
Abstract
Advances on materials' research for tissue engineering (TE) applications have shown that animal cells respond directly to the material physical, chemical, mechanical, and electrical stimuli altering a variety of cell signaling cascades, which consequently result in phenotypic and genotypic alterations. Gellan gum (GG) spongy-like hydrogels (SLH) with open microstructure, mechanical properties, and cell performance have shown promising results for soft TE applications. Taking advantage of intrinsic properties of GG-SLH and polypyrrole (PPy) electroactivity, we developed electroactive PPy-GG-SLH envisaging their potential use for skeletal muscle TE. Three different methods of in situ chemical oxidative polymerization were developed based on the availability of pyrrole: freely dissolved in solution (method I and III) or immobilized into GG hydrogels (method II). PPy was homogeneously distributed within (method I and III) and on the surface (method II) of GG-SLH, as also confirmed by Fourier Transform infrared spectra. PPy-GG-SLH showed higher conductivity than GG-SLH (p < 0.05) whereas PPy-GG-SLH (method I and II) showed the best conductivity among the 3 methods (∼1 to 2 × 10−4 S/cm). The microarchitecture of PPy-GG-SLH (method I) was similar to GG-SLH but PPy-GG-SLH (method II and III) presented smaller pore sizes and lower porosity. PPy-GG-SLH (method I and II) compressive modulus (∼450–500 KPa) and recovering capacity (∼75–90%) was higher than GG-SLH, nevertheless the mechanical properties of PPy-GG-SLH (method III) were lower. The water uptake of PPy-GG-SLH was rapidly up to 2500% and were stable along 60 days of degradation being the maximum weight loss 20%. Mechanically stable and electroactive PPy-GG-SLH (method I and II) were analyzed regarding cellular performance. PPy-GG-SLH were not cytotoxic for L929 cells. In addition, L929 and C2C12 myoblast cells were able to adhere and spread within PPy-GG-SLH, showing improved spreading in comparison to GG-SLH performance. Overall, PPy-GG-SLH show promising features as an alternative electroactive platform to analyze the influence of electrical stimulation on skeletal muscle cells.
Introduction
H
It has been frequently reported that cells respond to chemical, physical, mechanical, and electrical stimulus.3,12–15 Moreover, electrical stimulation occurs in all development and regeneration processes of animal tissues. Nevertheless, its existence and potential effect over soft tissue repair and regeneration are commonly disregarded. 16 Thus, three-dimensional (3D) electroactive biomaterials potentially play a significant role in tissue regeneration and can also act as excellent platforms to understand the effect of electrical stimulation on cell behavior. In fact, electroactive polymers aiming to understand the relationship between microstructure, electrical stimulus, and cell behavior have been proposed.17–22 In this context, different structures have been developed, such as electroactive films or 3D biomaterials.17–20 Polypyrrole (PPy) films are commonly synthesized by electrochemical reaction using specific dopants and have been associated with different components, as ECM components, to obtain cell adhesive surfaces.22–24 For example, laminin-coated PPy films doped with dodecylbenzenesulfonate were used to evaluate the differentiation of human neural stem cells (hNSCs) under the influence of electrical stimulation. Those results showed that hNSCs cells cultured under electrical stimulus expressed the differentiation marker β-III tubulin while unstimulated hNSCs kept the expression of the undifferentiated glial fibrillary acidic protein marker. 23 Indirectly, this study also confirmed that the chemical and biological properties of PPy polymer are altered due to the use of dopants or oxidizing agents, in particular, if the synthesis is performed by chemical or electrochemical reactions.23,25,26 In a different context, electrochemical reactions have also been used to synthesize pure PPy films, as electrical components and sensors,27–30 but not 3D electroactive scaffolds. Nevertheless, PPy films are considered mechanically rigid and brittle and these features represent a limitation for their use in TE applications.31,32 PPy-GG hydrogels electrode coatings on a neuronal sensor supported electrical stimulation over an extended period of time without losing performance, although its ability to direct cell fate was not shown. 29 Recently, chemical synthesis has emerged as one of the most promising method to obtain 3D electroactive hydrogels as an alternative to electrochemical reactions.33–35 The combination of PPy with other polymers has also been proposed not only to improve the chemical stability but also to allow cell adhesion, migration, and growth.36,37 Synthesis of PPy over hydrogels, such as bacterial cellulose hydrogels, 34 a composite of acrylamide and acrylic acid 38 and methylmethacrylate [poly(methyl methacrylate)], have been explored for drug delivery 39 but there are few reports about PPy electroactive hydrogels developed to study soft tissue regeneration. 40
Considering the evidence regarding the cell adhesive nature of GG-SLH 9 and the electroactive features of PPy, we developed electroactive spongy-like PPy-GG by incorporating PPy and creating electroactive SLH for soft and electrical tissue regeneration, such as skeletal muscle regeneration. Three different methods of synthesis based on in situ chemical oxidative polymerization using ammonium persulfate (APS) as the oxidizing agent were explored. Exploring the different methods developed to produce electroactive SLH, we aimed to synthesize PPY-GG-SLH based on the homogeneous distribution of PPy into GG microstructure associated with relevant physicochemical and electrical features and cellular performance to be further used as electroactive platforms for studying the behavior of skeletal muscle cells cultured with the appliance of the electrical stimulus. Combining the advantageous features of both, GG-SLH adhesive microstructure and electroactive properties of PPy, we expect to produce a helpful scaffold to further analyze the influence of electrical stimulus on skeletal muscle tissue regeneration.
Materials and Methods
Synthesis of PPy-GG-SLH
GG-SLH were prepared as previously described 9 with modifications that included three different approaches of in situ chemical oxidative polymerization of pyrrole (Py) monomers over the GG backbone, using APS as oxidizing agent (Fig. 1). Briefly, low-acryl gelzan powder (Mw ∼1000 kg/mol; Sigma) was dissolved in deionized water (1.25%, w/v), under stirring and at 90°C during 20 min. GG solution was allowed to cross-link with calcium chloride (0.18%) during 1 h.
