
Abstract
With the aim to obtain an injectable bioactive scaffold that can accelerate bone formation in sinus lift augmentation, in bony void and fracture repair, we have developed a three-dimensional (3D) jelly collagen containing lysophosphatidic acid (LPA) and 1α,25-dihydroxyvitamin D3 (1,25D3). Using an in vitro 3D culture model of bone fracture, we show that the contraction of the collagen gel is mediated by Rho-kinase activation in osteoblasts. The gel contraction showed dependence on cell concentration and was increased by LPA, which favored apposition and fastening of bone fragments approach. LPA was shown to act through actin cytoskeleton reorganization and myosin light chain phosphorylation of human primary osteoblasts (hOB). Moreover, LPA conferred osteoconductive properties as evidenced by the induction of proliferation, differentiation, and migration of hOB. The addition of 1,25D3 did not enhance cell-mediated gel contraction, but stimulated the maturation of hOB in vitro through the production of extracellular matrix of higher quality. On the basis of these observations, the collagen gel enriched with LPA and 1,25D3 described herein can be considered an injectable natural scaffold that allows the migration of cells from the side of bone defect and a promising candidate to accelerate bone growth and fracture healing.
Introduction
T
The bioactive lipid lysophosphatidic acid (LPA) has recently emerged as a highly significant regulator of bone cell biology by inducing chemotaxis, 10 growth, 11 maturation, 12 and survival 13 of osteoblasts, by promoting dendrite outgrowth in osteocytes, 14 and by stimulating osteoblastic differentiation of human mesenchymal stem cell (hMSC). 3 Moreover, by triggering actin stress fiber accumulation and cytoskeletal reorganization, LPA could therefore stimulate collagen matrix contraction and drive juxtaposition of bone fragments. For these reasons, we considered LPA to be a good candidate to functionalize injectable ECM scaffold with the objective to reduce the time required for fracture healing and speed bone regeneration in bone deficiencies such as nonunions or sinus lift augmentation. We hypothesize that, when injected in the bone defect, LPA-supplemented collagen gel can not only act as substrate for cells from vicinity of the defect to attach and populate but also as a mechanical framework for the reorganization and transformation of the matrix into a new bone. To explore this hypothesis, we developed a 3D collagen gel and studied if human primary osteoblast (hOB) can mediate gel contraction and by which mechanism, and then, we have tested in vitro the effect of LPA on the proliferation, migration, and differentiation of hOB-like cells. Furthermore, we have tested the in vitro efficiency of this scaffold containing LPA in modulating cell-mediated gel contraction rate and bone regeneration by itself and in conjunction with 1α,25-dihydroxyvitamin D3 (1,25D3).
Materials and Methods
Cell culture and reagents
Bone trabecular fragments (taken at surgery after informed consent and provided by the Orthopedic Institute, Major Hospital, Novara, Italy) were used in the 3D culture model of bone fracture or digested with collagenase/elastase as described previously
15
to obtain hOB. Where not specified, reagents were from Sigma-Aldrich (Milan, Italy). The osteoblasts were cultured in Iscove's modified Dulbecco's medium (IMDM; EuroClone) supplemented with 10% fetal calf serum (Hyclone), 2 mM
Collagen matrix contraction
Osteoblasts in FAFA-IMDM without serum were mixed at a ratio 1:1 in the collagen solution before gel formation at different cell densities to understand if gel contraction was cell mediated and if it was cell concentration mediated. The range of hOB concentration studied was as follows: 102, 103, 104, 105, 106, and 107 cells/mL. A density of 5 × 104 cells/100 μL gel was used to test the activities of 1,25D3 (10−7 M) and LPA (1 μM) or the combination of both on collagen contraction. Cell/collagen mixture cultured without 1,25D3 or LPA was used as basal control; and in the case of experiments to understand the role of ROCK aliquots of Y27632 (10 μM), a Rho-kinase antagonist were added. After the polymerization (60 min at 37°C), the polymerized collagen matrices were cultured with 1 mL of FAFA-IMDM added with the factor to be tested and the media were renewed every 3 days. To determine the extent of matrix contraction, two methods, one direct and the other indirect, were used: (1) in the direct approach, the matrices were measured and contraction data presented as the change in diameter after 24 h (and expressed as percent contraction starting-final); and (2) in the indirect approach, two fragments of human spongiosa (Puros; Zimmer, Carlsbad, CA) were embedded in each gel before polymerization and the change in distance between the edge of the bone fragments (represented by arrow heads in Fig. 1) was measured as a function of time. Images of the gels were taken at time 0, 1 h, 1, 3, 6, 12, and 24 days and distances between the bone fragments quantified. Images were acquired using an inverted microscope equipped with a digital camera connected to an image analysis system (all from Leica Microsystems) and analyzed using Qwin Plus V Image Analysis software (Leica Microsystems).
