
Abstract
Human perivascular stem/stromal cells (hPSC) are a multipotent mesenchymogenic stromal cell population defined by their perivascular locale. Recent studies have demonstrated the high potential for clinical translation of this fluorescence-activated cell sorting (FACS)-derived cell population for autologous bone tissue engineering. However, the mechanisms underlying the osteogenic differentiation of PSC are incompletely understood. The current study investigates the roles of canonical and noncanonical Wnt signaling in the osteogenic and adipogenic differentiation of PSC. Results showed that both canonical and noncanonical Wnt signaling activity transiently increased during PSC osteogenic differentiation in vitro. Sustained WNT3A treatment significantly decreased PSC osteogenic differentiation. Conversely, sustained treatment with Wnt family member 16 (WNT16), a mixed canonical and noncanonical ligand, increased osteogenic differentiation in a c-Jun N-terminal kinase (JNK) pathway-dependent manner. Conversely, WNT16 knockdown significantly diminished PSC osteogenic differentiation. Finally, WNT16 but not WNT3A increased the adipogenic differentiation of PSC. These results indicate the importance of regulation of canonical and noncanonical Wnt signaling for PSC fate and differentiation. Moreover, these data suggest that WNT16 plays a functional and necessary role in PSC osteogenesis.
Introduction
M
Bone marrow MSC (BMSC) are most commonly studied for bone repair, yet have significant impediments for clinical translation. These include low cell frequency, low yield, harvest-site morbidity, and culture requirement. In contrast, adipose tissue is readily available by routine liposuction procedures with minimal morbidity.4–6 The stromal vascular fraction (SVF) has been previously used for bone repair, but formed bone tissue unreliably 7 or with a low efficacy, 8 compared with culture-expanded adipose-derived stromal cells.
To address the issue of heterogeneity within the SVF, we previously purified a population of MSC termed perivascular stem/stromal cells (PSC) from the SVF of human white adipose tissue. 9 PSC are purified by fluorescence-activated cell sorting (FACS) and are a comparatively homogenous MSC population for regenerative medicine applications.10,11 PSC are abundant in human white adipose tissue and are present in clinically relevant numbers (approximately 40% of the viable SVF). 11 Importantly, PSC are identified and derived without culture expansion.10,12
PSC originate in the vessel wall, which has now been recognized and well established as a rich source of mesenchymal progenitor cells.13,14 PSC are composed of two distinct yet related cell populations, including pericytes (CD34−CD146+CD45−) and adventitial cells (CD34+CD146−CD45−).14,15 Both perivascular cell populations express canonical MSC markers in situ, after FACS purification, and after in vitro expansion (including CD44, CD73, CD90, and CD105).14,15 Compared with cells of the SVF from the same patient, PSC have shown a significantly greater potential for bone formation by their ability to form bone in an intramuscular pouch model in SCID mice.11,16 In addition, PSC have been shown to promote in vivo bone regeneration across other animal models, including a rat spinal fusion model10,16 and a calvarial defect model. 17
The commitment of MSC to an osteogenic cell fate relies on many signaling pathways and transcription factors, including Hedgehog signaling,18–20 NEL-like molecule-1 (NELL-1) signaling,20,21 β-catenin-dependent canonical Wnt signaling, and β-catenin-independent noncanonical Wnt signaling.22–24 Ample studies have examined the importance of Wnt signaling on osteogenesis, regulation of bone mass, and skeletal healing.25,26 Although the influence of canonical and noncanonical Wnt signaling in MSC osteogenesis has been studied, PSC are a distinct MSC subtype defined by their perivascular residence. In our prior studies, the novel differentiation factor NELL-1 has been previously shown to predispose PSC to an osteogenic cell fate,11,16,27 potentially via canonical Wnt signaling activation. 28 The current study aims to investigate the role of canonical and noncanonical Wnt signaling in PSC osteogenesis.
The canonical β-catenin-dependent pathway is initiated in MSC by the binding of extracellular Wnt ligands such as Wnt3a, 29 Wnt6, Wnt10, or Wnt10b 30 to the transmembrane frizzled (Frz) receptors on the cell surface. 31 This binding induces complex formation with the low-density lipoprotein receptor (LRP5/6) and intracellular disheveled (DSH) proteins, 32 which then inhibits an intracellular complex to inhibit the phosphorylation and subsequent degradation of β-catenin, leading to its accumulation. β-catenin translocates to the nucleus and subsequently binds with lymphoid enhancer-binding factor/T cell factor 32 to promote transcription of genes that stimulate MSC osteogenic differentiation. The ability of canonical Wnt signaling to enhance osteogenesis in MSC is controversial and is thought to depend on several factors such as the precise level of Wnt signaling, stage of cell differentiation, and microenvironment.23,33 Low levels of canonical Wnt signaling are known to predispose MSC to cell cycle entry, preventing osteogenesis. 34 However, high levels of β-catenin have been shown to impede osteogenic differentiation, potentially via noncanonical Wnt signaling inhibition. 33
Like the canonical pathway, noncanonical Wnt signaling pathways have also been known to promote an osteogenic fate in MSC, 35 in particular the C-Jun N-terminal kinase (JNK) pathway.36–38 Similar to the canonical pathway, the noncanonical pathway is also activated by binding of a Wnt ligand to an Frz receptor. Subsequent recruitment of Rho-GEF proteins then activates GTPases, which in turn activate the JNK pathway. 39 JNK then translocates to the nucleus, where it can activate multiple transcription factors. Known noncanonical Wnt ligands such as Wnt4, Wnt5, and Wnt11 have been shown to enhance osteogenesis in MSC in vitro.35,40 JNKs have also been known to bind to β-catenin itself in crosstalk between the canonical and noncanonical Wnt signaling pathways. 39
Since both the canonical and noncanonical JNK Wnt signaling pathways have been found to play an important role in the process of osteogenic differentiation of a diverse variety of MSC, the current study investigates the role of two prominent Wnt signaling ligands in PSC under osteogenic and adipogenic differentiation conditions: WNT3A, a potent canonical Wnt ligand and WNT16, a mixed canonical and noncanonical Wnt signaling ligand, which has previously been analyzed by RNA Seq revealing a 21-fold increase in PSC compared with the unpurified SVF. As a comparison, we examined the effects of the canonical Wnt signaling inhibitor Dickkopf-1 (DKK1). Overall, our findings suggest that WNT16 plays a functional and necessary role in PSC osteogenesis.