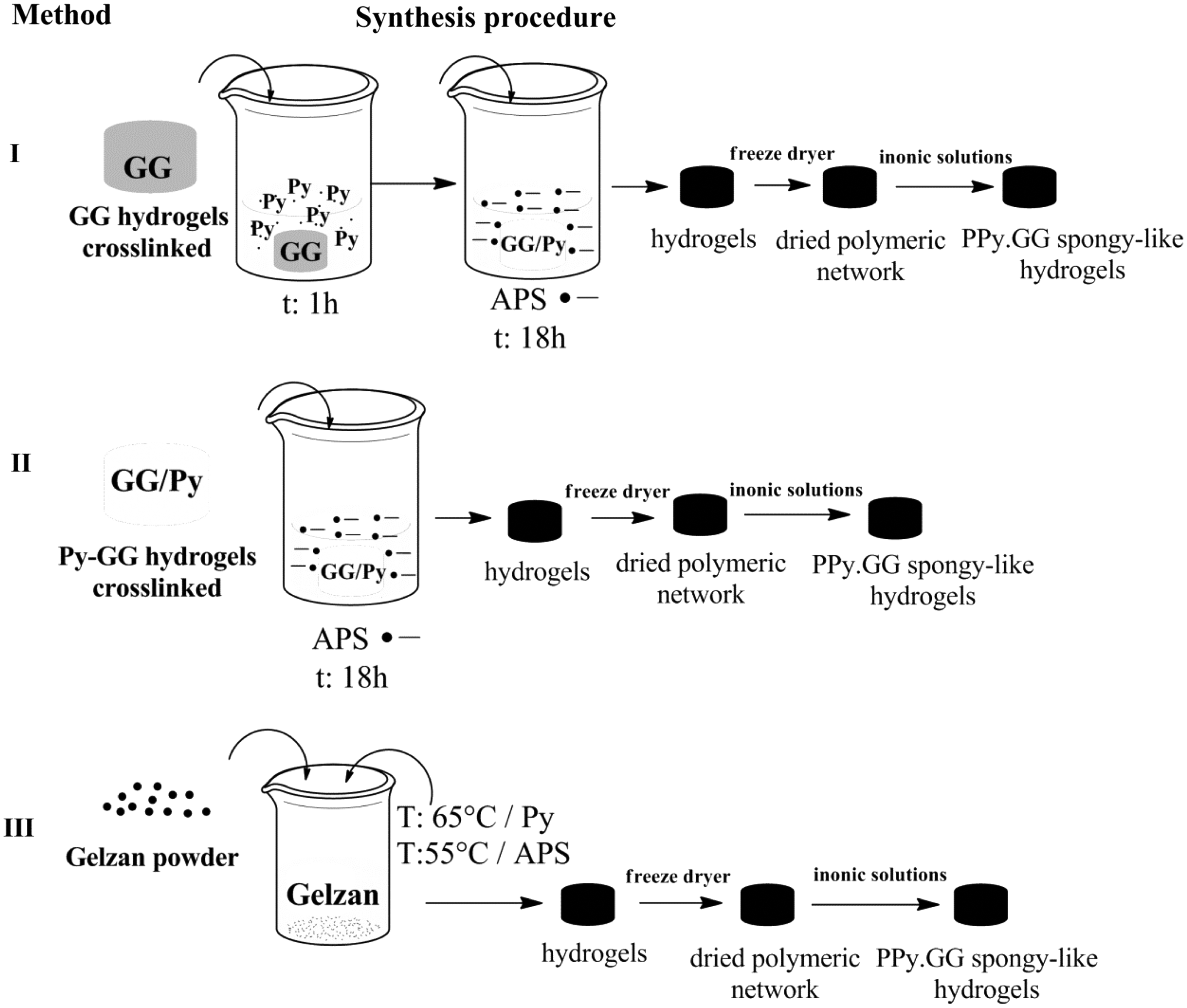
Schematic illustration of the methods followed to produce GG and PPy three-dimensional electroactive spongy-like hydrogels. GG, gellan gum; PPy, polypyrrole.
After that, GG hydrogels were immersed in Py (Sigma) solution (0.1 M of PPy in phosphate-buffered saline [PBS]) and subsequently oxidized with APS (Sigma) (method I). In method II, Py (0.1 M in PBS) was incorporated into the GG solution at 65°C and cross-linking and oxidation with APS were promoted as described for method I. In method III, Py and APS were added to the GG solution at 65°C and 55°C successively before cross-linking. At 50°C calcium chloride was added into the solution to start the cross-linking process for 1 h. The formed GG and PPy-GG hydrogels were punched of 16 mm of diameter and 5 mm of height, subjected to successive washings with PBS, frozen at −80°C (48 h), and freeze-dried (72 h) to obtain dried polymeric networks of GG and PPy-GG. Samples were sterilized by ethylene oxide and re-hydrated, as described for the respective analysis, to give rise to SLH. 9
Fourier transform infrared analysis
The presence of PPy over GG dried polymeric networks was evaluated by Fourier Transform infrared (FTIR). FTIR analysis was performed using an IR-Prestige-21 spectrophotometer (Shimadzu Scientific Instruments) with an attenuated total reflectance (ATR) accessory in the range of 2000–800 cm−1 by accumulating 70 individual scans. Three specimens per condition were analyzed and a representative spectrum of each sample was interpreted.
Conductivity measurement
A four probe standard method, with a Keithley 6220 current source and a Keithley Model 6517A electrometer, was used to determine the electrical conductivity of both GG and PPy-GG dried polymeric networks. Four specimens from each group of biomaterials were analyzed and the statistical analyses were performed.
Micro computed tomography and scanning electron microscopy
To evaluate the microstructure of the GG and PPy-GG dried polymeric networks, samples were analyzed by Scanning Electron Microscopy (SEM) and micro computed tomography (μCT). For SEM analyses, samples were submersed in liquid nitrogen for 3 min for transversal fracture, placed on stubs, sputter-coated (208HR; Cressington Company) with gold, and further analyzed using NanoSEM-FEI Nova 200 (5 kV). In parallel, samples were scanned by μCT (Skyscan 1072) in a high-resolution mode using a pixel size of 11.31 μm and 35 keV of energy and 215 μA of current. Representative data sets of 150 slices were transformed into a binary image using a dynamic threshold of 45 and 255 (gray values) to distinguish polymer material from pore voids. These data were used for morphometric analysis (CT Analyzer v1.5.1.5; SkyScan), which included quantification of the pore wall thickness, structure porosity, and pore size. Three-dimensional virtual models of representative regions in the bulk of the structures were also created, visualized, and registered using the image processing software (CT-vox; SkyScan). One specimen per condition was analyzed.