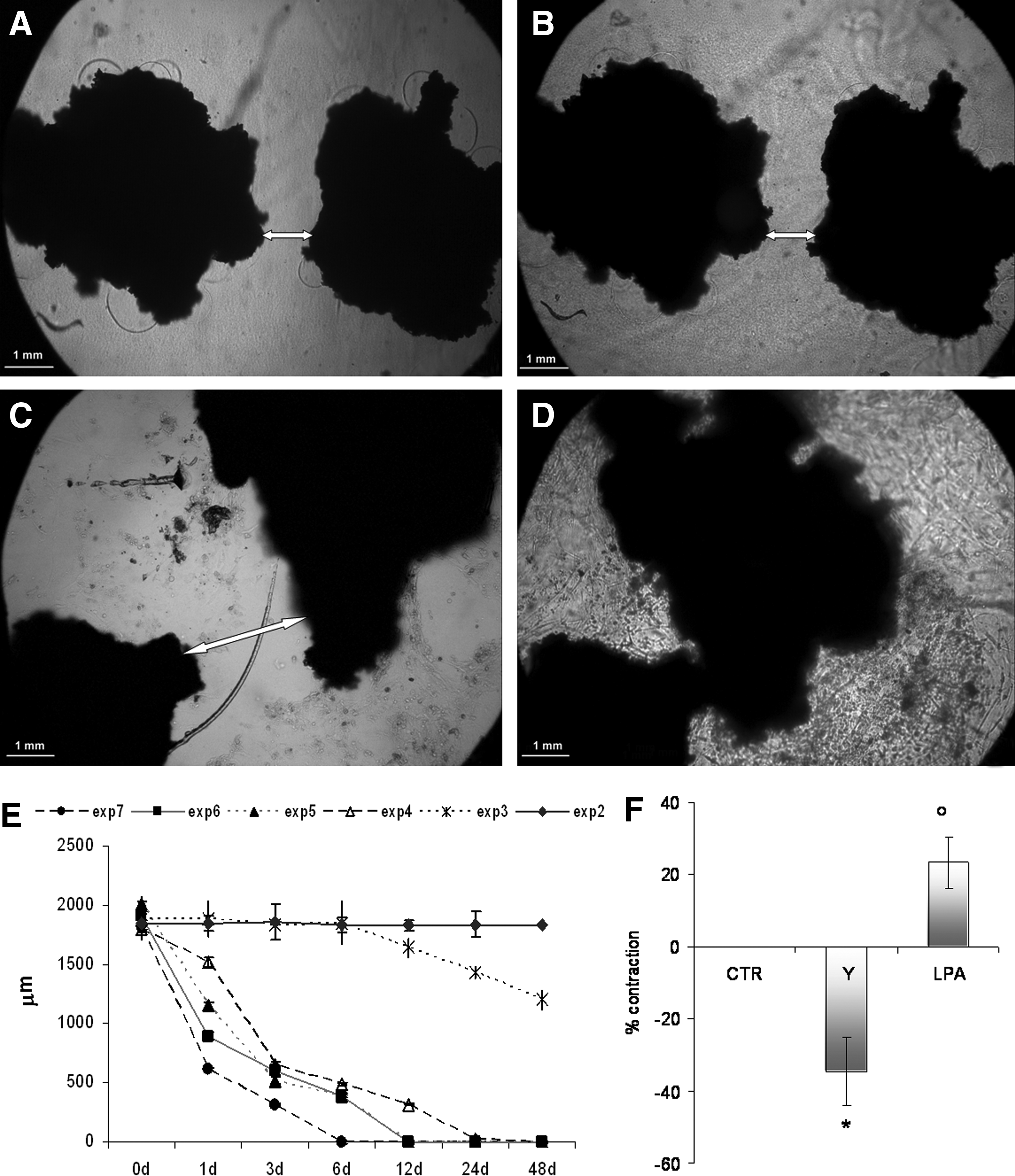
Collagen matrix contraction is hOB mediated and ROCK mediated.