Materials and Methods
PSC isolation
PSC were isolated from human subcutaneous adipose tissue via FACS. Lipoaspirate was stored for no more than 48 h at 4°C before processing. The human stromal vascular fraction (hSVF) was obtained by collagenase digestion. Briefly, lipoaspirate was diluted with an equal volume of phosphate-buffered saline (PBS) before digestion with Dulbecco's modified Eagle's medium (DMEM) containing 3.5% bovine serum albumin (Sigma-Aldrich, St. Louis, MO) and 1 mg/mL type II collagenase for 70 min under agitation at 37°C. Next, adipocytes were separated and removed by centrifugation. The pellet was then resuspended in red cell lysis buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) and incubated for 10 min at room temperature. After centrifugation, pellets were resuspended in PBS and filtered at 70 μm. The resulting hSVF was further processed for cell sorting (to isolate PSC). hSVF was incubated with a mixture of the following directly conjugated antibodies: anti-CD34-phycoerythrin (1:100; Dako, Glostrup, Denmark), anti-CD45-allophycocyanin (1:100; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and anti-CD146-fluorescein isothiocyanate (1:100; AbD Serotec, Raleigh, NC). All incubations were performed at 4°C for 15 min in the dark. Before sorting, 4′,6-diamidino-2-phenylindole (DAPI; 1:1000; Invitrogen, Carlsbad, CA) was added for dead cell exclusion; the solution was then passed through a 70-μm cell filter and then run on FACSAria cell sorter (BD Biosciences, San Diego, CA). Sorted cells were plated for in vitro studies. In this manner, distinct microvessel pericytes (CD34−, CD146+, and CD45−) and adventitial cells (CD34+, CD146−, and CD45−) were isolated and combined to constitute the PSC population. Prior publications have characterized canonical MSC marker expression among PSC, including CD44, CD73, CD90, and CD105.14,15 Cells were cultured at 37°C in a humidified atmosphere containing 95% air and 5% CO2. The expansion of cells was performed in DMEM, 20% fetal bovine serum (FBS), and 1% penicillin/streptomycin. Routinely, medium was changed every 3 days unless otherwise noted.
Assays in growth medium
PSC were seeded in six-well plates at a density of 1 × 105 cells per well and allowed to adhere overnight. Cells were cultured in DMEM +10% FBS +1% penicillin/ streptomycin and treated with recombinant WNT16 or WNT3A (50 ng/mL) for 6 days, at which time RNA isolation was performed. Medium with Wnt ligands was refreshed every 3 days.
Osteogenic differentiation assays
Assays for PSC differentiation are adapted from our prior publications.27,28 The osteogenic differentiation medium (ODM) was constituted with 10 mM β-glycerophosphate and 50 μM ascorbic acid in DMEM +20% FBS. ODM with Wnt ligand supplementation was changed every third day of differentiation. In select experiments, small-molecule inhibitors of MAPK/JNK signaling were added, including SP600125 and PD98059 (25 μM). In small-molecule inhibitor experiments, DMSO served as vehicle control.
Alkaline phosphatase (ALP) staining was performed using the leukocyte alkaline phosphatase kit (Sigma-Aldrich). Briefly, cells were seeded in 24-well plates at a density of 1 × 104 cells/well. Cells were cultured under osteogenic differentiation conditions for 12 days before staining. Cells were then washed with PBS and fixed with formalin for 10 min at room temperature. Following fixation, cells were stained using the leukocyte alkaline phosphatase kit (Sigma-Aldrich) according to the manufacturer's protocol. Cells were incubated in ALP stain for 15 min at 37°C and then washed with PBS. Cells were allowed to dry and pictures were taken at 100× magnification using the Olympus IX71 inverted system microscope (Olympus, Cypress, CA). Relative staining was quantified using Adobe Photoshop CC2015.