Mechanical testing
Universal mechanical testing machine Instron 4505 (Instron Int. Ltd.) was used to perform compression tests on GG and PPy-GG-SLH (hydrated in culture medium for 4 h, at 37°C). A crosshead speed of 2 mm/min was used and each sample was compressed up to 60% of strain. The compressive modulus was determined in the most linear region of the stress–strain graph until 10% of strain in X-axis. At least six specimens were used for each condition.
Recovery after deformation
The recovery capacity of GG and PPy-GG-SLH were assessed by measuring, using a caliper, the specimen height before (Hi), immediately (HDef) and after 0.2, 1, and 24 h (Ht) of being submitted to 60% of strain, according to the following equations (1, 2):
For the calculations, deformation of 60% was set as 0% of recovery. Four specimens of each condition were used and the experiments were repeated three times. The recovering test was conducted in Dulbecco's modified Eagle medium (DMEM) bathing solution.
Water uptake
The water uptake ability was evaluated by immersing both, GG or PPy-GG dried polymeric networks in DMEM (Sigma-Aldrich) at 37°C up to 7 days. Samples were weighed before (Wi) and after (Wf) immersion in DMEM. Water uptake capacity was determined by the following equation:
Four specimens of each condition were used and the experiments were repeated three times.
Weight loss
Weight loss was determined after immersing both GG and PPy-GG dried polymeric networks in PBS (0.01 M, pH 7.4; Sigma-Aldrich) solution containing 0.02% of sodium azide (Sigma) at 37°C with stirring, for up to 60 days. Samples were weighted before (Wi) and after 1, 3, 7, 14, 28, and 60 days. At least four specimens were used for each experiment. In the end of each experimental point, samples were freeze-dried and weighed (Wf). The weight loss for each sample was calculated using Equation (4).
Cell viability and proliferation
L929 cell line was used to evaluate any cytotoxicity of the prepared electroactive PPy-GG-SLH. Cells were cultured in DMEM culture medium containing 10% of fetal bovine serum (FBS; Life Technologies) and 1% of antibiotic-antimycotic (Life Technologies). Cells were plated in 24-well tissue culture plates at a density of 25,000 cells/well. After 24 h, GG and electroactive PPy-GG dried polymeric networks were placed on top of the cells and incubated for further 1, 3, and 7 days without changing the culture medium to assess the effect of any potential toxic leachable. After each time point, cell metabolic activity was determined by MTS assay (Promega Corporation) and dsDNA quantified using the Quant-iT™ PicoGreen® dsDNA kit (Life Technologies), following manufacturer's instructions. Cell morphology was daily observed in an inverted microscope (Axiovert 40 CFL; Zeiss).
Cell cytoskeleton staining
L929 cells (100,000 cells × specimen) were seeded on GG and PPy-GG dried polymeric networks (samples were not soaked with culture medium prior seeding) and kept in culture for 3 and 7 days. After 3 and 7 days of culture, cells were fixed with formalin (4%; Bio-Optica Milano S.p.a.) and the cytoskeleton of the cells stained with Phalloidin-TRITC (1:500; Sigma-Aldrich). Nuclei were stained with 4′,6-diamidino-2-phenylindole (1:1000; Sigma-Aldrich), samples were observed in an Axio Imager Z1 m fluorescence microscopy (Zeiss) and images were acquired with the ZEN 2.1 blue edition software.
C2C12 cells adherence
C2C12 cells were grown in DMEM supplemented with 10% FBS and 1% pen/strep. C2C12 cells were seeded (25,000 cells × mm3) on GG and PPy-GG dried polymeric networks (samples were not soaked with culture medium prior seeding) and kept in an atmosphere of 37°C and 5% of CO2. The adhesion of C2C12 cells on PPy-GG samples was analyzed by SEM and by confocal microscopy. For SEM analysis, samples (PPy-GG) were fixed with glutaraldehyde (2.5%), washed with PBS solution, and dehydrated with ethanol solutions (30%, 50%, 70%, 80%, 90%, and 100%). Posteriorly, samples were dried using critical point dryer equipment (Autosamdri-815 [Series A]; Tousimis) and they were mounted on a carbon tape under aluminum stubs and sputter-coated with thin gold layer under vacuum using sputter coater (Cressington model 108auto; TED PELLA, Inc.) before being analyzed by SEM (SEM; Jeol JSM-6010LV). For confocal microscopy analysis, cells were fixed with formalin (4%; Bio-Optica Milano S.p.a.) and the cytoskeleton of the cells stained with Phalloidin-TRITC (1:500; Sigma-Aldrich). Nuclei were stained with DAPI (1:1000; Sigma-Aldrich), samples were observed in an Axio Imager Z1m fluorescence microscopy (Zeiss) and images were acquired with the ZEN 2.1 blue edition software.
Statistical analysis
Statistical analyses were conducted using a one-way analysis of variance and Tukey's test assuming unequal or equal variances for n (p < 0.05 was considered statistically significant and represented by *). The results from at least three independent experiments were expressed as the mean ± standard error of mean. The software Statistics 8.0 (StatSoft Software) was used to perform the analysis.
