosphate (Sigma), and 100 nM dexamethasone (Sigma). After 14 days of induction, the cells were fixed with 95% ethanol and stained with 1% Alizarin Red S (ARS) for 30 min at 37°C. To detect adipogenic differentiation, the cells were fixed with 4% paraformaldehyde (Polysciences) and stained with Oil Red O (Cyagen) after 14 days of osteogenic induction.
Lentiviral transduction of ADSCs
Osx-overexpressing lentiviruses were purchased from Cyagen Technologies. ADSCs were transduced with lentiviruses expressing Osx (Lenti-Osx) or control lentiviruses (Lenti-GFP) at a multiplicity of infection of 50 using 8 mg/mL polybrene. Transduction efficiency was determined by analyzing Osx mRNA and protein levels at 48 h and 72 h post-transduction.
Protein extraction and western blotting analysis
Cells were lysed in RIPA lysis buffer containing 10 mM phenylmethylsulphonyl fluoride (Beyotime) on ice for 30 min. Equal amounts of protein samples were electrophoresed on 10% SDS-PAGE gels (Beyotime), transferred onto PVDF membranes (Millipore), and immunoblotted with antibodies specific to the following proteins: rat Osx (Abcam), 1:800 dilution; Runx2 (Bioworld Technology), 1:1000 dilution; special AT-rich binding protein 2 (Satb2) (Abcam), 1:1000 dilution; alkaline phosphatase (ALP) (Abcam), 1:800 dilution; osteocalcin (OCN) (Biosynthesis Biotechnology), 1:300 dilution; osteopontin (OPN) (Bioworld), 1:800 dilution; Dkk1 (ABclonal), 1:800 dilution; and β-catenin (ABclonal), 1:1000 dilution. The membranes were developed using an ECL detection kit (Millipore) on an ABI system.
RNA extraction and real-time quantitative PCR analysis
Total RNA was extracted from cells at 80% confluence using TRIzol reagent (Invitrogen). Reverse transcriptions were performed using a PrimeScript RT reagent kit (Takara). Primers for each target gene were synthesized according to the sequences listed in Table 1. Real-time PCR reactions were performed on an Applied Biosystems 7300 Real-time PCR system using the SYBR Green PCR kit (Takara) according to the manufacturer's recommendations. Relative mRNA expression was quantified compared to GAPDH as an internal control.
Immunofluorescence microscopy
ADSCs were seeded and grown on glass coverslips 24 h before immunofluorescent staining. After fixation with 4% paraformaldehyde for 20 min at room temperature and two washes with ice-cold PBS, the cells were permeabilized with 0.1% Triton X-100 (Sigma) in PBS for 15 min at room temperature. The cells were washed thrice with PBS and blocked in 3% bovine serum albumin for 1 h at room temperature. The cells were then incubated with primary antibodies diluted in 1% bovine serum albumin to 5 mg/mL (anti-Osx) or 10 mg/mL (anti-Runx2) overnight at 4°C. After three washes with 1% bovine serum albumin for 10 min, the cells were incubated with appropriate secondary antibodies and subjected to cytoskeletal actin staining for 1 h at room temperature. After three washes with 1% bovine serum albumin, the cells were incubated with DAPI (Invitrogen) at a 1:1000 dilution for 5 min in the dark, rinsed with PBS, and mounted on coverslips.
Immunofluorescence was analyzed on a confocal LSM700 microscope (Carl Zeiss) using a Plan-Apochromat 63 × 1.4NA oil objective (Carl Zeiss) at room temperature with ZEN2009 software. DAPI, Alexa Fluor® 488 and Alexa Fluor 647 fluorescence was induced by exciting with 405, 488, 488, and 639 nm lasers. Minimal adjustments to brightness and contrast were applied to all images using ZEN2009 software.
Alkaline phosphatase and Alizarin Red S activity assays
An ALP activity assay was performed as previously described using an ALP activity kit (Sigma) and was normalized to total cell protein content. 27 After ARS staining, images were acquired, and nodules were destained by incubation with 10% cetylpyridinium chloride (Sigma) in 10 mM sodium phosphate for 30 min at room temperature. Calcium concentration was determined by measuring the absorbance at 520 nm with a universal microplate reader (Bio-Tek Instruments). This experiment was performed in triplicate; the results are presented as the mean ± SD.
Lithium chloride treatment
LiCl (Sigma) was dissolved in sterile PBS at 10 mM and stored at 4°C. In subsequent experiments, cells were cultured in either complete medium or osteogenic medium supplemented with 10 mM LiCl.