For the detection of mineralization, cells were seeded in growth medium in 24-well plates at a density of 1 × 104 cells/well. Twenty-four hours after cell seeding, the basal medium was replaced with ODM containing WNT3A, DKK1, or WNT16 in triplicate per treatment for 12 days (50 ng/mL). Cells were washed with PBS and fixed with 4% paraformaldehyde. Following fixation, cells were stained with 2% alizarin red (Sigma-Aldrich) at room temperature for 15 min, then washed with deionized water, and allowed to dry. Pictures were taken at 100× magnification using the Olympus IX71 inverted system microscope (Olympus). To quantify bone nodule deposition, 10% v/v acetic acid was added and cells were incubated at room temperature for 30 min with shaking. Cells were then scraped from the wells and vortexed for 30 s. Next, cells were overlaid with mineral oil and heated to 85°C for 10 min. Briefly, cells were cooled on ice for 5 min and then centrifuged at 20,000 g for 15 min. Ten percent ammonium hydroxide was added to adjust the pH to between 4.1 and 4.15. Absorbance was measured in triplicate at 405 nm in 96-well plates using Epoch microspectrophotometer (Bio-Tek, Winooski, VT).
Adipogenic differentiation assays
PSC were seeded in six-well plates at a density of 1 × 105 cells per well and allowed to adhere overnight. Medium was then replaced with the MesenCult adipogenic differentiation medium (StemCell Technologies, Inc., Vancouver, BC) supplemented with exogenous WNT16 or WNT3A (50 ng/mL). Cells were cultured under adipogenic differentiation conditions for 7 days for gene expression analysis. Differentiation medium with exogenous Wnt ligands was changed every 3 days. For visualization by oil red O (ORO) staining, after 10 days of adipogenic differentiation, cells were washed with PBS and fixed with 10% formalin for 30 min. ORO stock solution was prepared from powder (Sigma, St. Louis, MO) by mixing 300 mg ORO powder with 100 mL of 99% isopropanol. Stock solution was diluted 3:2 stock solution: deionized water and allowed to sit at room temperature for 10 min. The working solution was then filtered by gravity filtration. After fixation by formalin, cells were washed with water and 60% isopropanol was added to wells for 5 min before staining. ORO working solution was then added to each well and incubated at 37°C for 30 min. Following incubation, the cells were washed with tap water and counterstained with hematoxylin for 1 min. Images were taken using Q capture software.
Ribonucleic acid isolation and quantitative real-time polymerase chain reaction
Gene expression was assayed by quantitative real-time polymerase chain reaction (RT-PCR), based on our previous methods.28,41 Primers are in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/tea). Time points for specific gene expression include 3, 6, 9, and 12 days of osteogenic differentiation, with additional details provided in the accompanying figure legends. Briefly, total RNA was extracted using the RNeasy Kit (Qiagen, Santa Clarita, CA). One microgram of total RNA from each sample was subjected to first-strand complementary deoxyribonucleic acid (cDNA) synthesis using the SuperScript III Reverse-Transcriptase Kit (Life Technologies) to a final volume of 20 μL The reverse transcription reaction was performed at 65°C for 5 min, followed by 50°C for 50 min and 85°C for 5 min. For qRT-PCR, the reaction was performed using 2 × SYBR green RT-PCR master mix and an ABI PRISM 7300 qRT-PCR system instrument (Applied Biosystems, Foster City, CA). The primers used are listed in Supplementary Table S1. qRT-PCR was performed using 96-well optical plates at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and at 60°C for 60 s. The relative quantification of gene expression was performed using a Comparative CT method according to the manufacturer's protocol and was normalized to the expression levels of the housekeeping gene, GAPDH or ACTB, in each sample.
Total protein extraction and western blot analysis
Protein extraction and western blot analysis were adapted from our prior publications. 28 Western blot analysis was performed using antibodies against nuclear β-catenin, JNK (1:1000), pJNK (1:2000) (Cell Signaling Technologies, Danvers. MA), GAPDH, or H3 (Santa Cruz Biotechnology). To measure cytoplasmic and nuclear β-catenin protein levels, cells were seeded in six-well plates at a density of 4 × 104 cells/well. After attachment, basal medium was replaced with starvation medium containing DMEM + 1% FBS for 16 h. Medium was then replaced with ODM for up to 12 days. Cells were then washed with PBS and resuspended in 150 μL of radioimmunoprecipitation assay buffer (RIPA) (Thermo Fisher Scientific) with 100× Halt protease/phosphatase inhibitor cocktail (Life Technologies) added. Nuclear and cytoplasmic protein was isolated using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific).
To measure JNK and phosphorylated JNK (pJNK), cells were seeded in six-well plates at a density of 4 × 104 cells/well and after attachment, basal medium was replaced with starvation medium containing DMEM + 1% FBS for 16 h. Cells were then treated with WNT16 for 15, 30, 60, and 120 min in starvation medium and then resuspended in 150 μL of RIPA buffer (Thermo Fisher Scientific) with Halt protease/phosphatase inhibitor cocktail (Life Technologies) added. For western blot analysis, 30 μg of total protein combined with 5× reducing loading buffer (Thermo Fisher Scientific) was boiled for 5 min, separated by SDS-PAGE, and electrotransferred to a nitrocellulose membrane (GE Healthcare, Piscataway, NJ) at 200 V for 1 h at 4°C. The membrane was blocked for 1 h in Superblock (TBS) Blocking Buffer (nuclear β-catenin, cytoplasmic β-catenin, ACTB, GAPDH, nuclear H3) (Thermo Fisher Scientific) or 5% BSA (JNK, pJNK).