Results
PPy-GG-SLH synthesis
Three different methods of in situ chemical oxidative polymerization were followed to produce PPy-GG-SLH. The main difference between each method was based on the Py availability before the oxidative reaction starts. During the oxidative reaction with APS, Py was available under the following conditions: (i) Py dissolved in PBS solution (method I); (ii) Py immobilized into GG hydrogels (method II); or (iii) dissolved in a heated GG:PBS solution (method III). All methods involved the polymerization of Py to

Hydrogels dried polymeric network
Chemical and electrical characterization
The chemical characterization of the dried polymeric networks was performed by FTIR-ATR (Fig. 3A). Chemical analyses confirmed the presence of PPy in the GG dried polymeric networks prepared by the different methods (method I, II, and III). The presence of PPy was confirmed by comparison with GG and PPy spectra. Despite the proximity of GG and PPy characteristic peaks, the presence of PPy was clearly identified in the PPy-GG dried polymeric networks, independently of the produced method used for synthesis (Fig. 3A). The spectrum of the GG samples showed a characteristic peak at 1608 cm−1 (carboxylate group) and the characteristic bands at 3442 cm−1 (O–H stretching), at 1640 cm−1 (carboxylate anion adsorption), and at 1076 cm−1 (pyranoside ring adsorption). 41 The characteristic peaks of PPy between 1561 and 1480 cm−1 related to asymmetric −C═C− and asymmetric −C–N− stretching vibration of typical PPy ring, were also observed. The fundamental PPy band in the spectra at 1198 cm−1 corresponds to ═C–H in-plane vibration. The presence of a band at 918 cm−1 can be attributed to ═C–H out of plane vibration involving the polymerization of Py.42,43 Upon confirmation of the presence of PPy in the produced structures, its electrical conductivity was measured in their dried polymeric networks state. The obtained results showed that PPy improved the electrical conductivity of the obtained materials (Fig. 3B). Moreover, there was a significant increase (p < 0.05) of the electrical conductivity values when comparing GG-SLH with all electroactive SLH (methods I, II, and III). Method I provided the most electroactive samples (2.10 × 10−4 S/cm) when compared with samples synthesized by method II.

GG and PPy-GG dried polymeric networks
Morphological and microstructural properties
Morphological features and respective microstructure of the three different dried polymeric networks formulations prepared by methods I, II, and III were studied by micro-CT analysis (Fig. 4A, B). Figure 4A shows the reconstructed microstructure (two-dimensional and 3D) of GG Fig. 4A(i) and (v), PPy-GG-method I Fig. 4A(ii) and (vi), PPy-GG-method II Fig. 4A(iii) and (vii), and PPy-GG-method III Fig. 4A(iv) and (viii) samples. The results obtained by μCT were analyzed using CT Analyzer software to obtain the morphometric parameters (pore size, pore wall thickness, and porosity). The obtained results indicate that, independently of the method used, polymerization of PPy promotes a reduction in pore size and porosity in comparison with GG dried polymeric networks. While the GG structures presented a porosity of 91%, the PPy-GG structures produced by the methods I, II, and III showed 83%, 83%, and 88% porosity, subsequently. Moreover, the pore size was smaller for the structures containing PPy (method I and II) than for the ones prepared only with GG (196 μm; Fig. 4B). The average of samples pore size was 194 μm (method I), 154 μm (method II), and 115 μm (method III) for the structures containing PPy, respectively, which represent a reduction of 1%, 21%, and 41% in comparison to the structure without PPy. Additionally, for samples prepared by methods I and II, the presence of PPy lead to an increase of the pore wall thickness.

