
Abstract
The loss of salivary gland function caused by radiation therapy of the head and neck is a serious condition and it affects a patient's quality of life. The current lack of effective therapies demands new options to be explored. This study tested whether human salivary gland epithelial cells (SGECs) could be successfully cultured on a decellularized porcine gut matrix (SIS-muc) in both mono- and coculture with microvascular endothelial cells (mvECs). By performing immunofluorescence imaging, transmission as well as scanning electron microscopy (SEM), quantitative polymerase chain reaction (qPCR), and an amylase enzyme assay, it was investigated as to what extent the three-dimensional (3D)-cultured cells could maintain their molecular differentiation and the production of working α-amylase (α-AMY) compared with two-dimensional (2D) culture. In both 3D mono- and coculture, SGECs were successfully cultured and formed acinar-like structures. Those findings were confirmed by SEM imaging. Immunofluorescence imaging revealed that 3D-cultured cells expressed α-AMY, Claudin-1 (CL-1), and water channel protein aquaporin-5 (AQP-5). Two-dimensional-cultured cells only were positive for α-AMY. Real time (RT)-qPCR analysis showed that α-AMY relative gene expression was higher in both 3D mono- and coculture than in 2D culture. In α-AMY enzyme assay, cocultured SGECs showed about 25 times increased enzyme activity compared with 2D-cultured cells. In conclusion, the SIS-muc combined with endothelial coculture seems a suitable culture setting for the tissue engineering of functional human salivary gland tissue.
Introduction
S
Materials and Methods
Salivary gland tissue collection and preparation
Salivary gland explants were collected during surgical excisions of benign parotid gland tumors and immediately transferred to ice-cold phosphate-buffered saline solution (PBS; Sigma-Aldrich, Munich, Germany). Informed written consent was obtained beforehand, and the study was approved by the institutional ethics committee on human research of the Julius-Maximilians-University Würzburg (study approval number 16/06). The data were analyzed anonymously and according to the principles expressed in the Declaration of Helsinki. The explants were washed three times with 10 mL Dulbecco's modified Eagle's medium (DMEM; Gibco, Darmstadt, Germany) supplemented with 1% penicillin streptomycin (PenStrep; Biochrom, Berlin, Germany), and the connective tissue was carefully removed. The samples were transferred into a petri dish and cut into pieces of ∼1 mm3 under sterile conditions.
Isolation and two-dimensional cell culture of SGECs
The salivary gland tissue pieces were transferred into a 24-well plate, and 250 μL of airway epithelial cell medium (AECM) was added per well (Fig. 1A). The SGECs were cultured under static conditions in an incubator (37°C, 5% CO2), and the medium was changed every 2–3 days. After the SGECs began sprouting from the salivary gland tissue pieces, cell confluency was investigated regularly by using phase-contrast microscopy. The cells were transferred to a 25 cm2 cell culture flask at a confluence level of about 80% and expanded for a further 4 weeks. After three passages, the cells were harvested and vitality was checked by trypan blue staining. The cells were seeded on SIS-muc at a concentration of 1.5 × 105/cm2. Cell isolation and the culturing procedure of mvECs have already been described elsewhere. 15 The mvECs in this study were used between passage 2 and 3 and derived from human foreskin.

Isolation and cell culture of SGECs derived from human salivary gland tissue explants.
Preparation of SIS-muc matrix and 3D cell culture
Segments of porcine jejunum (30–50 cm length) were taken from land race pigs (∼20 kg; Niedermayer, Dettelbach, Germany). The sections were explanted containing their supplying arterial and venous pedicle, then rinsed with physiologic sodium buffer solution, and finally stored in PBS containing 1% gentamycine (PAA, Cölbe, Germany) for transportation. Afterward, a multi-step decellularization procedure was applied, leading to a decellularized small intestinal scaffold with maintained mucosal structures (SIS-muc). The exact procedure of SIS-muc preparation has been described earlier.16,17 All procedures were performed in compliance with the German Animal Welfare Act. The decellularized matrix was cut into appropriate sections and fixed between two metal cylinders (i.e., cell crowns). For the monoculture model, SGECs were seeded on the matrix with a concentration of 1.5 × 105 per crown; whereas for the coculture, 4 × 105 mvECs per crown were added. Monoculture was maintained by using AECM (PromoCell, Heidelberg, Germany); whereas for the coculture of SGECs and mvECs, a 1:1 mix of AECM and VascuLife®-Medium (Lifeline, Carlsbad, CA) was applied to the models. Tissue models were cultured for either 7 or 14 days before collecting supernatants and fixation of the models with 4% paraformaldehyde (PFA) for histological analysis. For both mono- and cocultures (i.e., two culture conditions), SGECs (n = 3 patients, 2 samples per experiment) were seeded on SIS-muc scaffolds (n = 3 donors, 8 samples per pig) for either 7 or 14 days (i.e., two time points), resulting in 24 individual samples.