Fabrication of the 3D scaffolds
Three-dimensional porous scaffolds were prepared through a combination of solvent casting/salt leaching and a thermal-induced phase separation (TIPS) technique according to methods previously described by Zhang et al. 26 For animal experiments, molds were designed into cylinders with a diameter of 5 mm and a height of 2–3 mm. A 5% gelatin solution supplemented with 0.02% genipin (w/v) as a cross-linker was used to coat the scaffold surfaces. The VEGF-containing scaffolds were prepared by immersing the primary scaffolds in the rhVEGF165 solutions (R&D Systems; 1 μg/mL) at 37°C under vacuum for 30 min and then dried. As a control, a scaffold without coating, designated Sf, was created, and the scaffolds coated with the 5% gelatin solution were designated Sf-g.
Characterization of the 3D scaffolds
The microstructures of the porous scaffolds were confirmed using a scanning electron microscope (SEM; JSM-6360LV, Jeol). The compressive modulus of each scaffold (diameter, 15 mm; height, 10 mm) was detected on an Instron universal testing machine (CMT5000). The crosshead speed was set at 1 mm/min with a 1 kN load cell. The compressive modulus was determined based on the slope of the initial linear portion of the stress–strain curve. The swelling ratio was determined using equation (1)
wherein
wherein
wherein
Scaffolds harboring ADSCs for in vitro studies
Scaffolds were sterilized by immersion in 75% ethanol for 2 h followed by UV irradiation for 24 h. Before cell seeding, the scaffolds were incubated in complete medium for 24 h. Then, 20 μL of a single-cell suspension with a density of 1.0 × 107 cells/mL was added to each scaffold. After ensuring cell adhesion following 2 h of incubation, the scaffolds were incubated with osteogenic medium (DMEM, 10% FBS, 50 μM ascorbic acid, 10 mM β-glycerophosphate, and 100 nM dexamethasone) for 7 days. ARS staining and an ARS activity assay were performed to detect the osteogenic effects of the cell-seeded scaffolds. After a series of procedures, such as immunofluorescent staining, the scaffolds were observed under a laser scanning confocal microscope (Olympus).
Animal experiments
A total of 18 male Sprague Dawley rats weighing 350–400 g were used in this study. Briefly, the animals were anesthetized by injection of pentobarbital (Nembutal 3.5 mg/100 g). After exposing the calvarium with a sagittal incision, a 5 mm critical-sized defect was created using a low-speed dental engine with a burr. The surgery was performed on both sides of the skull in 18 rats. The 36 defects of 18 rats were randomly divided into six groups, each receiving one of the following implants: (1) empty scaffold (n = 6), (2) ADSC+Sf/g scaffold (n = 6), (3) LeV-GFP+Sf/g scaffold (n = 6), (4) LeV-Osx+Sf/g scaffold (n = 6), (5) LeV-Osx+Sf/g-VEGF scaffold (n = 6), and (6) LeV-Osx+Sf-VEGF scaffold (n = 6). Then, the incision was closed in layers with 4–0 resorbable sutures. Eight weeks later, the cranial bones were harvested, fixed, and used for microcomputed tomography (micro-CT) and hematoxylin and eosin staining.
Microcomputed tomography and histology
The morphologies of the reconstructed calvarial bones were assessed using a micro-CT system (μCT-80; Scanco Medical). The following CT settings were used: pixel matrix, 1024 × 1024; slice thickness, 20 μm. After scanning, the micro-CT images were segmented using a nominal threshold value of 225 as reported previously, 28 and 3D histomorphometric analysis was performed automatically. The parameter of volume of newly regenerated bone was used for comparison in this study. The scaffolds were also fixed, decalcified, and cryosectioned into a series of 8-μm sections using a microtome (CM1100; Leica) and then subjected to hematoxylin and eosin staining. For Micro-CT analysis, the entire “region of interest” was chosen and multiple slices were chosen for histological analysis. The resolution for the images analyzed is 2448 pixel × 1920 pixel. To avoid bias, the samples from different groups were collected, processed, and analyzed under the same condition, and four randomly selected cross sections from each implant were observed using standard methods as previously described.11,29 Bone formation was analyzed using Image J software to assess histological images.
Statistical analysis
All data are expressed as the mean ± SD of three or more independent experiments. Statistical analyses were conducted using Student's t-test or ANOVA unless otherwise stated. p values <0.05 were considered statistically significant.