Representative
Physical properties
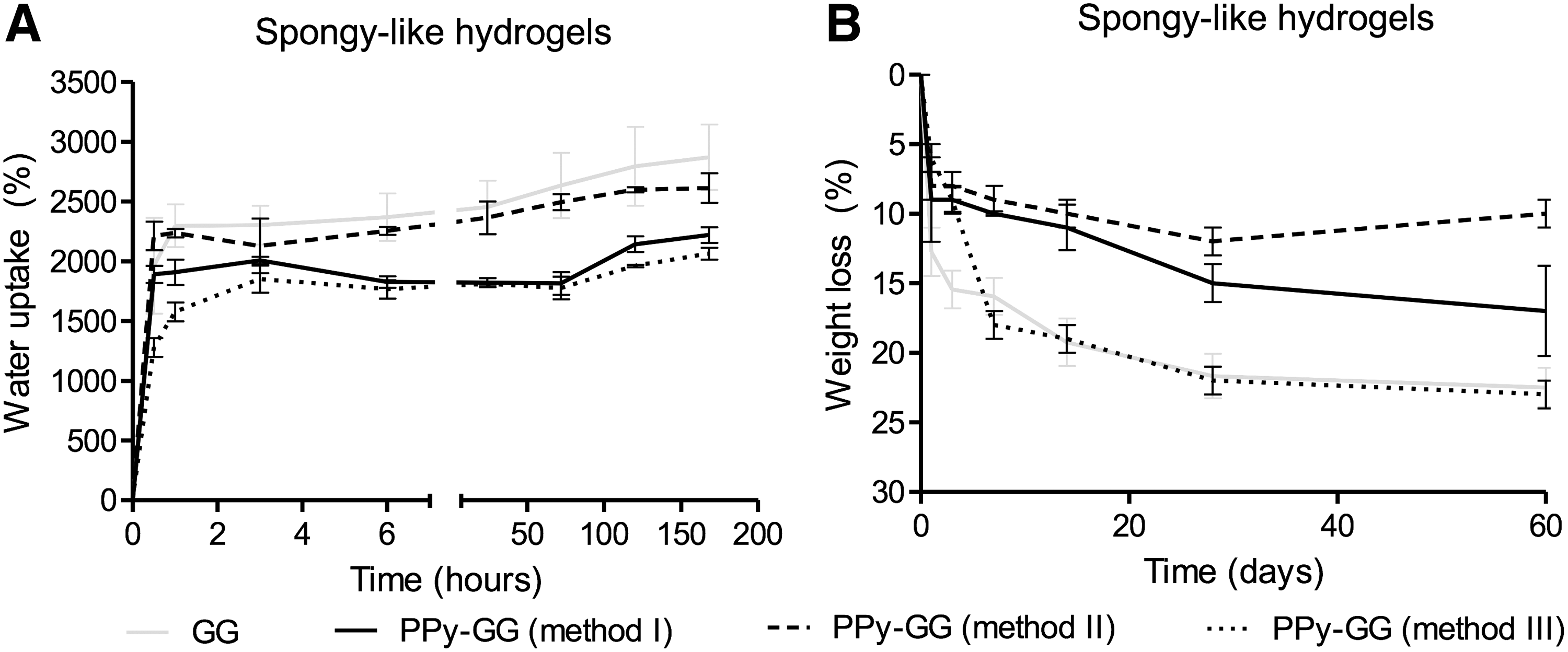
The mechanical performance of the developed electroactive materials was analyzed considering their stiffness (Fig. 5A–C) and recovering capacity after deformation (Fig. 5D). The strain × stress curves of GG and the electroactive PPy-GG samples in dried (dried polymeric networks) and wet (SLH) state are shown in Figure 5A and B subsequently. The presence of PPy resulted in an increase in the compressive modulus of dried polymeric networks samples prepared by methods I and II (Fig. 5C). In opposition, the compressive modulus of the dried polymeric networks prepared by method III decreased in comparison with the respective GG samples. After hydration, the presence of PPy resulted in a decrease of stiffness of all SLH formulations, more precisely, ∼20–30% for samples prepared by methods I and II and ∼70–80% for samples prepared by method III. The statistical analyses showed that significant differences (p < 0.05) were observed between GG dried polymeric networks and all electroactive dried polymeric networks. GG-SLH showed a significant difference when compared to PPy-GG-SLH produced by method III. To assess the recovery capacity of the SLH after deformation, samples were submitted to 60% of compressive strain and its recovery measured after 10 min, 1 and 24 h (Fig. 5D). The electroactive SLH prepared by methods I and II were able to recover almost entirely the 3D structure after 10 min, and 85% and 88%, respectively, after 24 h. Interestingly, the recovery capacity of the samples with PPy was slightly faster than the ones of GG only. In opposition, samples prepared using method III showed the lowest recovery capacity. There were significant differences in the recovery capability between GG-SLH and all PPy-GG-SLH (methods I, II, and III) after 10 min and 1 h of compression. After 24 h of compression, statistical differences were detected between GG-SLH and PPy-GG-SLH produced by method III. PPy-GG-SLH produced by methods I and II were able to recover the microstructure after compression as GG-SLH. PPy-GG-SLH produced by method III were very fragile and difficult to handle. Consequently, due to its weak mechanical properties, SLH prepared by method III were discarded for the following biological experiments. The water uptake capability of the dried polymeric networks showed that the production method affects the amount of water retained in the structure along the time (Fig. 6A); although independently of the processing method used, the PPy-GG samples showed lower water uptake capability than GG samples. This feature was more evident for SLH produced by methods I and III, after 7 days of hydration, which showed ∼30% less water content than the structures without PPy due to the hydrophobic character of PPy distributed within the structure.
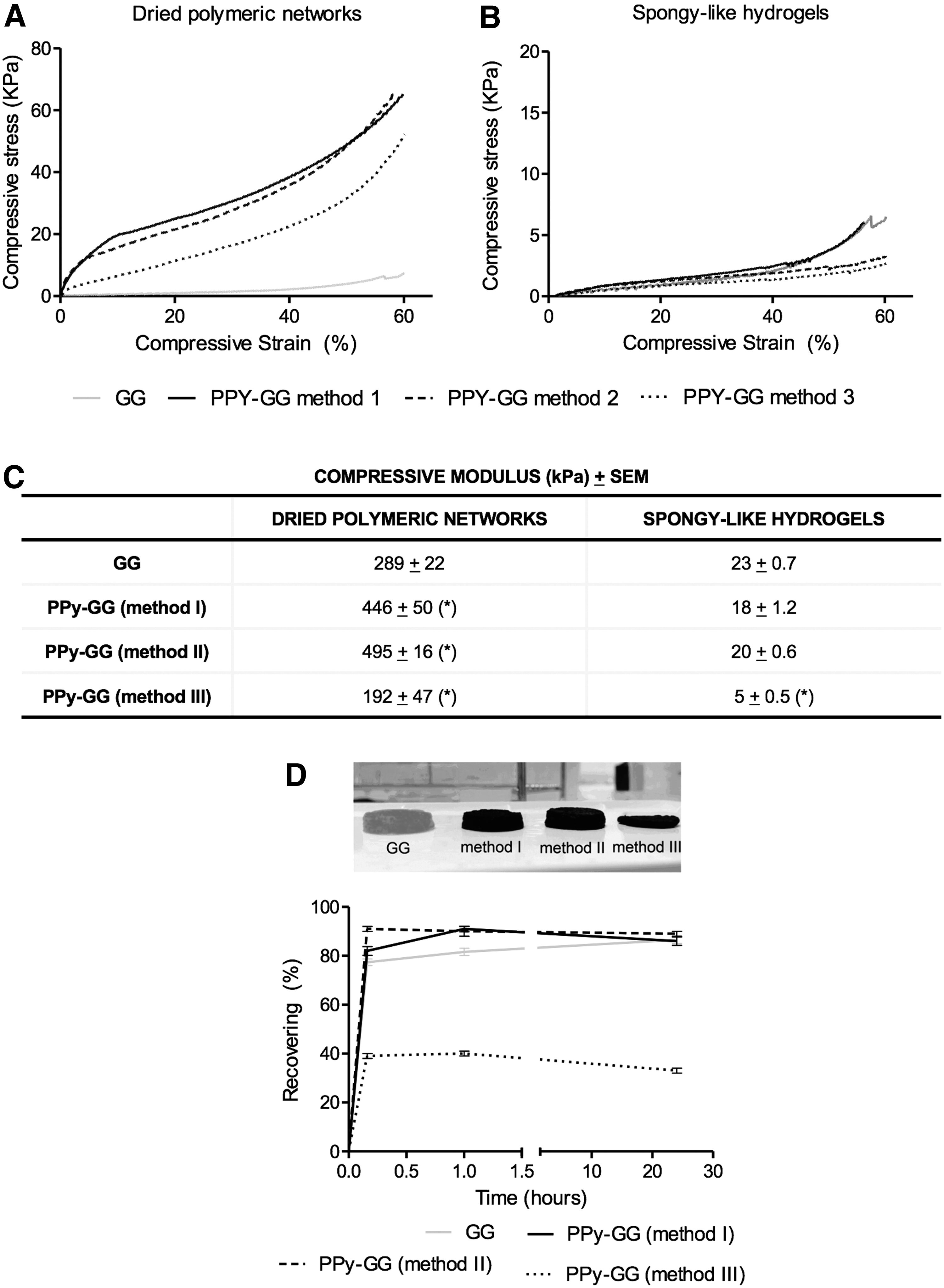
Mechanical characterization of GG and PPy-GG dried polymeric networks and spongy-like hydrogels. Stress–strain curves of
GG and PPy-GG spongy-like hydrogels
The weight loss measured after 1, 3, 7, 14, 21, 28, and 60 days of immersion in PBS (Fig. 6B) showed that the presence of PPy improved the stability and prevented the degradation of samples produced by method I and II. Thus, after 60 days samples synthesized by method I and method II were more stable than GG-SLH and lost respectively 8%, 14%, and 22% of their initial weight. PPy-GG-SLH produced by method III and GG samples showed a similar weight loss profile.