Immunofluorescence imaging
For immunofluorescence staining of two-dimensional (2D)-cultured SGECs, cells of the third passage were seeded in a 24-well dish (10.000 cells/well) and incubated overnight. On the next day, cells were fixed with 4% PFA (AppliChem, Darmstadt, Germany) and subsequently incubated with blocking solution (0.3% Triton X, 5% bovine serum albumin, 5% Donkey Serum) for 30 min at room temperature. To prepare the colonized SIS-muc, cell culture was stopped at days 7 and 14; the inner diameter of the scaffold was cut out; and one quarter was fixed in 4% PFA for 1 h at room temperature, embedded in paraffin, sliced, and stored. To perform immunofluorescence staining, the paraffin sections were deparaffinized and then incubated in citrate buffer, pH 9.5 at 95°C for 15 min. Finally, the slides were incubated with blocking solution for 30 min. Salivary gland cryosections served as positive controls, whereas fibroblasts were used as negative controls. All samples were incubated overnight with primary antibodies (diluted 1:100) at 4°C. To characterize the SGECs in both 2D and 3D culture, the following primary antibodies were applied: rabbit anti-Claudin-1 (CL-1; Life Technologies, Carlsbad, CA), rabbit anti-α-Amylase (α-AMY), mouse anti-pan-Cytokeratin (Sigma-Aldrich, St. Louis, MO), and mouse anti-aquaporin-5 (AQP-5; Abcam, Cambridge, United Kingdom). Endothelial cells in the coculture samples were identified by using rabbit anti-vWF (Abcam). After incubation overnight, secondary antibodies alexa-555 goat anti-rabbit and alexa-647 goat anti-mouse (Life Technologies) were applied in a 1:400 dilution for 1 h in darkness at room temperature. Finally, samples were embedded with Mowiol mounting medium (Carl Roth, Karlsruhe, Germany) containing 0.1% DAPI (Invitrogen, Darmstadt, Germany) for staining of cell nuclei. Images were acquired by using a BZ-9000 fluorescence microscope (Keyence, Osaka, Japan). The images shown in Figures 3 and 4 represent three separate experiments. Every object slide included three serial sections of different areas of the scaffold.
Transmission electron microscopy
For transmission electron microscopy (TEM) sample preparation, parts of the colonized SIS-muc were fixed in 2.5% glutaraldehyde (Merck, Darmstadt, Germany), then rinsed in 50 mM cacodylate buffer (Carl Roth) for 15 min, postfixed first in 2% osmium tetroxide (Science Services, Munich, Germany) for 1 h, and finally, after rinsing again in H2O, stained with 0.5% uranylacetate (Merck) overnight. The next day, samples were dehydrated through an ethanol series and subsequently embedded in a propylenoxide/epon-mix (Sigma-Aldrich) overnight. After 48 h of polymerization, the samples were cut into 60 nm sections by using an EM UC7 Microtome (Leica, Wetzlar, Germany). Examination and documentation was performed on an EM 900 transmission electron microscope (Carl Zeiss, Jena, Germany).
Scanning electron microscopy
To perform scanning electron microscopy (SEM) analysis of the colonized SIS-muc, samples were first fixed in 6.25% glutaraldehyde (Merck) at 4°C overnight and rinsed in Sorensen's phosphate buffer (81.8% 0.1 M Na2HPO4,12.2% 0.1 M KH2PO4; Applichem) five times to completely remove the fixative. After dehydration in an aceton series, samples were dried by using the critical point drying method and finally, a gold/palladium coating was applied with Sputter Coater SCD 005 (Leica). Samples were stored in a desiccator (Novus NS-Tubus; DURAN Group, Wertheim, Germany) and examined on a DSM 950 scanning electron microscope (Carl Zeiss).
Amylase activity test
In addition to immunostaining, the respective amylase enzyme activity in the supernatants was analyzed by using an Amylase Assay Kit (Abcam). Samples of 300 μL of the supernatant were collected from the apical compartment of the cell crowns at days 3, 6, 9, and 12 of cell culture and stored at −80°C until use. As a control, the same amount of medium was taken from the cell culture flasks of the SGEC 2D culture. After thawing on ice, samples were centrifuged to remove insoluble parts. The supernatants were collected and kept on ice. For the assay, materials and samples were brought to room temperature; 50 μL of nitrophenol was added for the standard curve dilutions, positive controls, and samples in a 96-well plate format, in duplicate. The reaction mix was added, and the absorbance was measured every 10 min at optical density (OD) = 405 nm in the kinetic mode of the Infinite M2000 micro plate reader (Tecan, Männedorf, Switzerland) for a period of 110 min. The measured absorption was estimated according to the equation of the standard curve. Amylase activity was detected by measuring the change in OD resulting from the amount of nitrophenol generated by the enzyme within the reaction time. One unit amylase was defined as the amount of amylase that cleaves ethylidene-pNP-G7 to generate 1.0 μmol of nitrophenol per min at pH 7.2 at 25°C.