Western blot analysis was performed using antibodies against nuclear β-catenin (Cell Signaling Technologies), GAPDH (Santa Cruz Biotechnology), H3, JNK, or pJNK (Cell Signaling Technologies) at dilutions of 1:1000 overnight at 4°C, then washed with TBST, and incubated in a secondary antibody (Abcam, Cambridge, MA) at 1:10,000 dilution in blocking buffer at room temperature for 1 h. Following incubation, the membranes were washed three times with TBST and proteins were visualized using the Peirce enhanced chemiluminescence western blotting substrate (Thermo Fisher Scientific) as per the manufacturer's instructions. Quantitation of western blot intensity was performed using NIH ImageJ.
Small interfering RNA and transfection
Knockdown of WNT16 was performed using Silencer Select chemically synthesized small interfering RNA (siRNA) (Thermo Fisher Scientific, catalog number: 4392420, S28065). Cells were seeded in six-well plates at a density of 4 × 104. At 50% confluence, basal medium was replaced with antibiotic-free basal medium. Transfection was performed using X-tremeGENE siRNA Transfection Reagent (Sigma-Aldrich) and 150 pM WNT16 siRNA or scrambled siRNA diluted in minimal essential medium (Opti-MEM). For the confirmation of siRNA efficiency, six hours after transfection, medium was replaced with basal medium and the efficiency of the knockdown was validated using qRT-PCR. For alizarin red (AR) and ALP staining, six hours after transfection, medium was replaced with ODM and cells were cultured under osteogenic differentiation conditions for 12 days.
In vivo intramuscular implantation
Intramuscular implantation of hPSC was performed so as to confirm their bone-forming effects, with adaptations from our previous methods. 11 An absorbable collagen sponge of defined dimensions (0.5 × 1 × 1.5 cm) was used (Medtronic, Minneapolis, MN). This carrier was chosen as a nonosteoinductive carrier, as intramuscular implantation of an acellular collagen sponge is known to have no bone forming effects. 42 2.5 × 105 hPSC in a 20 μL PBS suspension were applied to the sponge carrier. Intramuscular implantation was performed in 8-week-old male SCID mice. Animals were anesthetized by isoflurane inhalation (5% induction, 2% maintenance) and premedicated with 0.05 mg/kg buprenorphine. Bilateral incisions in the hind limbs were made, and pockets were cut in the biceps femoris muscles by blunt dissection, parallel to the muscle fiber long axis. Implants were placed bilaterally. The fasciae overlying the muscle were sutured with a simple continuous pattern, and the skin was closed in a separate layer using 5–0 Vicryl (Ethicon, San Angelo, TX) sutures in a subcuticular pattern. Animals were postoperatively treated with buprenorphine for 48 h. Animals were housed and experiments were performed in accordance with guidelines of the Chancellor's Animal Research Committee of the Office for Protection of Research Subjects at the University of California, Los Angeles. Live high-resolution radiographs were taken at 2 weeks postoperative using a cabinet radiography system (Faxitron Bioptics, Lincolnshire, IL). Mice were anesthetized using isoflurane (5% induction, 2% maintenance).
Statistical analyses
All results are expressed as mean ± standard deviation (SD). Statistical analyses were performed using the SPSS16.0 software. All data were normally distributed. Student's t test was used for two-group comparisons, and one-way ANOVA test was used for comparisons of three or more groups, followed by Tukey's post hoc test. Differences were considered significant when p < 0.05.
Results
Perivascular stem/stromal derivation
First, PSC were purified from human lipoaspirate using FACS to detect a population of pericytes and adventitial progenitor cells based on expression of CD146 and CD34 (Fig. 1). Briefly, using previously established protocols, the SVF of lipoaspirate was processed so as to remove DAPI+ nonviable cells (Fig. 1A), as well as CD45+ hematopoietic cells (Fig. 1B). Next, pericytes were defined as a CD146+CD34−CD45− cell population while adventitial progenitor cells are CD34+CD146−CD45− cell population (Fig. 1C). When combined, this bipartite population is termed PSC and is characterized by expression of characteristic MSC markers (CD44, CD73, CD90, and CD105).14,15 Prior studies have confirmed that PSC have multilineage differentiation potential, including an ability to differentiate down osteogenic, adipogenic, myogenic, and chondrogenic lineages.14,15 In this study, the osteogenic differentiation potential of hPSC was further confirmed in vitro and in vivo (Fig. 1D, E). After 5 days under standard osteogenic differentiation conditions, hPSC in monolayer culture demonstrated robust enzymatic activity for ALP (Fig. 1D). PSC-induced ectopic bone formation was next assessed, using an intramuscular implantation model in the hind limbs of SCID mice (Fig. 1E) using our previously published methods. 11 Over the course of two weeks, spontaneous, ectopic ossification was noted in the hPSC-loaded implant site (arrow, Fig. 1E).

hPSC isolation and osteogenic differentiation.