Biological performance
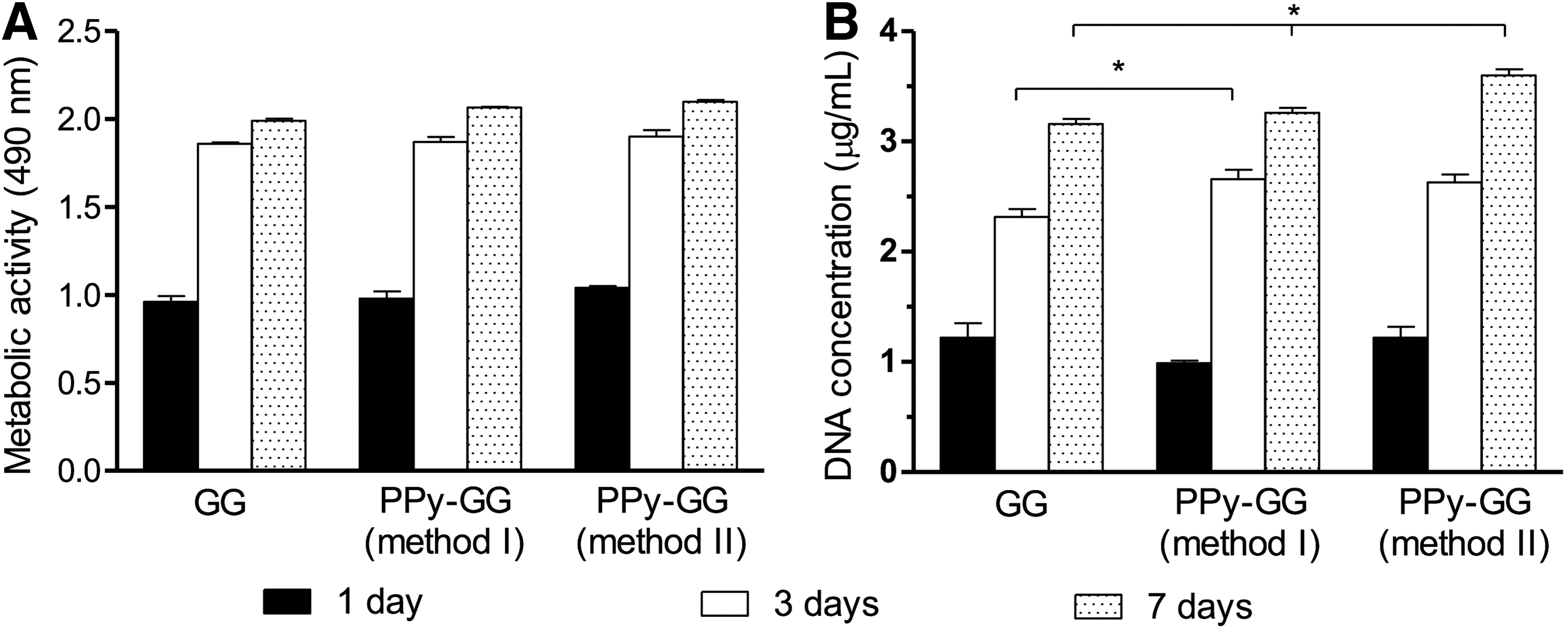
In light of the results regarding the electrical, mechanical, and physical characterization of the different formulations, just samples synthesized by method I and II were evaluated for its potential cytotoxicity using L929 cells according to ISO 10993-5 (Fig. 7A, B). The in vitro results showed that the electroactive SLH prepared by both methods did not negatively affect the metabolic activity of L929 along the time (Fig. 7A). In addition, statistical analyses showed that there were no statistic differences on L929 metabolic activity between GG-SLH and the electroactive SLH (method I and II) considering the same experimental time points. L929 cells proliferation was also investigated to observe whether the presence of PPy could interfere with cells proliferation (Fig. 7B). After 3 days of in vitro culture, a significantly higher amount of DNA from L929 cells was detected when seeded in PPy-GG-SLH produced by method I when compared to GG-SLH (Fig. 7B). At the end of 7 days of in vitro culture, PPy-GG-SLH prepared by both methods stimulated L929 proliferation and statistic differences were detected when they were compared to GG-SLH (* indicates statistical differences when p < 0.05). After entrapping L929 cells within GG and PPy-GG (method I and II)-SLH, L929 cells showed morphological spreading for the 7 days of cell culture (Fig. 8A). L929 cells were able to adhere, acquire, and then maintain their typical morphology, with an organized cytoskeleton along the 7 days of in vitro culture within GG and PPy-GG (method I and II)-SLH (Fig. 8A). The cellular morphology of L929 cells cultured within GG and PPy-GG (method I and II)-SLH after 3 and 7 days of in vitro culture were quantified, as shown on Figure 8B. L929 cells cultured within GG-SLH presented an average of the morphological area of 31.74 μm2 after 3 days of culture and 58.73 μm2 after 7 days. L929 cultured within PPy-GG-SLH produced by method I and II induced L929 spreading after 3 days (127.27 μm2 [400%] and 215.12 μm2 [677%] and 7 days (87.87 μm2 [150%] and 167.47 μm2 [285%]) of in vitro culture. Statistical analyses have shown statistical differences on L929 morphology cultured within GG-SLH and the electroactive PPy-GG-SLH (method I and II). Statistical differences of L929 cells morphology were also observed between cells cultured within PPy-GG-SLH produced by method I and PPy-GG-SLH produced by method II.