Real time-quantitative polymerase chain reaction analysis
RNA of 2D cell cultures and 3D tissue models was isolated by using the sRNeasy Micro Kit with in-column DNA digestion (Qiagen, Venlo, NL). At days 7 and 14 of cell culture, samples for real time (RT)-quantitative polymerase chain reaction (qPCR) analysis were collected and transferred immediately to Buffer RLT (Qiagen) containing 10% β-mercaptoethanole to inactivate RNAses and stored at −80°C. Accordingly, 2D-cultured SGECs underwent cell lysis protocol before RNA isolation started. RNA concentration was measured by using an Infinite M2000 micro plate reader (Tecan). RNA was transcribed into cDNA by using the iScript™ Reverse Transcription Supermix (BioRad, Hercules, CA). To determine optimal annealing temperatures, gradient PCR was performed and the related RNA bands were detected by using gel electrophoresis. For qPCR, exon-overlapping primers for α-AMY and cytokeratine 18 (CK18) were designed with Primer-BLAST tool and manufactured by Thermo Fisher (Waltham, MA). Primer specifications are given in Table 1.
Amplified cDNA was detected with the TaqMan probe system. All PCRs were performed as duplicate by using the TaqMan Universal Master Mix (Thermo Fisher) and a CFX96 thermocycler (BioRad) at the annealing temperature of 61°C. Expression levels of α-AMY were normalized to the values of CK18, and the results were shown as an x-fold change in gene expression, relative to the 2D culture. The relative normalized expression levels reflect three biological replicates of the SGEC 3D culture assay. Prevalidated housekeeping genes EF1α and GAPDH were selected for qPCR analysis.
Statistical analysis
All experiments were repeated with at least three independent patient samples per group (n = 3), which were used for statistical analysis. The level of statistical significance was set at p < 0.05, which was indicated with an asterisk (*). The nonparametric Kruskal-Wallis test (with post hoc Dunn's test) was used for statistical analysis. All statistical tests were performed by using SigmaPlot 12.5, including SigmaStat tools. Results are given as mean ± standard deviation (SD).