Results
Identification of ADSCs
The isolated ADSCs had the typical fibroblast- or spindle-like morphology (Fig. 1A). The results from the cell counting kit-8 assay revealed that, after the third passage, the ADSCs proliferated into a logarithmic phase on the third day and reached a plateau period on the seventh day; the proliferation curve had an “S” shape (Fig. 1B). Cell cycle analysis by flow cytometry revealed that approximately 26.34% of cells were in S-phase (Fig. 1C), indicating the vigorous proliferation potential of the harvested ADSCs. The acquired cells were negative for CD45 and CD11b, but positive for CD29 (Fig. 1D), indicating the mesenchymal nature (vs. hematopoietic) of these stem cells. The colony-forming assay results demonstrated that a mass of cell colonies (more than 50 cells) formed (Fig. 1E), reflecting the stemness of the ADSCs. The adipogenic and osteogenic differentiation capacities of the ADSCs as detected by the Oil Red O and ARS staining were evident from the numerous lipid droplets and calcium nodules formed compared to the ADSCs cultured in the control medium (Fig. 1F, G). Moreover, as shown in Figure 1H, the fluorescence intensities of Runx2 and OPN were higher for the cells cultured in the osteogenic medium compared to those cultured in the control medium, indicating the osteogenic differentiation capacity of the isolated ADSCs.
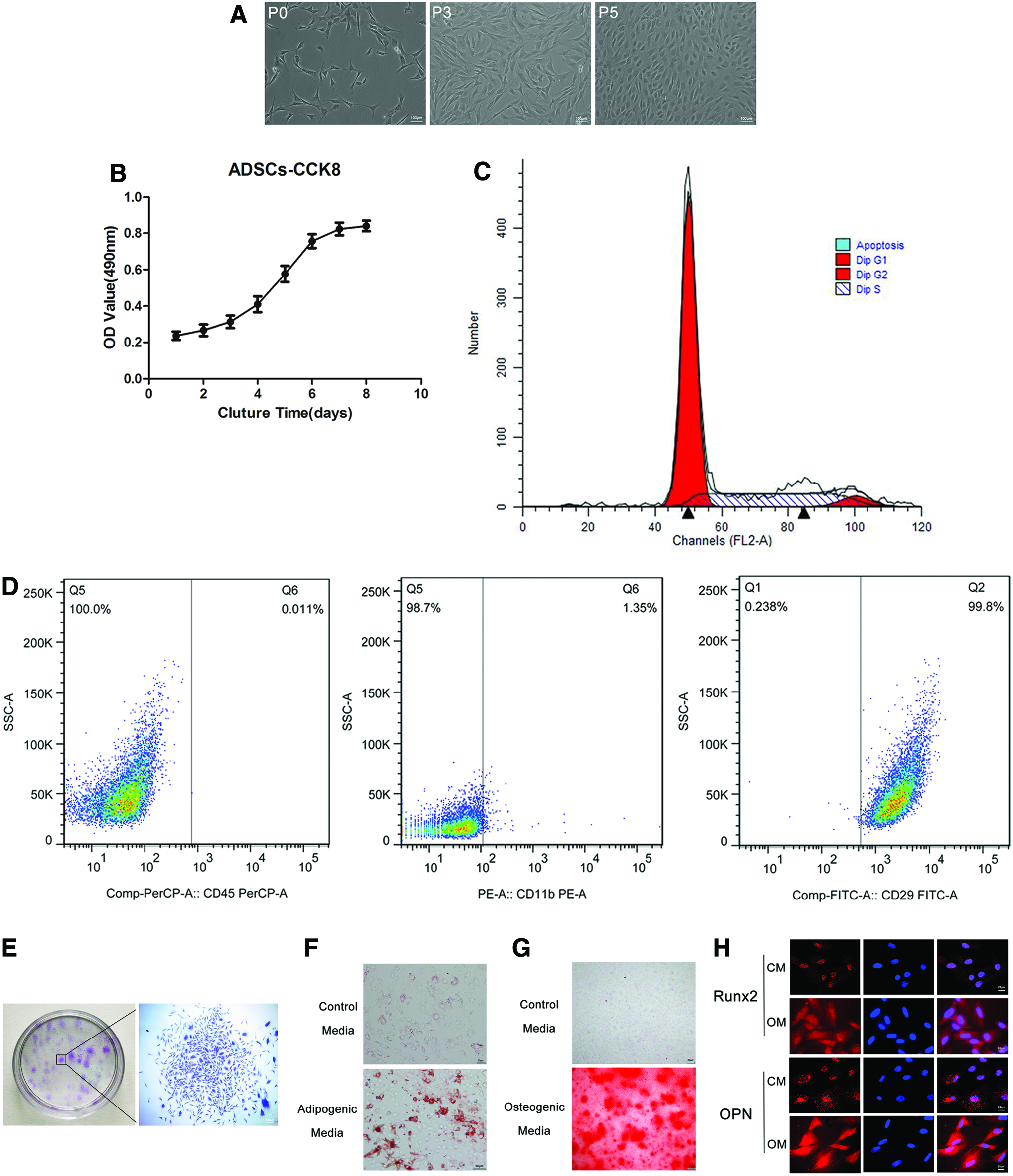
Identification of ADSCs.