PSC undergo osteogenesis associated with changes in Wnt signaling
We next sought to correlate the process of PSC osteogenic differentiation with changes in Wnt signaling activity overtime in culture. As expected, under osteogenic differentiation conditions, PSC demonstrated increased gene expression of osteogenic markers, RUNX2, ALP, and OCN, with peak expression on days 9, 9, and 12, respectively (Fig. 2B–D). Bone nodule deposition in differentiated PSC, as observed by AR staining, increased approximately eightfold over PSC cultured in basal medium (Fig. 2A). Expression of gene markers associated with Wnt signaling was observed to increase during early osteogenic differentiation (Fig. 2E–J). AXIN2, CMYC, and DKK1 expression peaked on days 6, 3, and 6, respectively, while accumulation of nuclear β-catenin occurred progressively throughout the observed time period. The gene expression of Wnt signaling ligand WNT16 peaked earlier (day 3) and decreased thereafter.

Regulation of Wnt signaling during hPSC osteogenic differentiation.
Sustained WNT3A treatment inhibits PSC osteogenic differentiation
Next, WNT3A was supplemented to ODM throughout PSC differentiation. As expected, treatment of PSC by WNT3A significantly increased expression of downstream Wnt signaling markers (AXIN2, CMYC, DKK1) but also led to decreased osteogenic marker expression (RUNX2, ALP, and OCN) and bone nodule deposition (Fig. 3A, B, G). Conversely, exposure to canonical Wnt signaling inhibitor DKK1 attenuated the expression of AXIN2 and CMYC (Fig. 3C). Interestingly, osteogenic marker expression (RUNX2, ALP, OCN) and bone nodule deposition were significantly increased by 17.6% in DKK1-treated PSC compared to control, as observed by AR staining (Fig. 3D, H).
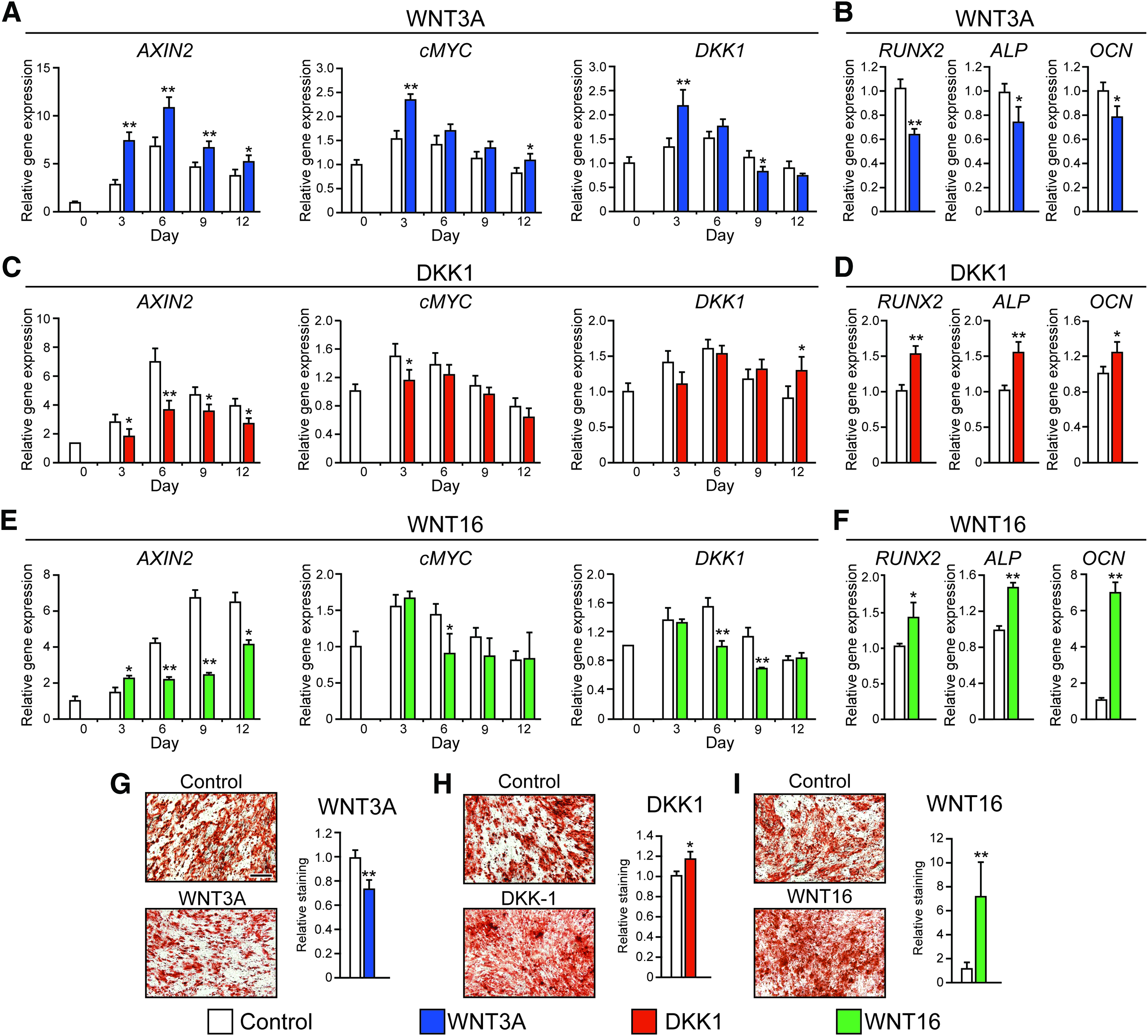
The effects of canonical and noncanonical Wnt signaling on osteogenesis of PSC.