Results
Establishment of salivary gland cell culture models
In 2D culture, SGECs emigrated from the MOCs after 7–10 days and formed an adherent confluent monolayer after 2–3 weeks of expansion (Fig. 1B, C). SGECs cultured on the SIS-muc formed a monolayer on the surface of the scaffold (Fig. 2A′, B′) and acinar-like cellular clusters (Fig. 2A′′, B′′) within the cavities of the scaffold that partially merged with the monolayer (Fig. 2D, E′, F′, H). Those findings did not vary significantly between monoculture and coculture and the length of culture time.
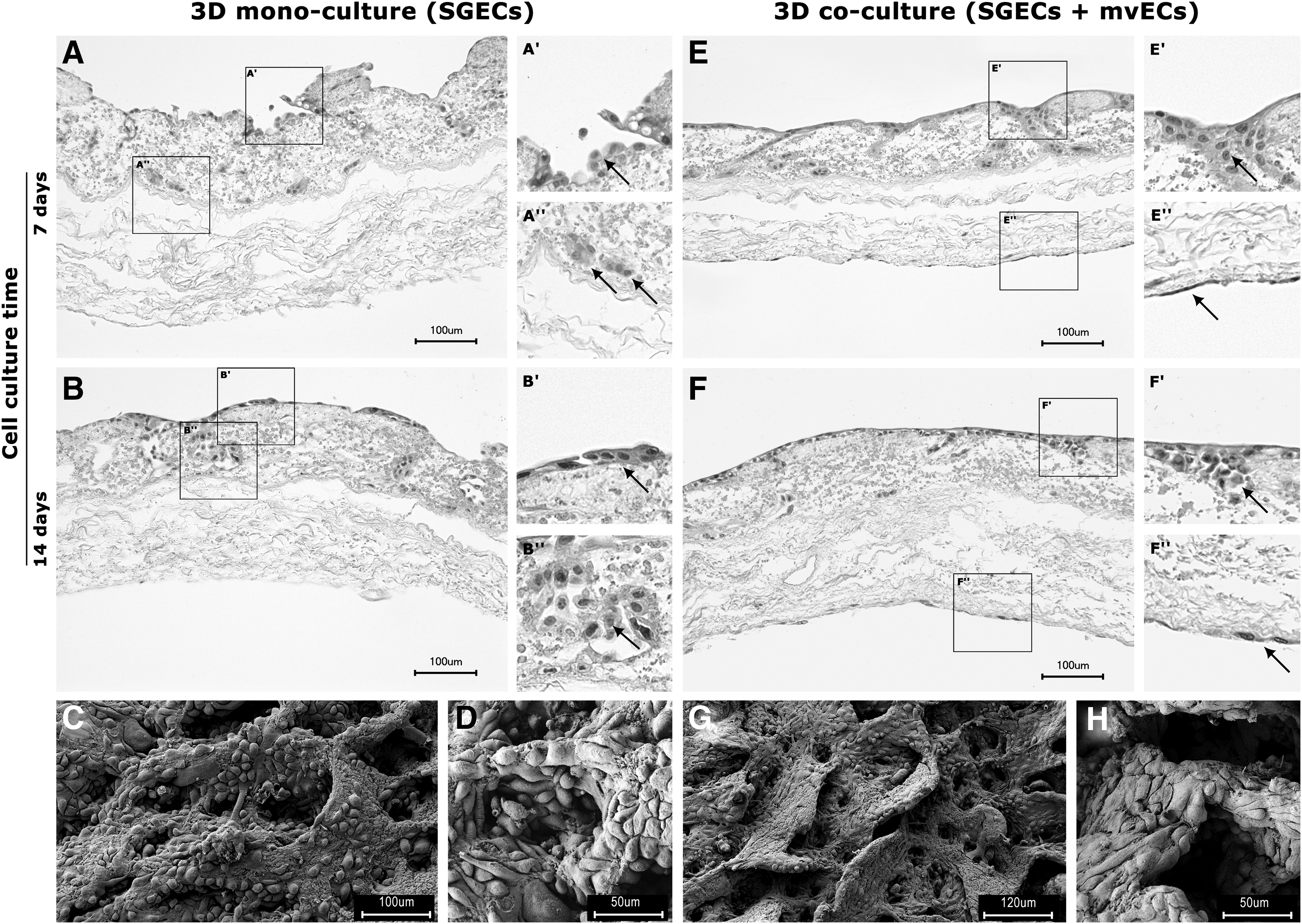
Monoculture and coculture on decellularized SIS-muc at different time points.
Salivary gland protein expression analysis
Characteristic protein expression for α-AMY, AQP-5, and CL-1 was analyzed by immunofluorescence microscopy. Native salivary gland tissue showed high expression of α-AMY within the acinar cell bodies (Fig. 3A), whereas AQP-5 (Fig. 3D) was present in the acinar cell membranes and CL-1 (Fig. 3G) in the ductal cells. SGECs in 2D culture were positive for α-AMY (Fig. 3B) while showing no expression for AQP-5 (Fig. 3E) and CL-1 (Fig. 3H). Three-dimensional monoculture of SGECs revealed α-AMY expression both in the monolayer on the surface and in the acinar-like cell clusters in the depth of the crypts (Fig. 3C, C′). There was no notable difference detected between the different time points. High expression of AQP-5 was primarily found in the depth of the crypts of the 3D culture (Fig. 3F, F′), whereas CL-1 expression was limited to the surface of the matrix cell layer (Fig. 3I, I′). In 3D coculture, mvECs cultured on the serosal side of the scaffold were positively stained for vWF. SGECs on the mucosal side were positively stained for pan-CK (Fig. 4).