Osx overexpression promotes ADSC osteogenesis
To verify the function of Osx in the osteogenic differentiation of ADSCs, ADSCs stably overexpressing Osx were generated using a lentiviral expression vector. The mRNA and protein levels of Osx were markedly increased in the LeV-Osx-transduced cells in comparison with the negative control (nontransduced cells) and LeV-GFP-transduced cells (Fig. 2A, B). Immunofluorescence staining confirmed these data (Fig. 2C). We next characterized the effects of Osx overexpression on the osteogenic differentiation of ADSCs. The ADSCs transduced with LeV-Osx viruses exhibited more retention of Alizarin Red S in comparison with the two controls (nontransduced and LeV-GFP-transduced ADSCs) after osteogenic induction for 10 days (Fig. 2D). As expected, the ARS and ALP activities in the Osx-overexpressing cells were significantly higher than in the two controls (Fig. 2E, F). Real-time PCR and western blotting data revealed that overexpression of Osx significantly promoted the expression of Satb2, ALP, OCN, and OPN (Fig. 2G, H). Together, these results show that Osx overexpression enhanced the osteogenic differentiation of ADSCs.

LeV-Osx transduction and Osx overexpression promotes osteogenesis in ADSCs.
Downregulated β-catenin expression in Osx-overexpressing ADCSs could be partially recovered by LiCl treatment
Several studies have indicated that Osx promotes osteogenic differentiation by downregulating the Dkk1-dependent Wnt-signaling pathway. Therefore, we studied the role of Osx in the osteogenic process of ADSCs. In Osx-overexpressing cells, β-catenin expression was obviously decreased, while Dkk1 expression was upregulated in comparison with controls (Fig. 3A, B). This result supported the published observation that Osx controls the expression of osteogenic genes partially through the Dkk1-Wnt/β-catenin pathway. 30 To further support this hypothesis, we examined the influence of LiCl, a canonical Wnt pathway activator, on the major players in the Wnt signaling pathway. First, Osx-overexpressing ADSCs were treated with increasing concentrations of LiCl (0, 2.5, 5, 10, and 20 mM), and the expression of β-catenin in these cells was analyzed by western blotting. β-catenin expression was upregulated with increasing LiCl concentration (Fig. 3C), while 20 mM LiCl inhibited ADSC growth. Therefore, we chose 10 mM as an optimal concentration for subsequent experiments. Osx-overexpressing ADSCs were treated with or without 10 mM LiCl for 10 days, and β-catenin expression in these cells was analyzed by immunocytochemistry (Fig. 3D). As expected, LiCl enhanced the expression of β-catenin. The LiCl-treated cells also exhibited fewer mineral nodules than untreated cells (compare lane 1 to lane 2 in Fig. 3E), a