Sustained WNT16 treatment enhances osteogenic differentiation
To assess the effects of WNT16, PSC were next exposed to exogenous WNT16 under osteogenic differentiation conditions. Sustained WNT16 treatment significantly decreased Wnt signaling activity compared to the control (Fig. 3E). In contrast, WNT16 treatment resulted in increased osteogenic differentiation marker expression, including a 40.3% increase in RUNX2 mRNA transcript and a 45.0% increase in OCN mRNA transcripts (Fig. 3F). AR staining confirmed increased PSC osteogenic differentiation with WNT16 treatment (Fig. 3I). This increase in osteogenic differentiation was accompanied by increased JNK signaling activity among WNT16-treated PSC, as shown by p46 and p54 phosphorylation assessed by western blot (Fig. 4A). To further test the importance of JNK signaling activity, WNT16 was next coapplied with either of the two inhibitors of MAPK/JNK signaling. SP600125 is a selective reversible inhibitor of JNK signaling, 43 while PD98059 is an MEK inhibitor. 44 Results showed that JNK inhibition via coapplication of SP600125 completely abrogated the WNT16-induced expression of the osteogenic marker type I collagen (COL1) (Fig. 4B), as well as ALP staining and quantification (Fig. 4C). In contrast, coapplication of the MEK inhibitor PD98059 with WNT16 resulted in only partial inhibition of osteogenic marker expression (Fig. 4B, C). Thus, the in vitro pro-osteogenic effects of WNT16 in PSC appear to be dependent on intact JNK signaling.
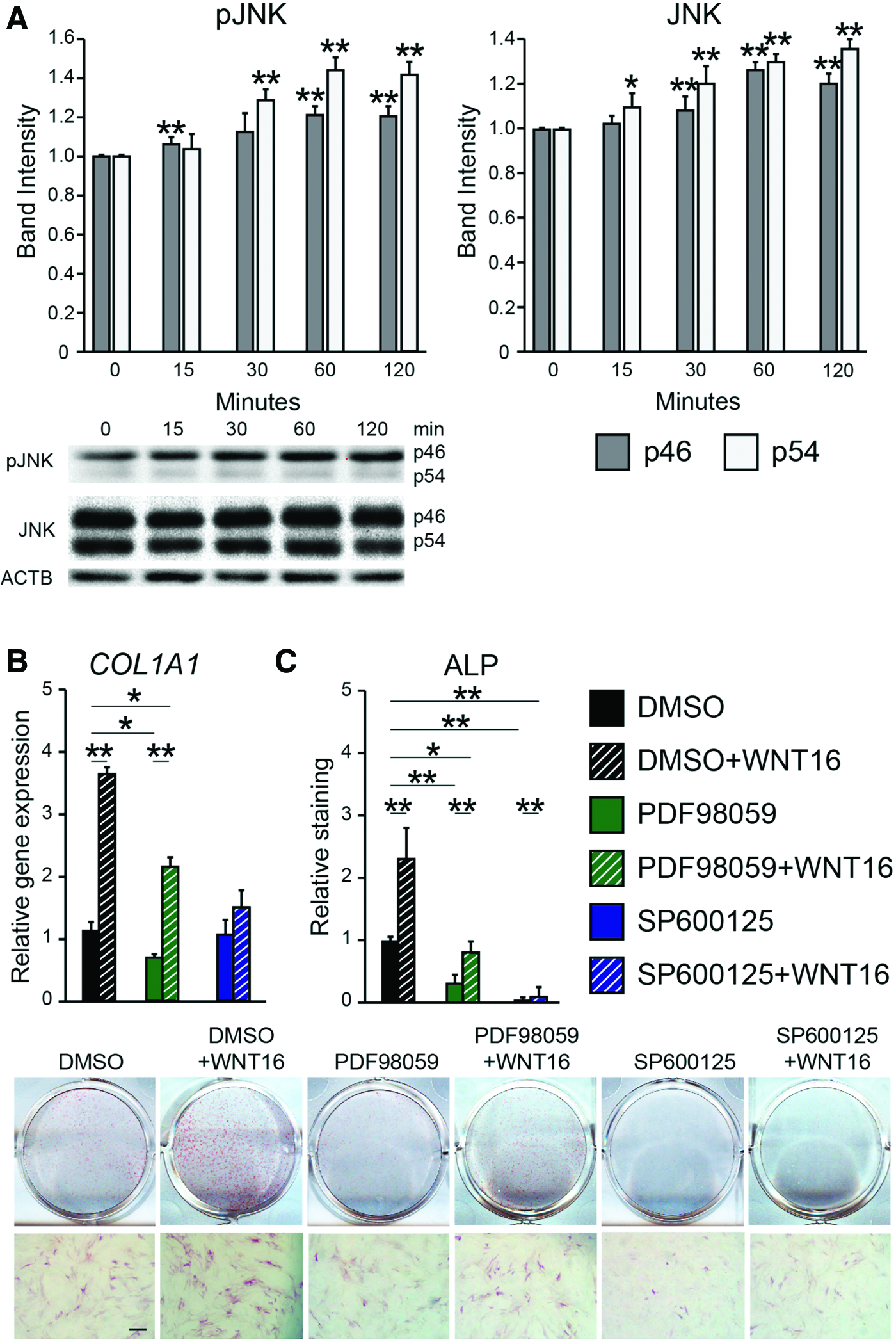
WNT16 and JNK signaling activity.