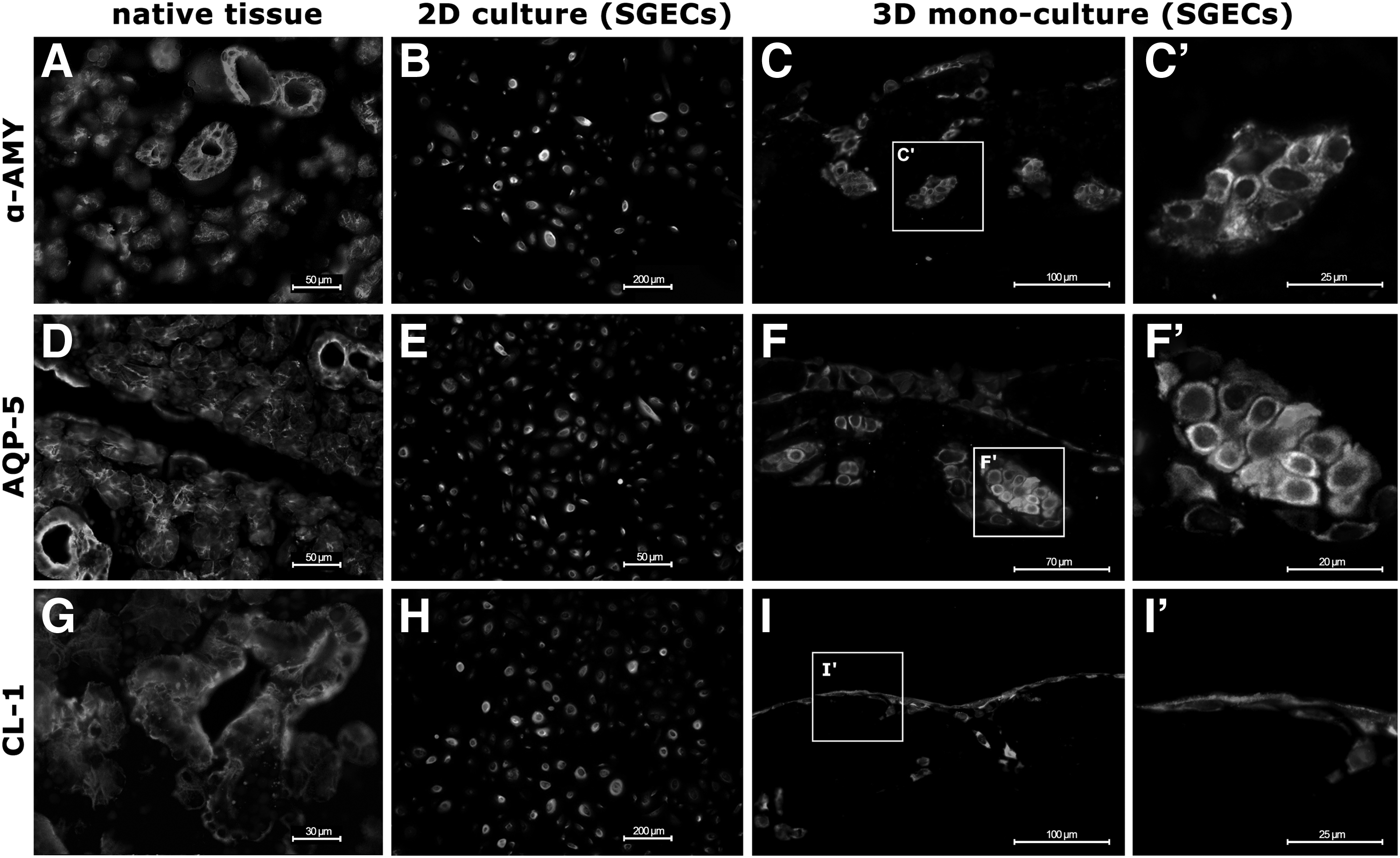
Immunohistological characterization of native SG tissue, 2D and 3D monoculture of SGECs. Immunofluorescence of SGECs with pan-cytokeratin (green) and typical target antigens (red).

3D coculture of SGECs on the mucosal side and mvECs on the serosal side of the SIS-muc.
Ultrastructural characteristics
Ultrastructural analysis on the SIS-muc by transmission electron microscopy (TEM) showed SGECs interconnected with junctional complexes (Fig. 5A′, A′′) featuring a dense cytoplasm with intact mitochondria (Fig. 5A′′) and microvilli (Fig. 5A′′, A′′′), indicating regular cell function. TEM analysis revealed electron-dense secretory granules (Fig. 5D) that were typical for native SG tissue. Within the cellular clusters mentioned earlier, preapoptotic cells around cell-free spaces were observed (Fig. 5B, C), suggesting the formation of lumina as similarly described by Wells and Patel. 18