Fluorescence and confocal microscopy
The 3D collagen cultures or cultures on collagen-coated coverslips, after 24 h of treatments, were fixed for 20 min at room temperature with 3% paraformaldehyde in phosphate-buffered saline (PBS), blocked with 1% glycine and 1% bovine serum albumin (BSA) in PBS for 30 min, and permeabilized with 0.2% Nonidet P-40 in PBS for 10 min. For actin staining, samples were incubated with fluorescein isothiocyanate (FITC)- or tetramethylrhodamine isothiocyanate (TRITC)-conjugated phalloidin (8 U/mL) for 45 min at 37°C in a humidified incubator in the dark. For the staining of myosin, samples were incubated with anti-myosin light chains (MLCs; MY-21; 1:200; Sigma-Aldrich) overnight at 4°C and then with secondary antibody conjugated to FITC (1:300; Santa Cruz). After washing in PBS, samples were observed and images captured using a Leica DM 2500 fluorescence microscope equipped with a DFC7000 camera or a Nikon A1 Rsi microscope using the software NisElements. Qwin Plus V 2,6 software was used to process and measure images. Ten cells for each treatment and each culture model were measured.
MLC phosphorylation
MLC phosphorylation was determined using the urea/glycerol-polyacrylamide gel electrophoresis (PAGE) method. 17 Cells were treated with ice-cold 10% (w/v) trichloroacetic acid containing 10 mM dithiothreitol and homogenized on ice. After centrifugation at 8,000 rpm (Beckman Microfuge) for 1 min at 4°C, the pellets were washed thrice with diethylether, dissolved in the urea sample buffer (10 mM dithiothreitol, 0.005% bromophenol blue, 8 M urea, glycerol 5%, 20 mM Tris-HCl, and 23 mM glycine, pH 8.6), and subjected to urea/glycerol-PAGE using 12% gels. Transfer to nitrocellulose membrane was carried out for 1 h at 100 V. Blots were blocked with 3% BSA in PBS-Tween and then incubated with anti-MLC monoclonal antibody (MY-21; Sigma-Aldrich) in blocking solution at 4°C for 12 h. After washing in PBS-Tween, membranes were incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (PerkinElmer) at room temperature for 1 h and then visualized by enhanced chemiluminescence. Quantitative evaluation of the nonphosphorylated, monophosphorylated (MonoP), and dephosphorylated (DiP) MLC was performed by densitometric analysis using NIH Image.
Cell proliferation and differentiation
To study the effect of LPA and 1,25D3 on hOB proliferation and differentiation, the same 3D culture model described for collagen contraction was used. Proliferation was tested using the ATP quantification Kit (ViaLight Cambrex). At time 0 and after 1, 3, 6, and 12 days of culture of cells in collagen gel enriched with 1,25D3 10−7 M, LPA 1 μM, or the combination of both, cells were lysed with cell lysis reagent and treated with ATP monitoring reagent. Light produced was measured by a luminometer and expressed as relative luminescence units (RLUs). For alkaline phosphatase activity (APA) detection, cells in the collagen gel containing 1,25D3 (100 nM), LPA (1 μM), or the combination of both, after 1, 3, and 6 days of culture, were rinsed and homogenized on ice for 2 × 10 s using a 250 sonifier (Branson Ultrasonics, Danbury, CT) in 100 μL PBS. To 50 μL of this solution was added 50 μL of substrate (1 mM paranitrophenyl-phosphate in 1 M diethanolamine +1 mM MgCl2, pH 9.8). The mixture was incubated at 37°C until the color was comparable with a standardized series of paranitrophenol and measured in a microplate spectrophotometer at 410 nm (Bio-Rad, Milan, Italy). For mineralization studies, hOB were cultured for 3 weeks in FAFA-IMDM supplemented with 10 mM β-glycerophosphate and 50 μg/mL
Migration study
The effect of LPA, 1,25D3, or LPA +1,25D3 on cell migration was investigated in a monolayer culture model and the 3D collagen-medicated scaffold. 1.5 × 105 cells were plated in 24-well plates in 300 mL of medium and incubated at 37°C in 5% CO2 until confluence and then serum starved for 20–24 h. The monolayers were wounded by the introduction of linear scratches with a sterile pipette tip, rinsed with PBS, and cultured in FAFA-enriched medium without or with the tested factors at the following concentrations: 1,25D3 100 nM and LPA 2,5 μM. Six selected fields (kept the same from time 0 to 3 days) for each experiment were tracked for distance travelled. The selected fields (30–40 μm) were at fixed distance from the border of the culture plate and for each experiment were acquired using a Leica digital camera connected to an inverted microscope (Leica Microsystems). Images were acquired at time 0, after 6 h and 1, 2, and 3 days after the addition of the stimuli. The distances travelled by the cells were measured using Qwin Image Analysis system (Leica Microsystems). In the 3D collagen scaffold, cells were seeded on a 24-well nonadhesive plate at 1 × 106 cells/well and then, the collagen scaffolds with or without LPA, 1,25D3, or both were placed in the plate and incubated at 37°C with gentle shaking. After 3 weeks, the samples were washed with PBS and some samples stained with FITC-phalloidin for confocal observations, and others after formaldehyde fixation, dehydration, and paraffin inclusion, were cut into sections and stained by Masson trichrome.