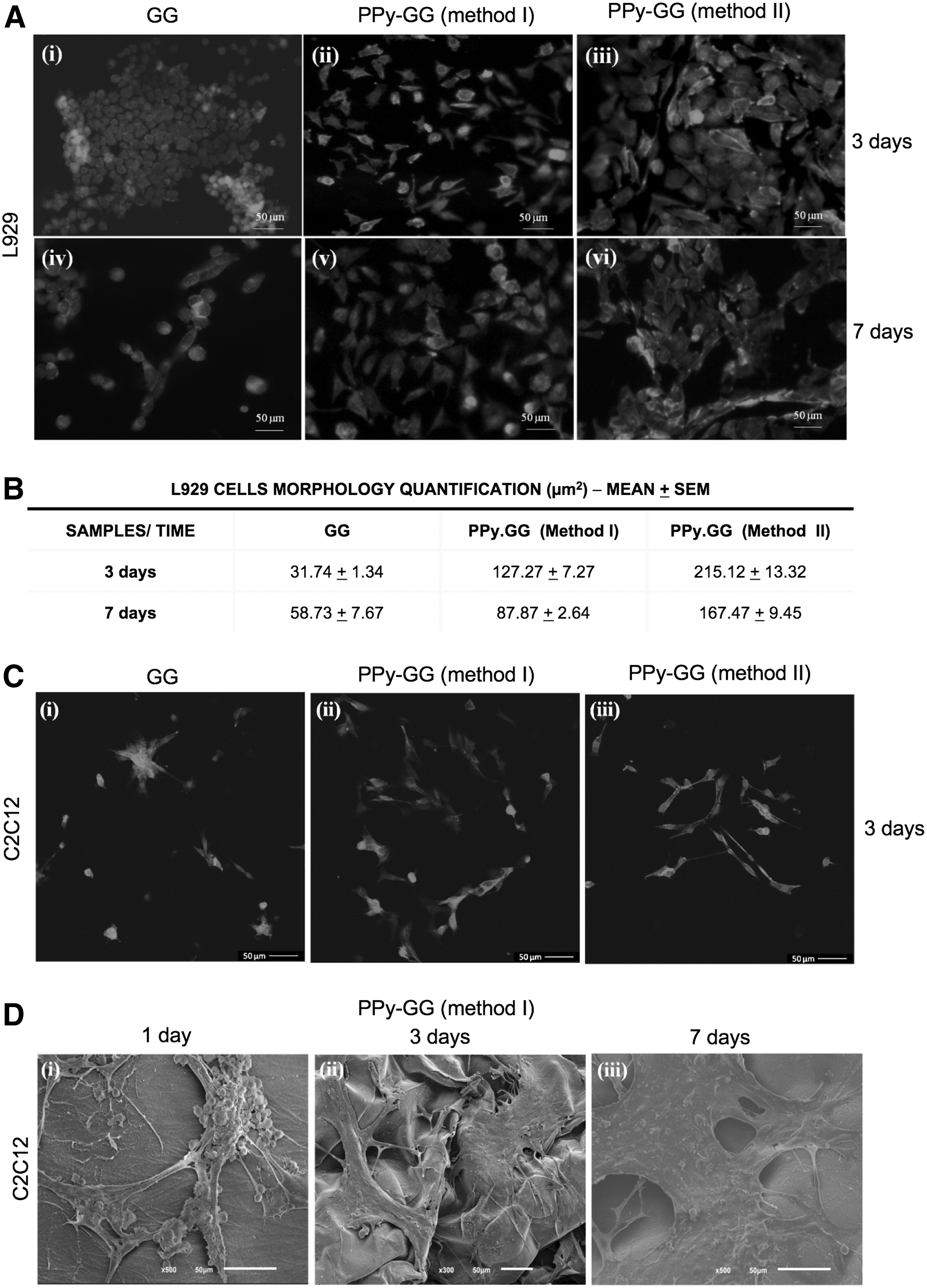
Representative fluorescence microscopy images after phalloidin-TRITC staining (red) and 4′,6-diamidino-2-phenylindole (blue) of
Cell adhesion and spreading were also evaluated with skeletal muscle cells (C2C12 cells) to confirm the adhesiveness surface of the novel electroactive matrices, as an early step to validate them for skeletal muscle TE applications (Fig. 8C, D). C2C12 cells were in vitro cultured within GG and PPy-GG-SLH during 3 days (Fig. 8C). Confocal images showed that C2C12 cells adhered and spread faster on the surface of PPy-GG-SLH produced by methods I (Fig. 8C[ii]) and II (Fig. 8C[iii]) than on GG SLH (Fig. 8C[i]).
Discussion
In this work, we describe the preparation of PPy-GG-SLH that combine the relevant properties of GG-SLH, such as water content, porosity, resilience to deformation, and cell adhesiveness, with the electroactive property of PPy. The chemical synthesis of PPy required the total control of the reaction (concentration, temperature, purity, reaction time, stirring rate, and other parameters) to keep the reproducibility of the samples.33,40 We tested three different methods for the in situ chemical oxidative reaction of Py over GG hydrogels, maintaining control of all those important parameters. The purpose of using different methods was to evaluate which method would be better to produce electroactive SLH without compromising their intrinsic properties and providing a homogeneous distribution of PPy. Chemical analysis confirmed the presence of PPy in the GG dried polymeric networks, independently of the method used (method I, II, and III). The clear identification of the PPy characteristic peaks is a clear indication of its presence in the polymer matrix. A little overlapping of the bands was observed and consequently, the C–C (1321 cm−1) and C–N vibration bands (1201 cm−1) of the PPy ring were shifted toward the absorption bands of pure GG. The shift in the peak position after the polymerization can be attributed to some chemical interactions between PPy and GG. Method I allowed a homogenous polymerization of Py within the GG dried polymeric networks resulting in samples with the highest conductivity (2.05 × 10−4 S/cm). This method was adapted from the one previously described by Muller et al. (2013) that also reported the production of electroactive PPy-bacterial cellulose hydrogels with PPy homogeneously distributed within the bacterial cellulose microstructure. 35 However, in method II, the polymerization of Py occurred just on the borders of the 3D structural networks and did not reach the material's bulk. That event probably occurred due to the physical immobilization of Py on the border of the cross-linked GG hydrogel that quickly reacted with APS forming an outer electroactive layer, which in turn acted as a barrier for the penetration of APS and consequently limited the polymerization of Py. The absence of PPy in the bulk of GG-SLH synthesized by method II might be responsible for the decreased electroactivity and significant differences in the physicochemical features in comparison to samples synthesized by method I. Results revealed that the distribution of PPy within the structure of PPy-GG dried polymeric networks interfered with their water uptake ability. Samples produced by method I and III showed ∼30% less water absorption capacity than GG-SLH due to the hydrophobic property of PPy that was homogeneously distributed within GG microstructure.