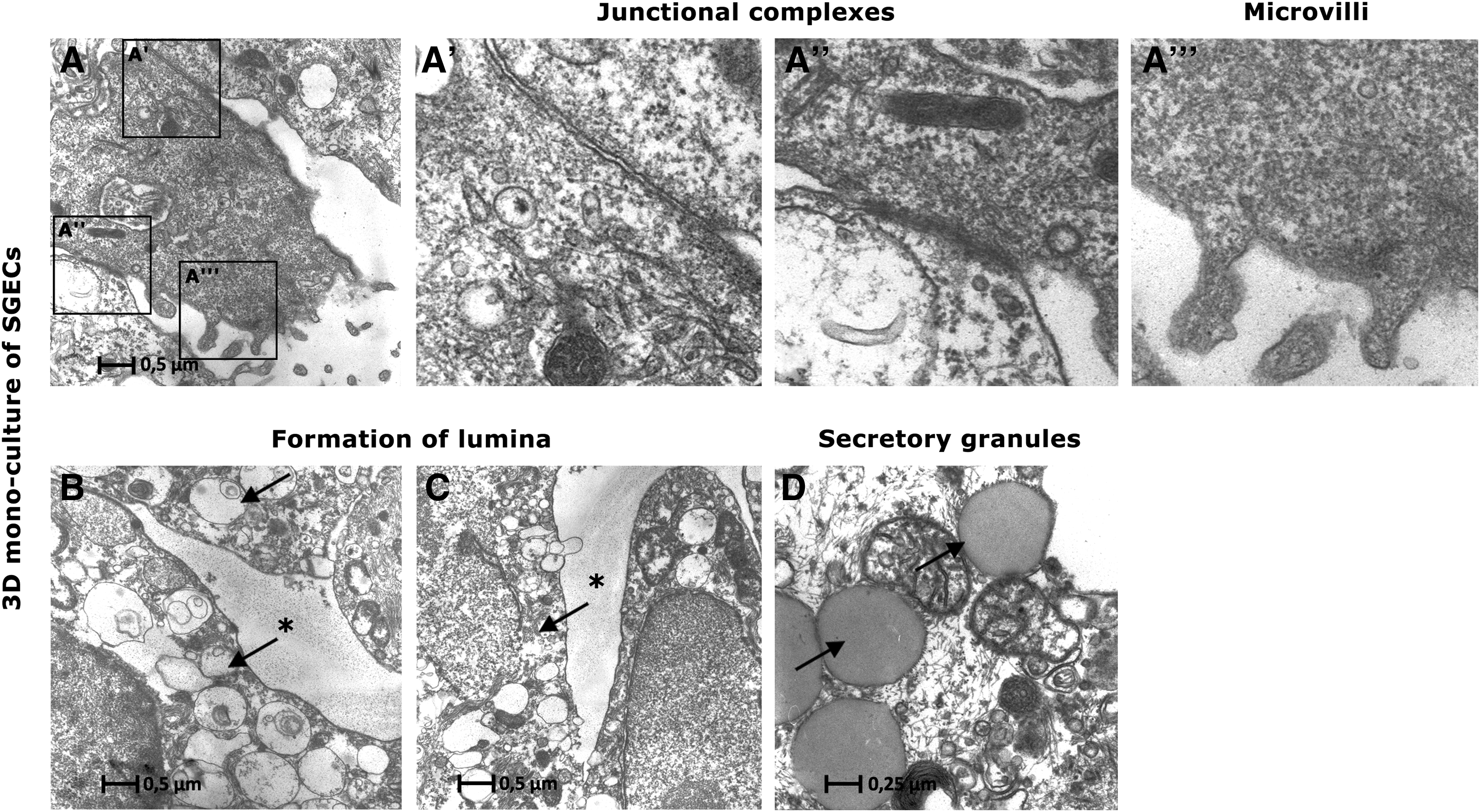
TEM images of 3D monoculture of SGECs.
Amylase gene expression and enzyme activity
Enzyme activity of α-AMY was examined in both 2D- and 3D cultures (mono- and coculture). The enzyme activity assay showed that 2D culture featured the lowest levels of α-AMY enzyme activity at all of the four analyzed time points (Fig. 6A). At cell culture day 3, 3D monoculture showed a notably higher activity than 2D culture (p < 0.1), but dropped to roughly the level of 2D culture in the course of cell culture. In contrast, enzyme activity in the supernatant of 3D coculture was around 25 times higher than the one in 2D culture at day 3. At the next two analyzed time points, 3D coculture α-AMY activity levels dropped to 0.38 mU × mL−1 at day 9 but at day 12 they reached an even higher level than the one measured at day 3.