In vitro culture model of bone fracture
Trabecular vital bone fragments 1–2 mm size in FAFA-IMDM without serum were mixed at a ratio 1:1 in the collagen solution before gel formation together or without the factor to be tested (1,25D3 10−7 M; LPA 1 μM), or the combination of both and compared after 3 weeks of culture in the calcification medium described in calcein staining paragraph. After the culture period, three samples from each stimulus were used to test mechanical properties of the newly formed tissue in the collagen matrix between the bone fragments, whereas other specimens were treated for microscopic observations. The tensile strength was measured with an Instron 5564 mechanical testing instrument (Instron Corporation, Canton, MA). The tensile test was carried out at a constant elongation speed of 0.05 mm/s. Since the machine relays real-time displacement of the load frame and strength employed on the sample, the strength at the breaking point was used as parameter. For histological observations, the specimens were fixed at 4°C for 2 days in 4% phosphate-buffered formaldehyde and dehydrated in ascending ethanol for one night before a three-step impregnation in methylmethacrylate (MMA) for at least 3 days. For embedding, specimen blocks were impregnated in 80% (v/v) stabilized MMA and 20% (v/v) Plastoid N (Rohm Pharma) for 2 h in uncapped vials under vacuum and imbedded in capped 10 mL glass vials at 37°C overnight. After the polymerization, the glass vials were removed and moistened sections (100 μm) were cut on a Leica SP 1600 Saw Microtome with a rotating diamond saw blade for high-quality sample preparation of hard materials for microscopic analysis, and mounted on polyethylene slides. The sections were stained using fuchsin/light-green.
Statistical analysis
For statistical evaluation regarding collagen contraction data, cell proliferation, differentiation and motility results, and mechanical properties, a nonpaired Student's t-test was used. Groups were compared by analysis of variance and Bonferroni Student's two-tailed t-test was applied (SPSS, Chicago, IL). The conventional p ≤ 0.05 level was considered to reflect statistical significance. All experiments were replicated two to four times and replicated twice for each experimental condition within each test. All data are presented as mean ± standard deviation.
Results
Gel contraction is mediated by osteoblasts with a Rho-dependent mechanism
The 3D collagen gel showed no evidence of contraction in the absence of hOB. While the diameter of the collagen gel matrices remains unchanged from their initial diameter of 10 mm without cells, in the presence of hOB, a contraction of 50% ± 8% (based on diameter of the gel) was observed. In studies where embedded bone fragments were used as an indirect measure of gel contraction, the bone fragments (inserted at 2 mm distance from each other) showed no evidence of movement toward each other in the absence of cells throughout the duration of the contraction experiment (Fig. 1A, B). In contrast, osteoblast-activated gel contraction led to apposition of the fragments with zero separation distance between the two fragments (Fig. 1C, D). The contraction of the collagen leading to a decrease in matrix diameter and appositional movement of the bone fragments showed dependence on hOB concentration, with increasing hOB numbers leading to a dramatic decrease in time required for the bone fragments to coalesce (Fig. 1E). Since the contraction of collagen gel by fibroblasts has been shown to be Rho-kinase mediated, 19 we compared an hOB-induced contraction in the presence of Rho-kinase antagonist (Y27632) and LPA, an agonist of Rho kinase. We saw that, at the same concentration of hOB in the gel (5 × 105 cells/mL), with the stimulation of LPA, there is an increase in the contraction of collagen compared to the control, while the contraction was decreased in the presence of the Rho-kinase inhibitor (Fig. 1F).