finding supported by the results from the ARS and ALP activity assays (Fig. 3F, G). Real-time PCR further revealed that the mRNA level of β-catenin increased and the mRNA levels of OCN and OPN decreased in the Osx-overexpressing ADCSs treated with 10 mM LiCl (Fig. 3H). Western blot analysis of the same cells produced similar results on the protein level (Fig. 3I). Collectively, our findings clearly demonstrate that Osx overexpression enhances osteogenesis by downregulating β-catenin and upregulating Dkk1. In addition, we found that treating Osx-overexpressing ADSCs with LiCl inhibited their osteogenic differentiation.
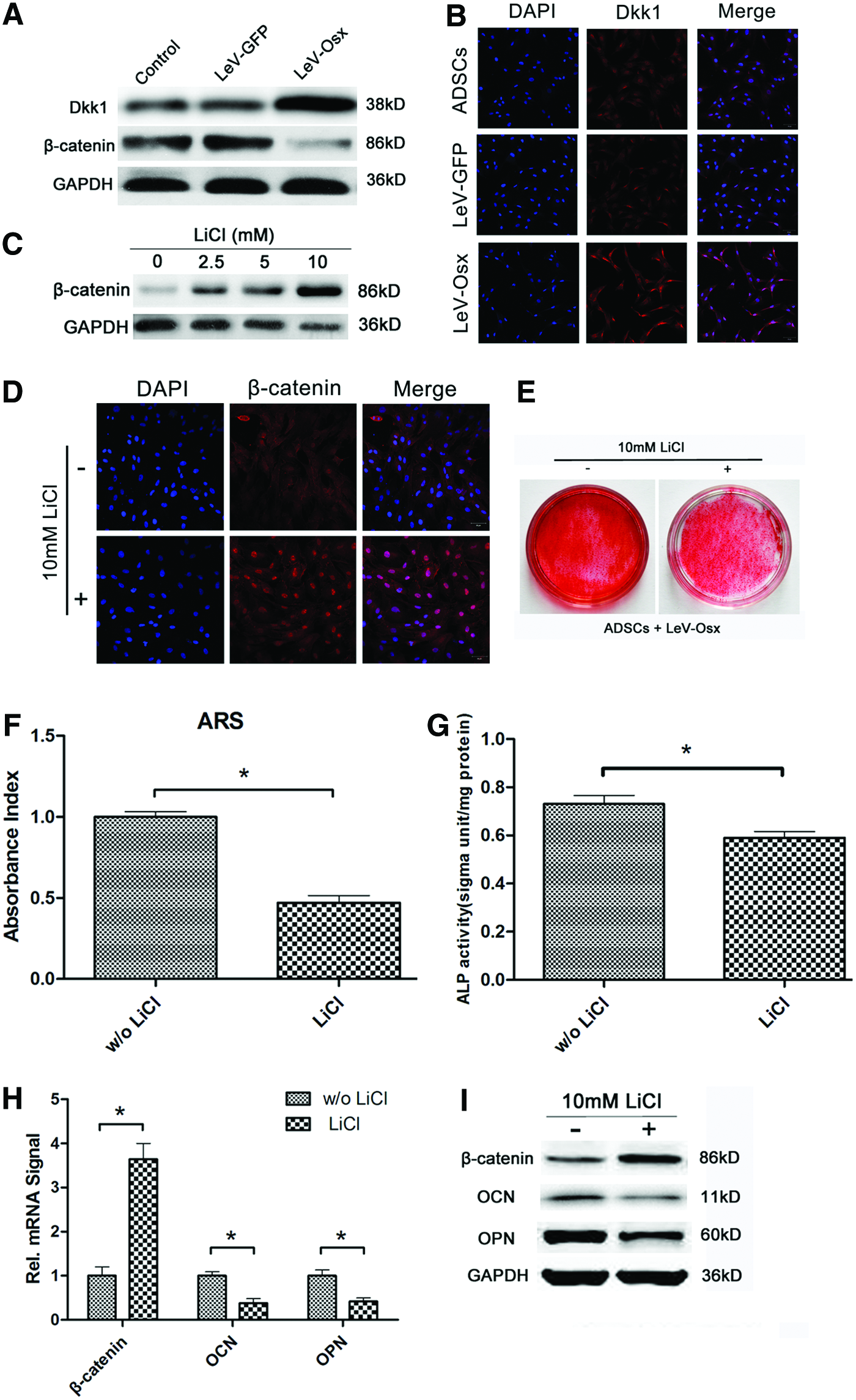
LiCl treatment recovered downregulated β-catenin expression in Osx-overexpressing ADSCs.
Controlled release of VEGF from 3D porous gelatin-coated/VEGF scaffolds
To develop 3D porous gelatin-coated/VEGF scaffolds, a combination of solvent casting, salt leaching, and the TIPS technique with gelatin surface coating was used. This approach resulted in scaffolds capable of the controlled release of VEGF (Fig. 4A). Scaffold morphology was observed under a SEM and revealed a hierarchical porous microstructure (Fig. 4B). Specifically, large pores (200–300 μm) and medium pores (40–50 μm) were generated through salt leaching with screened NaCl particles (200–300 μm) and NaHCO3 particles (45–50 μm), while small pores (<10 μm) were produced by TIPS. In detail, the large pores, whose diameter was nearly 200–300 μm, were measured through SEM in accordance with the diameter of the screened NaCl particles, and so on. The pores were well organized and mutually connected, laying a foundation for cell adhesion and proliferation. The compressive modulus test results revealed that the scaffolds had an approximately five-fold higher compressive modulus after gelatin coating (Fig. 4C). The swelling capacity of the Sf-g scaffolds was higher than that of the Sf scaffolds (Fig. 4D), indicating that the water-intake ability of the porous scaffolds improved with gelatin coating. Comparing the VEGF release kinetics between the Sf and Sf-g scaffolds showed a burst release pattern in the case of the Sf scaffolds and a biphasic release pattern in the case of the Sf-g scaffolds, the latter being characterized by a small burst release followed by a sustained release (Fig. 4E). Comparing the relative release of VEGF from the Sf and Sf-g scaffolds demonstrated that approximately 88% of the VEGF in the Sf scaffolds was released within the first 24 h, while only approximately 35% of the VEGF in the Sf-g scaffolds was released within the first day, followed by a steady release of 75% of the VEGF by 96 h (Fig. 4F). These data indicate that controlled release of VEGF from the 3D porous scaffolds was achieved.