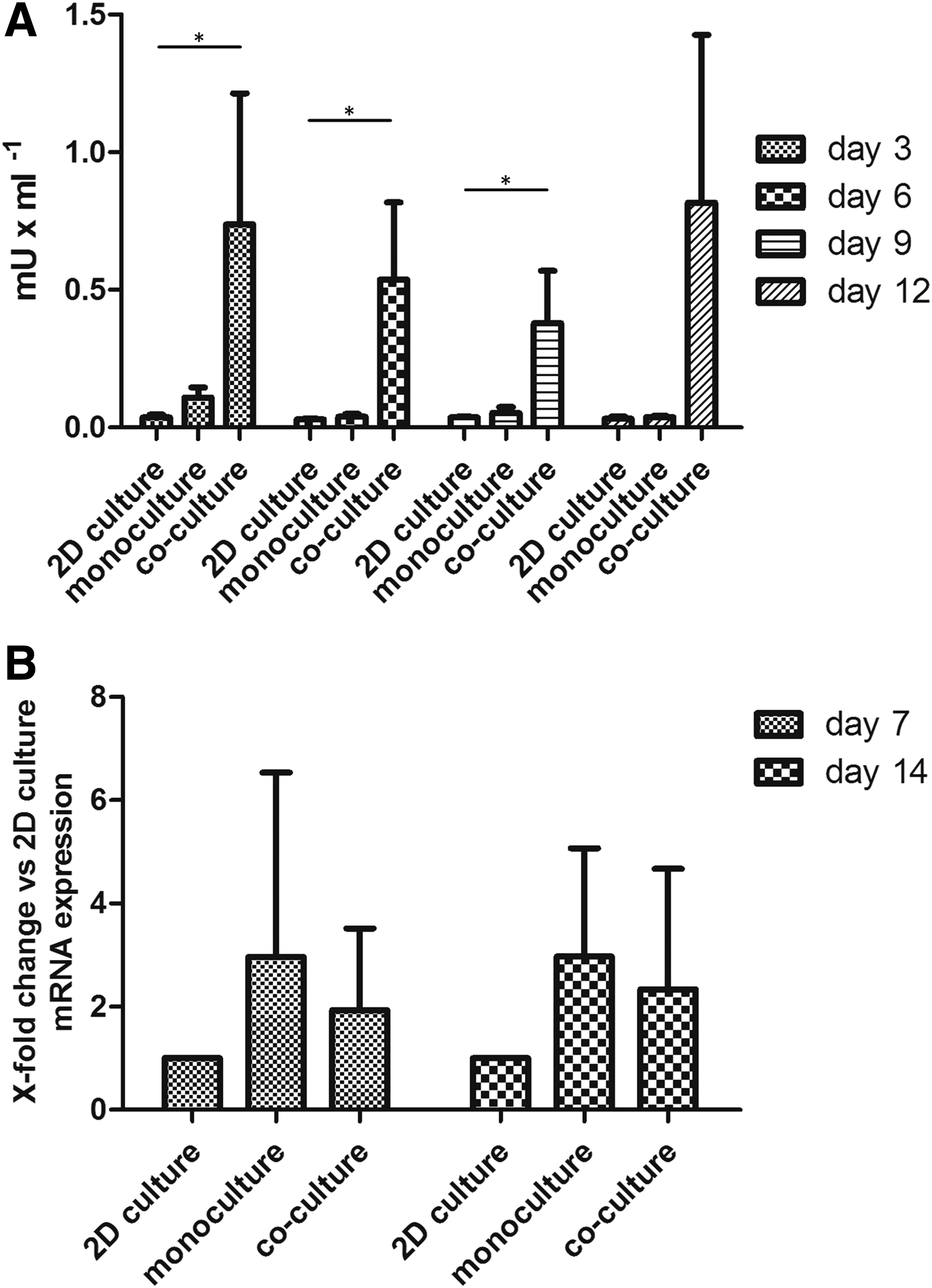
α-AMY enzyme activity and gene expression at different cell culture times.
Gene expression analysis performed with RT-qPCR revealed that in 3D monoculture, normalized expression of α-AMY was 2.96 times higher than in 2D culture after 7 and 14 days (Fig. 6B). In 3D coculture, the SGECs showed a 2-fold and 2.3-fold increased expression level at days 7 and 14 compared with 2D control, respectively (Fig. 6B). Those trends were clearly visible in the mean data of the n = 3 examined biological replicates but were not statistically significant due to high variability.
Discussion
Radiotherapy of the head and neck induces fibrosis of the salivary glands, thus indicating that once the salivary gland tissue is severely damaged it will hardly recover. The resulting xerostomia is still very common in daily clinical practice and reduces a patient's quality of life to a great extent. Different approaches exist to reduce damage of the salivary glands during radiation. IMRT is described as one possibility to minimize radiation dosage to the salivary glands.4–6 Drug treatment also reduced xerostomia after radiotherapy but it came up with side-effects.7,8 One promising new approach could be the injection of botulinum toxin into the salivary glands before radiation. 9 Further, surgical methods can be protective to some extent.10–14 The aim of tissue engineering is to create a functional artificial salivary gland from autologous glandular cells. In future clinical applications, patient-derived cells could be propagated before radiation is initiated and seeded on biocompatible matrices to create a new artificial gland ex vivo. The successful isolation and culture of salivary gland acinar cells has previously been reported,19–21 but there is no method for engineering a functional salivary gland that is capable of producing saliva in a sufficient, physiological extent at the moment.