Evidence for Rho-mediated actin cytoskeleton modifications in human osteoblasts
Representative images of hOB when cultured in collagen-coated coverslips and collagen 3D matrices are shown in Figures 2A–F; Table 1 summarizes measurements. Cells in monolayer were well spread with lamellipodia and actin stress fibers. In the control (Fig. 2A), cells had more elongated and compact morphology with actin filaments that cross the cell cytoplasm following the long axis toward the anchor points at the two opposite poles of the cell. Cells treated with the Rho-kinase agonist (Fig. 2B) had a more compact cell and isotropic morphology, and cover a smaller area, but exhibited an increased number of cellular processes. Actin in the extensions of cellular processes departed from spherical actin spots present in the cytoplasm and shows greater thickness compared to the control filaments. In the presence of Rho-kinase antagonist, Y27632 cells had a stellate-like shape and cytoplasmic projections, and actin-containing ruffles only along the cell margins (Fig. 2C). Even though the cell area appeared to be comparable to the control, the cells had shorter long axis and not spindle shaped, and had a greater number of pseudopodial processes that were not just localized to the poles. Furthermore, actin filaments in cell extensions were long, branched, and less thick. A markedly different appearance was seen in 3D collagen matrices where osteoblasts projected a dendritic network of extensions (Fig. 2D) that retracted when LPA was added to the gel (Fig. 2E), and in presence of the Rho-kinase antagonist, an overall increase in length and branching of the dendritic network, together with a reduced thickness of the actin filaments was observed (Fig. 2F). These findings suggest that hOBs in 3D floating collagen matrices developed neuron-like dendritic morphology distinct from osteoblasts on coverslips and responded more clearly to LPA by reducing cell dimensions, increasing extension number that showed reduced length and increased thickness of stress fibers. Actin localized cortically and was concentrated at the tips of cell extensions in control and Y27632-treated gels or in ruffles and at the tips of cell extensions in LPA-treated cells. Studies of myosin distribution in hOB revealed its localization at the center and end of the cells (Fig. 2G), and an organization definitely inferior compared to that present in the cells treated with LPA where the myosin is well localized along the stress fibers (Fig. 2H). Western blot analysis revealed that LPA seems to decrease the total MLC present in the cell. Experiments carried out to ascertain if LPA stimulation of contraction correlates with MLC phosphorylation (Fig. 2I) show that after 30 min of treatment, the MonoP and DiP forms of MLC were readily distinguished based on the faster molecular mobility in urea gels of the phosphorylated molecules. 20 Using densitometric analysis of the urea/glycerol-PAGE stained with anti-MLC, we carried out semiquantitative analysis of the nonphosphorylated and the phosphorylated forms of MLC. In the absence of LPA, the percent of phosphorylated forms of MLC reaches a quantification ±50% compared to the densitometric value of total MLC. In response to Y27632 stimulation, MLC phosphorylation decreased slightly, whereas incubation of hOB with LPA resulted in a stimulation of MLC phosphorylation with increased levels particularly evident for the DiP form of the molecule. Therefore, the LPA-stimulated MLC phosphorylation argues for a role for MLC kinase in LPA-dependent stimulation of collagen matrix contraction.

Cytoskeletal modification of hOB could be responsible for gel contraction. TRITC-phalloidin staining evidenced actin filaments in fluorescence microscopy.
Osteoblasts fixed and stained for actin with representative images shown in Figure 2A–F were measured using Leica imaging software (Qwin Plus V 2.6; Leica Microsystems). Ten cells for each treatment and for each culture model were measured. For the area, cell outlines were traced using images acquired at 20 × magnification and the longer distance from the border inside the cell was used to measure the long axis. For the extension measurements, the distance from the cell body to the tip of each extension was marked using images acquired at 40 × magnification, while for actin filament thickness measurements, 100 × magnification images were used. Data shown are average ± SD.
p < 0.05 compared to control.