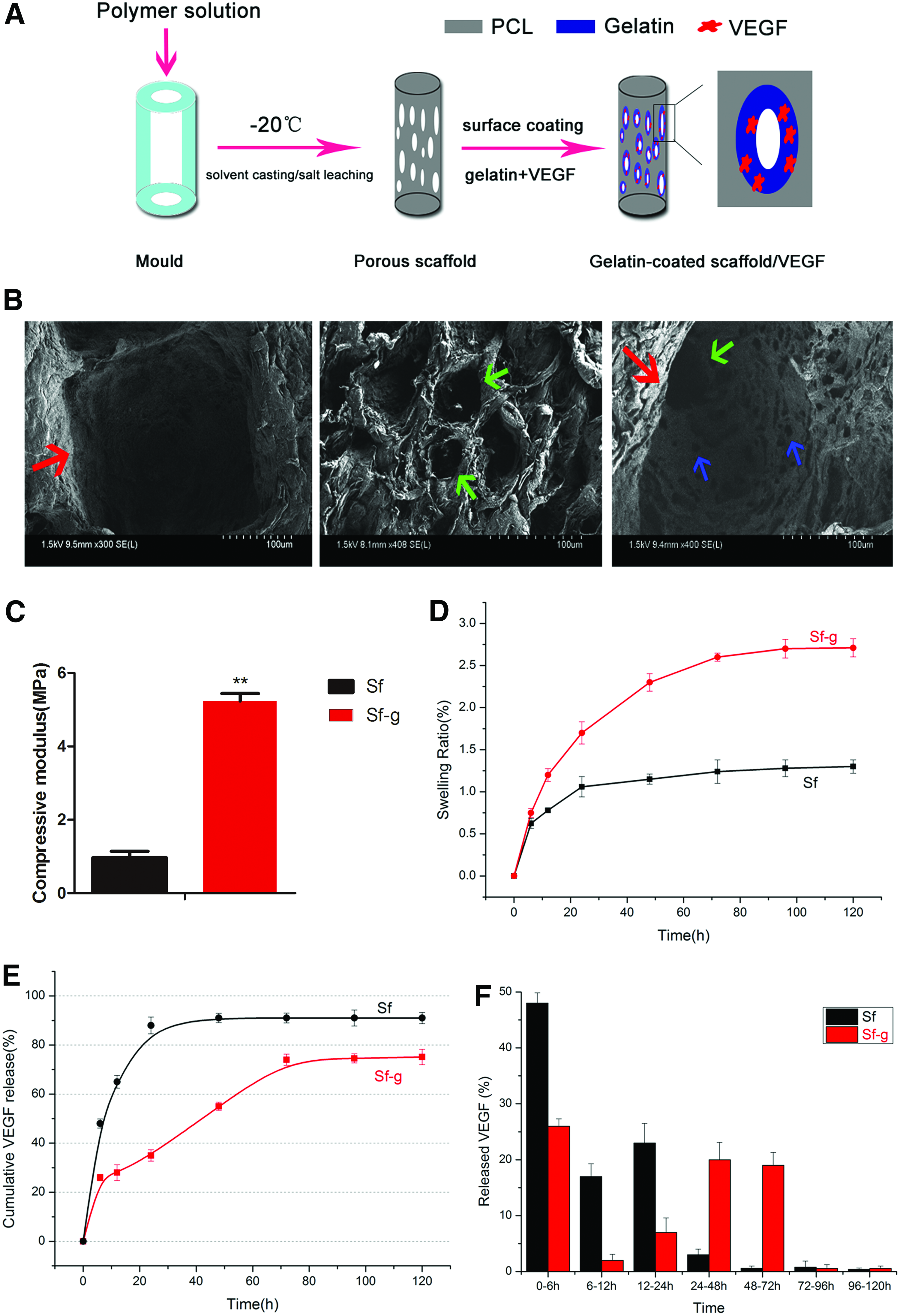
Fabrication and characterization of the 3D porous gelatin-coated/VEGF scaffolds.
Gelatin-coated/VEGF scaffolds accelerated the osteogenic process in Osx-overexpressing ADSCs
To evaluate the biocompatibility and osteoconductivity of Sf, Sf/VEGF, and Sf-g/VEGF scaffolds, Osx-overexpressing ADSCs were seeded on these scaffolds and cultured in osteogenic medium for 10 days in vitro. Under confocal microscope observation, the spatial distributions of cells on all three scaffold types were basically uniform (Fig. 5A, B), indicating the scaffolds had similar biocompatibility. However, the ADSCs grew better on the Sf-g/VEGF scaffolds compared to the others. As shown in Figure 5C, ARS staining revealed that the Sf-g/VEGF scaffolds displayed the best osteoconductivity. As expected, the ARS activity assay results confirmed the staining results (Fig. 5D). These data indicate that the Sf-g/VEGF scaffolds had markedly better osteoconductivity than the others.

Gelatin-coated/VEGF scaffolds supported the growth and osteogenic differentiation of Osx-overexpressing ADSCs.