Three-dimensional in vitro models have been proven to mimic the physiological conditions much better than standard 2D cell systems, which was also supported by the results of this study. Thus, the expression of AQP-5 and CL-1 via immunofluorescence was only detected in a 3D environment and the α-AMY enzyme activity was significantly higher under 3D and coculture conditions. In addition, PCR results emphasize the advantage of 3D over 2D culture regarding α-AMY gene expression levels, although those values were not statistically significant due to high variability. There are various possible reasons for the observed discrepancy between RNA and protein expression. It is well known that protein expression levels often do not correlate to RNA expression, which could be due to post-transcriptional or post-translational regulations, for example, by miRNA, RNA degradation, and proteolytic processing.22,23 Coculturing gains increasing influence in many fields of tissue engineering since it creates an optimized individual environment and, thus, affects cell differentiation, proliferation, and cell activity.24–26 In this study, mvECs seem to have a positive effect on SGECs since under coculture conditions α-AMY enzyme activity is about 25 times higher than under monoculture conditions. The molecular mechanism of this cell-cell interaction remains unclear and was out of the scope of this study. Previously, it was described by Cotrim et al. that endothelial cells play an important role in salivary gland function; however, a direct effect on α-AMY enzyme has not been described. 27 There are various epithelial-endothelial interactions that could possibly be involved. Endothelial cells may promote pro-acinar differentiation of salivary gland epithelial cells during morphogenesis via VEGFR2-dependent signaling 28 and, therefore, increase α-AMY production by the epithelium. Further, it is well known that parenchymal cells such as fibroblasts can induce gene expression of α-AMY. 29 Therefore, a similar mechanism could be assumed within the coculture of SGECs with mvECs.
One further step toward clinical applicability in tissue engineering is the analysis of human-derived cells and not animal-derived cells as in many earlier studies.30–32 The primary human salivary gland cells in this study were harvested from pieces of healthy parotid gland tissue that was obtained during routine operation on benign salivary gland tumors. For 3D culture approaches, different scaffolds, for example, Matrigel or matrices made from synthetic material,20,32–36 were applied to propagate salivary gland cells in vitro. It is well known that synthetic materials, such as polylactide, polyglycolide, or polyurethane, fulfill certain critical demands such as porosity, elasticity, appropriate cell adhesion, and viable degradation rates. But at the same time, disadvantages often occur during culture-like cell dedifferentiation, low cell-seeding efficiency, and poor retention of extracellular matrix.37,38 Polyurethane has been applied in clinical studies, whereas Matrigel is derived from a mouse sarcoma and, therefore, cannot be used for clinical application. The advantage of decellularized biological porcine matrices such as the SIS is the maintenance of major ECM components in vitro. Thus, the SIS was previously characterized as a biocompatible scaffold that is completely absorbable, 39 allowing cellular migration and differentiation.40,41 Clinical applicability has also been proven in various animal studies and human clinical trials.42–45 The small intestinal submucosa (SIS-muc; SIS, including mucosal villus/crypt structures) used in this study poses a preliminary test system for the biological vascularized scaffold (BioVaSc). The BioVaSc consists of a porcine decellularized small-bowel segment with remaining vascular structures that can be integrated into a dynamic culture system. 26 The vascular structures can be reseeded with an immunocompetent patient's own mvECs supporting the 3D growth of the tissue of interest within the lumen. The combination of the BioVaSc, sophisticated bioreactor technology, and coculturing of SGECs with mvECs could be considered a promising method to generate fully functional vascularized glandular tissue. Interestingly, Pradhan-Bhatt et al. describe the implantation of hyaluronic acid scaffolds seeded with human salivary gland cells into athymic rats 46 showing signs of vascularization after 1 week postimplantation. However, scaffolds of the size of a human submandibular gland would ideally need sufficient blood supply from the beginning of implantation. Without intrinsic vessels that guarantee sufficient nutrient supply, necrosis of cells in the center of the scaffolds is likely. As a first but important step, our study proves that the SIS-muc offers ideal conditions for the culture of SGECs. Conventional and immunofluorescent staining demonstrated viable SGECs on the SIS-muc expressing typical proteins such as α-AMY and AQP-5. These proteins have been described as essential functional acinar markers47,48 and could be primarily found in cells within the crypts, whereas CL-1 as a specific ductal marker 49 was more expressed on cells located toward the villus structures of the SIS-muc. Therefore, this construct may provide an in vivo-like excretory duct for future clinical applications. Immunohistological findings were further confirmed by SEM analysis, which indicated cell cluster formation especially in the crypts of the scaffold. TEM suggested formation of differentiated acinar cells with multiple secretory granules and formation of lumen-like spaces between preapoptotic cells. These observations are comparable to previous findings.18,50 Moreover, TEM revealed tight junctions that are crucial for unidirectional secretion since they are selective barriers for fluids, salts, and proteins. Together, these findings suggest an intact cellular function of SGECs and a high level of differentiation. Nonetheless, further studies should be conducted on duct functionality after pharmacological stimulation as well as an analysis of saliva composition needs to be addressed in follow-up studies.
In conclusion, in this study, it could be shown that human SGECs in coculture with human mvECs can be successfully cultured on the SIS-muc and might have the potential for future clinical applications in the field of salivary gland regeneration.
Footnotes
Disclosure Statement
No competing financial interests exist.