2D, two dimensional; 3D, three dimensional; LPA, lysophosphatidic acid; SD, standard deviation.
Human osteoblast proliferation, differentiation, and migration in response to LPA
To understand if LPA added to collagen may work to improve osteoconductivity in collagen gels, we tested its activity on proliferation, differentiation, and migration of hOBs when it was used alone or in combination with 1,25D3, which has been shown to provide differentiation stimulus to osteoblasts. 21 Our data confirmed an inhibition of proliferation of hOB treated with 1,25D3, while LPA produced a significant increase in proliferation at 6 and 12 days as shown in Figure 3A. However, when LPA was added in combination with 1,25D3, the proliferative response of the cells was inhibited. Furthermore, while 1,25D3 and LPA increased APA in hOB after day 1, at a longer incubation time, 1,25D3 had no effect on APA, whereas LPA induced a statistically higher activity also at 3 and 6 days of culture. The costimulation with both factors led to a synergistic increase in APA that was time dependent (Fig. 3B), together with an increased matrix mineralization (Fig. 3C). LPA resulted also to be chemotactic for hOB (Fig. 3D). The LPA group showed greater filling of the denuded areas on the dish with cell than untreated controls, and cells treated with 1,25D3 showed no migratory activity. When compared to control, LPA-treated hOB exhibited a 1.4-fold increase in the average distance that the cells moved over the course of the experiment as shown in the bar graph of Figure 3. The 3D collagen model of cell migration study showed that cells are present in greater quantities in the external part of the gel and then on the edge and the upper and lower images of the z planes acquired by confocal microscopy. It was also seen that cells were present even at a distance of 100 μm from the edge of the gel, indicating that the cells had migrated and grown in 3D in the structure (representative images shown in Fig. 4A–F). No differences in terms of cell quantity were seen among untreated collagen or collagen embedded with LPA or 1,25D3. In all samples, we saw that cells appeared more relaxed in the peripheral part of the gel (Fig. 4A, D acquired at z stacking position in the lower part of the gel), while more we move inside the gel, the cell morphology changed and they appeared more elongated and threadlike compared to cells in outer zone (Fig. 4C, F acquired at z stacking position in the central part of the gel). Using the longer axis of the cell as baseline for cell orientation, we have seen a high concentration of cells oriented perpendicular to the outer surface of the gel on the edge (Fig. 4B, E). The primary difference seen between the differently treated collagen was in the central part of the gel where in the LPA-medicated one (Fig. 4F), cells appeared to orient each other in the same direction as if there were collectively experiencing the same directional force, whereas in collagen without LPA, hOBs appeared randomly oriented (Fig. 4C). Masson trichrome staining of paraffin-embedded gels from the migration study in 3D collagen (collagen in green and cells in blue/red) is shown in Figure 4G. In all samples, cells were evenly distributed in the collagen matrix even if, as shown in confocal microscopy, a higher cell concentration was seen at the periphery of the gel. In the microscope images, a high cell concentration in the center of the collagen 3D matrix is clearly evident in both LPA- and LPA +1,25D3-treated samples. Moreover, in the central part of the sections, particularly in LPA +1,25D3-enriched gel, areas with higher green intensity are clearly visible, probably due to a greater amount of ECM production in this area or a greater contraction of collagen in this area, which could also increase the density of collagen bundles.