Enhancement of bone healing by Osx overexpression and VEGF release in vivo
A critical-sized defect of 5 mm in diameter was created on both sides of the cranium and filled with Sf/g-VEGF porous scaffolds containing genetically modified ADSCs (Fig. 6A). To evaluate new bone formation within the defects, micro-CT was performed after explantation of the cranium. Representative images showing the morphology of the newly formed bone on each type of scaffold are presented in Figure 6B. Only a small amount of new bone was visible on the peripheries of the defect edges in the empty scaffold group. The implantation of ADSC-complexed and LeV-GFP-transduced ADSC-complexed Sf/g scaffolds led to the formation of scattered patches of new bone across the defect areas, although less bone was formed on these scaffolds compared to the Sf/g scaffolds complexed with Osx-overexpressing ADSCs (LeV-Osx+Sf/g). The major portion of each critical-sized defect was filled with a substantial amount of new bone tissue in the LeV-Osx+ Sf/g-VEGF group 8 weeks postoperation, and the new bone tissue showed better regeneration compared to that in the LeV-Osx+Sf/VEGF group. The volume of regenerated bone (BV) was analyzed to quantify new bone formation. As shown in Figure 6C, it was significantly higher for the LeV-Osx+Sf/g-VEGF group compared to the controls.

Micro-CT evaluation of healed critical-sized calvarial defects 8 weeks postoperation.
Moreover, the histological evidence further supported the radiographic findings, indicating that the newly formed bone in the LeV-Osx+Sf/g-VEGF group had the typical organization and morphology of mature bone. In contrast, more fibrous connective tissue than bone tissue was observed in the other groups (Fig. 7A). The percentage of new bone area determined by hematoxylin and eosin staining was 45.53% ± 5.57% in the LeV-Osx+Sf/g-VEGF group, which was significantly higher than that measured in the other five groups.

Histological analysis of new bone formation.
Discussion
Craniofacial bones are formed by intramembranous ossification, a process characterized by the direct conversion of mesenchymal tissue into bone. To repair craniofacial bone defects, bone-tissue engineering has been considered a promising technique. The goal of this method, namely, the induction of functional bone regeneration, can be achieved using a combination of biomaterials (osteoconductive scaffolds), osteoprogenitor cells, and osteoinductive factor therapy. In this study, we repaired critical-sized defects in rat calvarial bone with Osx-overexpressing ADSCs seeded onto gelatin-coated PCL scaffolds, which were covered with recombinant human VEGF165 protein.
The groundbreaking study of Zuk et al. in 2001 demonstrated that ADSCs represent an alternative source of bone marrow-derived mesenchymal stem cells for cell-based therapies. In bone regeneration research, ADSCs have been widely used as seed cells.4,17,18,31 Recently, several studies reported that bone marrow-derived mesenchymal cells have superior bone-forming potential in comparison with adipose-derived cells.32,33 However, we still uphold the view that ADSCs are more promising for regenerative applications because these cells generally cause less damage to hosts. To obtain more data about ADSC-induced bone regeneration, ADSCs were isolated, characterized, genetically modified, and transplanted into scaffolds. Both in vitro and in vivo studies showed that the ADSCs exhibited favorable proliferation ability and osteogenic capacity. Although we obtained very promising results, additional studies aimed at understanding the fate and management of such ADSCs are required before their clinical application.
Osteogenic differentiation and bone regeneration are coordinated by a multi-step molecular pathway regulated by various transcription factors and osteoinductive proteins, including Osx and VEGF. Osx, a key osteoblast-specific transcription factor, is an important regulator of osteogenic differentiation in mesenchymal stem cells. To maximize ADSC capacity for bone regeneration, ADSCs were transduced with Osx via lentiviruses (Fig. 2). The cells transduced with Osx exhibited remarkably accelerated calcium nodule formation as shown by ARS staining and ALP activity measurements (Fig. 2E, F). Meanwhile, the mRNA (Fig. 2G) and protein (Fig. 2H) levels of four critical osteogenic factors, specifically Satb2, ALP, OCN, and OPN, were upregulated in the Osx-overexpressing ADSCs in contrast with the two controls (nontransduced and GFP-expressing ADSCs). As a downstream gene of Osx, Satb2 belongs to a family of special AT-rich binding proteins that bind to nuclear matrix attachment regions.34,35 Our previous study showed that Satb2 facilitated the differentiation of induced pluripotent stem cells (iPSCs) toward osteoblast-lineage cells. 29 In addition, silk scaffolds seeded with Satb2-modified iPSCs successfully repaired critical-size mouse calvarial bone defects. 29 The presence of ALP, another early-stage marker of osteoblastic differentiation, indicates the commitment of stem cells toward an osteoblastic phenotype. Conversely, OCN is considered a marker for late-stage bone formation, 36 which is characterized by bone mineralization via calcium ion homeostasis. OPN plays a critical role in the maturation of osteoblasts. 37 In this study, Osx-overexpressing ADSCs showed higher osteogenic capacity compared to nontransduced and GFP-expressing ADSCs, as verified by micro-CT and hematoxylin and eosin staining results (Figs. 6 and 7). Collectively, these data indicated that overexpression of Osx in ADSCs promotes osteoblastic differentiation and that such cells might be used to repair bone defects.