To further confirm the importance of WNT16 in PSC osteogenic differentiation, knockdown was performed using WNT16 siRNA (Fig. 5). qRT-PCR confirmed the knockdown efficiency of WNT16 mRNA at around 60.0% (Fig. 5A). In WNT16 siRNA-treated PSC, expression of osteogenic markers was reduced by 43.0% for RUNX2 and 60.0% for both ALP and OCN (Fig. 5B). Further confirmation by ALP and AR staining indicated reduced osteogenic differentiation with WNT16 siRNA (Fig. 5C, D). ALP staining was reduced by 64.1%, while bone nodule deposition as examined by AR staining was reduced by 70.0%. In summary, these data demonstrate that WNT16 expression is necessary for osteogenic differentiation in PSC.

WNT16 knockdown inhibits osteogenic differentiation in PSC.
Sustained WNT16 treatment enhances adipogenic differentiation
An inverse correlation between the osteogenic and adipogenic differentiation of mesenchymal progenitor cells is often observed (see James 45 for a review). However, notable exceptions to this exist, including BMP and IGF that may be both pro-osteogenic and proadipogenic in a context-dependent manner. 45 Next, PSC were exposed to exogenous WNT3A or WNT16 under adipogenic differentiation conditions (Fig. 6). At 10 days, sparse lipid accumulation was observed under control conditions, while WNT16-treated samples showed a significant increase in lipid droplets as visualized by ORO staining (Fig. 6A). Quantification showed a near four-fold upregulation of ORO staining intensity among WNT16 treatment, while WNT3A-treated PSC showed a modest 0.5-fold increase in comparison to control (Fig. 6B). Specific gene expression for the PPARG (peroxisome proliferator-activated receptor gamma) and CEBPA (CCAAT/enhancer-binding protein alpha) confirmed the marked proadipogenic effects of WNT16 treatment (Fig. 6C, D).

Effects of WNT3A and WNT16 on the adipogenic differentiation in PSC.
Additives to osteogenic and adipogenic medium may have influenced the availability or activity or exogenous Wnt ligands. To mitigate this possibility, assays for gene expression for osteogenic and adipogenic markers were replicated under growth medium conditions (Supplementary Fig. S1). Consistent with prior observation, sustained WNT16 treatment led to a significant increase among all osteogenic and adipogenic markers. In contrast, WNT3A treatment showed predominantly nonsignificant changes in comparison to control, excepting a modest increase in ALP and modest decrease in PPARG expression.
Discussion
Our findings reinforce the importance of canonical and noncanonical Wnt signaling in the process of osteogenic differentiation of human PSC. WNT3A, the most studied canonical Wnt ligand, inhibits PSC osteogenic differentiation when applied in a sustained manner. This is similar to previous reports in other MSC subtypes, where constitutive exposure to WNT3A has been found to suppress osteogenic differentiation, decrease levels of ALP mRNA, and inhibit matrix mineralization in BMSC.29,46 In addition, in fully differentiated BMSC, WNT3A exposure has been known to reduce mRNA expression of osteoblastic markers. 29 These findings are in contrast to WNT3A-induced osteogenesis when applied transiently, reported in bone grafts47,48 and across MSC subtypes, including BMSC-, ASC-, and MSC-like cell lines.49,50
DKK1 is a well-known inhibitor of Wnt signaling,51,52 which acts by two main mechanisms: in one mechanism of inhibition, DKK1 binds and antagonizes the Wnt signaling transmembrane receptor LPR5/6 by competitive inhibition.53–55 In another inhibition mechanism, DKK1 has been shown to form a ternary complex with LPR6 and another transmembrane protein termed Kremen2. The formation of this complex results in rapid endocytosis of LPR6 and hence complete removal from the outer cell membrane, rendering it inaccessible to extracellular Wnt signaling molecules. 55 In the vast majority of prior studies, DKK1 application has been shown to inhibit RUNX2 expression, activity, and subsequent osteogenic differentiation of MSC. 56 This includes studies in diverse MSC subtypes, including human amniotic fluid MSC 22 and BMSC. 57 Therefore, the pro-osteogenic effects of DKK1 in adipose-derived PSC were partially unexpected. Of note, there has been a similar account of DKK1-induced osteogenic differentiation in inflamed human periodontal ligament stromal cells (PDLSC) in contrast to healthy PDLSC. 58 As well, a prior study in unpurified mouse ASC showed no change with DKK1 application to osteogenic differentiation.57,58 Therefore, the effects of DKK1 on osteogenic differentiation must be more dependent on MSC source and purity than previously realized.