It was also confirmed, by μCT, that the use of different approaches for PPy polymerization affected samples' microstructure (pore size, pore wall thickness, and porosity). PPy-GG (method I) samples showed similar pore size to GG ones, as PPy-GG synthesis was performed from GG cross-linked precursor hydrogels, and hence the microstructure was only slightly altered by the presence of nonpolymerized Py. Nevertheless, PPy-GG (method II and III) samples have shown lower pore size in comparison to the GG materials. In both cases, precursor hydrogels were formed by mixing simultaneously both GG and PPy. Thus, Py could act as nucleating agent for the formation of ice crystals during freezing, resulting in smaller size crystals and, consequently, pores of smaller size postfreeze-drying.
Moreover, it was demonstrated that the use of different PPy polymerization methods affected the mechanical properties of obtained samples. As previously described, the presence of PPy resulted in an increase in the stiffness of dried polymeric networks samples prepared by methods I and II. As described in the literature, the mechanical properties of porous materials are highly dependent on their microstructure. 44 In accordance, PPy-GG dried polymeric networks samples prepared by methods I and II had lower porosity and higher pore wall thickness, consequently, they showed higher compressive moduli when compared to GG samples. The weaker mechanical properties of samples produced by method III can be explained by the reduced ionic cross-linking of the GG polymeric chain due to the addition of Py and APS directly in a heated GG solution, as Py may form hydrogen bonds with GG.
Consequently, the freezing/freeze-drying originated a different microstructure. PPy-GG-SLH synthesized by both methods I and II were capable of recovering almost 90% of their microstructure after suffering a 60% compressive strain. In opposition, samples synthesized by method III were very fragile and less prone to recover, showing structural instability and difficult handling.
Due to the described limitations of samples prepared by method III, just PPy-GG-SLH synthesized by method I and II were submitted to in vitro tests with L929. The results have shown that these materials did not negatively alter the L929 cells viability and proliferation compared to GG-SLH. A recent work also showed a negligible toxicity of PPy on human adult retinal pigment epithelium (ARPE-19) cells. 45 On the other side, L929 cells proliferation was significantly increased after 3 days when cultured in direct contact with PPy-GG-SLH produced by method I and after 7 days the improvement on L929 proliferation was observed on both electroactive PPy-GG-SLH when compared to GG-SLH. Thus, the quantification of proliferative and metabolic activity of L929 cells cultured with PPy-GG-SLH (method I and II) showed that the developed hydrogels were not cytotoxic. Moreover, L929 cells showed the highest spreading after 3 days of culture within PPy-GG-SLH in comparison to GG-SLH. Briefly, L929 cells spread faster within PPy-GG-SLH than on GG-SLH surfaces as showed by the statistically significant differences observed by morphological cells quantification (μm2). Previous results showed that GG-SLH microstructure allowed the entrapment and the spreading of human adipose stem cell and human osteoblast-like cells (SaOs-2) within GG-SLH, but under the standard culture conditions, dermal microvascular endothelial cells were not able to adhere on GG-SLH.9,46 Moreover, hyaluronic acid were conjugated with GG to produce PPy-HA-SLH that supported the formation of a monolayer of human keratinocytes that organized themselves in a pavement-pattern manner upholding their typical cell–cell contacts after 7 days of culture. 46 PPy-GG-SLH' cell adhesive features were improved for L929 cells by the incorporation of the electroactive polymer (PPy) that significantly reduced the water content, in comparison to GG-SLH. Regarding skeletal muscle applications, PPy-GG-SLH allowed the adherence and spreading of C2C12 cells during 7 days of culture, which were confirmed by confocal and SEM analysis.
In conclusion, electroactive PPy-GG-SLH were successfully synthesized using different methods to perform the in situ Py chemical oxidative reaction. PPy-GG-SLH synthesized by method I were more electroactive than the samples synthesized by method II and III. The method I allowed the homogeneous distribution of PPy within GG spongy-like hydrogel microstructure. It was also demonstrated by μCT analysis that the use of different polymerization methods allowed to obtained samples with different microstructures. Dried polymeric networks prepared by methods I and II have shown smaller porosity and, consequently, higher stiffness in comparison with GG ones. PPy-GG-SLH were not cytotoxic for L929 cells. Skeletal muscle cells (C2C12) adhered and spread faster within PPy-GG-SLH than on GG-SLH. Thus, we were able to engineer electroactive and skeletal muscle cell-compatible SLH comprising the essential physicochemical features of GG SLH and the potential electroactive nature of PPy. We expect that PPy-GG-SLH could become a useful platform to study the behavior of skeletal muscle cells over electrical stimulation.
Footnotes
Acknowledgments
The authors gratefully thank Elvira Fortunato, Daliana Muller and Guilherme Mariz de Oliveira Barra for performing the conductivity measurements of the samples produced using four probe methods equipment. The Researcher F.V.B. was supported by the grant of National Council for Scientific and Technological Development (CNPq) of Brazil through the program Science without borders. P.S. was financially supported by Khon Kaen University, Thailand and L.P.d.S. was financially supported by Fundação para a Ciência e Tecnologia (FCT, SFRH/BD/565 78025/2011). The FCT fellowship distinction attributed to V.M.C. under the Investigator FCT program (IF/01214/2014) is also greatly acknowledged.
Disclosure Statement
No competing financial interests exist.