VEGF, a key angiogenic factor, has the strongest and most significant biological activity in enhancing blood vessel formation.38,39 As osteogenesis is closely coupled with angiogenesis, we hypothesized that treatment with VEGF could promote the osteoblastic differentiation of Osx-ADSCs and enhance bone formation. To verify this hypothesis, a porous PCL scaffold fabricated through solvent casting/salt leaching and TIPS was used as a VEGF delivery vehicle. Porous PCL scaffolds were chosen because of their highly precise pore size, interconnectivity, structure, and mechanical properties. 26 To facilitate cell adhesion and growth, the PCL scaffolds were surface-coated with a gelatin layer. Importantly, in contrast to Sf scaffolds, the Sf/g scaffolds exhibited an enhanced compressive property and better hydrophilicity, satisfying the needs of bone-tissue engineering. The hierarchical pore structure observed on the Sf/g scaffolds could be key to bone generation.
Many studies have shown that a large initial burst release of drugs or proteins can lead to a transient supra-physiological concentration that causes severe inflammatory responses and even deformities.40,41 Therefore, slow controllable release of drugs or proteins is a subject of great interest to mimic the endogenous osteogenic micro-environment. Previous experiments have shown that VEGF and morphogenetic protein-2 can be bound to and released in a controllable manner from mineral-coated PCL scaffolds. 42 In this study, the gelatin-coated scaffolds showed a biphasic release profile for VEGF after the gelatin was cross-linked (Fig. 4E), which is consistent with findings reported by Zhang et al. 26 Specifically, approximately 35% of the VEGF contained within the Sf/g scaffolds was released in the first 24 h, whereas nearly 88% of the VEGF contained within the Sf scaffolds was released in the same period. We speculate that phase I of this release profile was due to the burst release of weakly entrapped VEGF, while phase II was due to the sustained release of VEGF resulting from the degradation of the gelatin matrix, as has been previously described. 43 As shown in Figures 5–7, the Sf-g/VEGF group showed more matrix mineralization and new bone formation compared to the Sf/VEGF group. These data indicate that the Sf/g-VEGF scaffold was more favorable for ASDC growth and osteoblastic differentiation due to the controlled release of VEGF.
To date, the specific mechanisms underlying Osx-mediated osteogenesis in ADSCs remain poorly understood. It has been demonstrated that Osx binds to and activates the Dkk1 promoter, while Dkk1 prevents the activation of Wnt signaling by binding to LRP5/6.12,44 Here, we provided evidence that Osx exerts its function through the Dkk1-β-catenin signaling pathway when ADSCs are undergoing osteoblast differentiation. First, Dkk1 and β-catenin were respectively upregulated and downregulated with the overexpression of Osx (Fig. 3A). Second, β-catenin expression increased following treatment with LiCl, a representative Wnt pathway activator (Fig. 3C). In conjunction with this, calcium nodule formation and ALP activity decreased sharply (Fig. 3E–G). Furthermore, the expression levels of OCN and OPN were markedly reduced according to ARS and ALP assay results (Fig. 3H, I). These findings highlight that Osx exerts its function during osteoblastic differentiation of ADSCs through the Dkk1-β-catenin signaling pathway, providing novel insight into the regulatory loops that are involved in bone formation. However, it should be noted that we did not evaluate the expression pattern of Dkk1/β-catenin in vivo. Nonetheless, we provide important information regarding the possible mechanism of Osx-mediated osteogenesis in ADSCs and provide a foundation for further investigations into the upstream and downstream molecular cues associated with Osx-mediated osteogenesis.
Conclusion
In summary, our data showed that Osx-overexpressing ADSCs exhibit robust osteogenesis both in vitro and in vivo. Furthermore, we showed that the immobilization of VEGF in porous PCL scaffolds enhanced the osteogenic ability of the seed cells. For the first time, we demonstrated that Osx overexpression affects ADSC osteogenic differentiation via the Dkk1-Wnt/β-catenin pathway, although the mechanism underlying this relationship requires further study. Overall, our work describes a promising method for bone-tissue engineering using Osx-overexpressing ADSCs and VEGF-coated PCL scaffolds.
Footnotes
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81371123 and 81302359), the Jiangsu Natural Science Foundation (BK2012844), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD, 2014–37), and the Qing Lan Project through grants awarded to J.H-Ye. This work was also partially supported by the National Natural Science Foundations of China (No. 81222013) with a grant awarded to J Gao and the Research Grants (15411950300) from Science and Technology Commission of Shanghai Municipality.
Disclosure Statement
No competing financial interests exist.