WNT16 is generally recognized as a mixed canonical and noncanonical Wnt signaling ligand in osteoblasts. 59 Although it has been hypothesized that WNT16 mainly stimulates bone formation through canonical signaling, while inhibiting osteoclast formation through noncanonical signaling, 59 there have also been claims supporting WNT16s ability to effect bone formation via noncanonical signaling. 60 Therefore, WNT16s ability to stimulate noncanonical activity via the JNK pathway and concomitant osteogenic differentiation was expected. In the current study, we demonstrate that WNT16 has the ability to stimulate JNK activity through noncanonical Wnt signaling by activation of both the p46 and p54 transcript variants of JNK, especially p54 activation. Moreover, use of the specific JNK inhibitor SP600125 abolished WNT16-induced upregulation of osteogenesis. Previous studies in the MC3T3-E1 cell line have suggested that p54 is more important in the induction of osteogenic differentiation. 61 Likewise, WNT16-mediated suppression of canonical Wnt activity was not unexpected, as studies in BMSC have found that WNT16 antagonizes canonical Wnt signaling of potent canonical ligands such as WNT3A and WNT1 both in vitro and in vivo.60,62 In this setting, it has been hypothesized that WNT16 suppresses the canonical pathway through competitive inhibition of canonical Wnt receptors. We further confirmed the importance of WNT16 in PSC by RNAi methods. Similar results have been previously shown where the knockdown of WNT16 by siRNA inhibited BMP-mediated osteogenic differentiation in human skeletal muscle MSC. 63 Interestingly, knockdown of β-catenin-dependent Wnt signaling has also been shown to reverse BMP-induced osteogenic differentiation, 64 again highlighting the dependence on model system and culture conditions.
Several studies have indicated that both noncanonical and mixed canonical and noncanonical Wnt signaling ligands such as WNT16 play a specific role in the regulation of adipogenic differentiation. 65 Several observations have led to the concept that noncanonical Wnt ligands induce adipogenesis primarily via blockade of canonical Wnt signaling. For example, canonical and noncanonical Wnt ligands demonstrate inverse expression patterns during adipogenic differentiation in MSC, 66 and forced upregulation of Wnt5b counteracts the inhibitory effects of Wnt3a on adipogenesis. 67 Importantly, the effects of mixed and noncanonical Wnt ligands on adipogenesis appear to be highly context dependent, as investigators observed positive 68 or negative effects 69 depending on the ligand, method of upregulation, cell type, and culture conditions. To our knowledge, this is the first examination of WNT16 effects on human mesenchymal progenitor cell adipogenesis.
Several limitations exist in the current study. First and foremost, the cell identity and potential for cell population heterogeneity within PSC are important to recognize. Increasingly, cell diversity within the perivascular niche has been recognized. Recently, the single-cell transcriptome of PSC suggested a spectrum of “primitive” to “differentiated” cell types within PSC derived from a single patient sample. 70 This heterogeneity within perivascular MSC reflects a broader observation within the literature, also observed by Sacchetti et al. in CD146+ MSC from different anatomic depots, 71 as well as Kramann et al. within Gli1+ adventitial MSC in inducible reporter mice. 72 Much remains to be understood regarding the developmental hierarchy and intrapopulation heterogeneity within PSC. Also important to recognize is the potential for an endothelial contaminant within PSC. Both the pericyte marker CD146 and the adventitial markers CD34 also mark endothelium, and removal of CD146+CD34+ cells allows for removal of most endothelial cells. As others have observed, 73 human white adipose tissue also contains a population of immature endothelial cells with a CD34+CD146−CD45− cell phenotype, which would be included in our adventitial cell gating. 73 In recent studies, we have confirmed the possibility of a minor endothelial contaminant in CD45−CD146−CD34+ SVF using quantitative RT-PCR. 12 Should absolute exclusion of all endothelial cells be desired for cell preparations, further use of the anti-CD31 antibody should be used.
Conclusions
In summary, PSC are a unique, purified population of MSC with high potential for osteogenic differentiation, regulated by canonical and noncanonical Wnt signaling. WNT16 appears to play an important and necessary role in the commitment and differentiation of PSC down an osteogenic lineage.
Footnotes
Authors' Contributions
Jia Shen: conception and design, collection and/or assembly of data, manuscript writing; Xuepeng Chen: collection and/or assembly of data; Haichao Jia: collection and/or assembly of the data; Carolyn A. Meyers: collection and/or assembly of data, manuscript writing; Swati Shrestha: collection and/or assembly of data; Greg Asatrian: collection and/or assembly of data; Catherine Ding: collection and/or assembly of data; Rebecca Tsuei: collection and/or assembly of data; Xinli Zhang: provision of study material; Bruno Peault: provision of study material; Kang Ting: provision of study material, financial support; Chia Soo: provision of study material, financial support; Aaron W. James: provision of study material, financial support, final approval of manuscript.
Acknowledgments
The present work was supported by the NIH/NIAMS (grants R01 AR061399, R01 AR066782, K08 AR068316) and the Orthopaedic Research and Education Foundation with funding provided by the Musculoskeletal Transplant Foundation.
Disclosure Statement
K.T. and C.S. are inventors of perivascular stem/stromal cell-related patents filed from UCLA. K.T and C.S. are founders of Scarless Laboratories, Inc., which sublicenses perivascular stem/stromal cell-related patents from the UC Regents, and who also hold equity in the company. C.S. is also an officer of Scarless Laboratories, Inc.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.