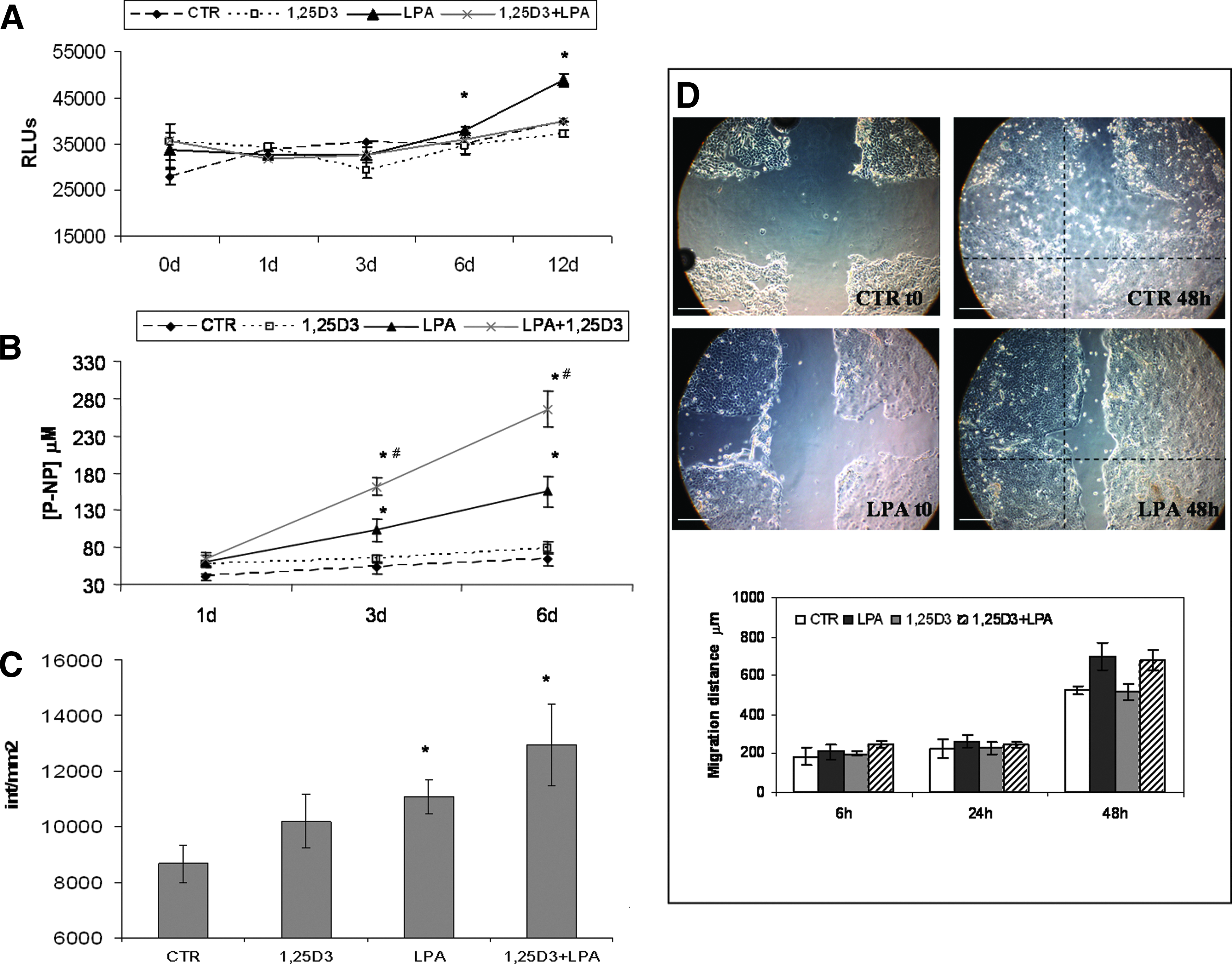
LPA induces hOB proliferation, differentiation, and migration. ATP quantification

LPA modifies cell orientation and ECM organization in the 3D collagen matrix. Confocal microscopy images
LPA and 1,25D3 cooperate to expedite the approach of bone fragments and bone regeneration
A time-course study to follow the apposition of bone fragments in the 3D collagen gel supplemented with LPA, 1,25D3, or LPA +1,25D3 showed that compared to the control, 1,25D3 does not speed up the apposition of the bone fragments. According to Parreno et al. 22 1,25D3 treatment did not affect collagen gel contraction, whereas LPA or LPA +1,25D3 increases the speed of contraction in the short-term (after 1, 3, and 6 days) and also provides to reach the total approach in 12 days instead of the 24 days required with 1,25D3 alone (Fig. 5A). The histological results of the newly formed tissue around bone fragments show the formation of a fibrous bone-like tissue within 3 weeks of culture (Fig. 5B, C). The addition of 1,25D3 to collagen increased the density of the newly formed tissue that also showed an increased tensile strength (Fig. 5D).
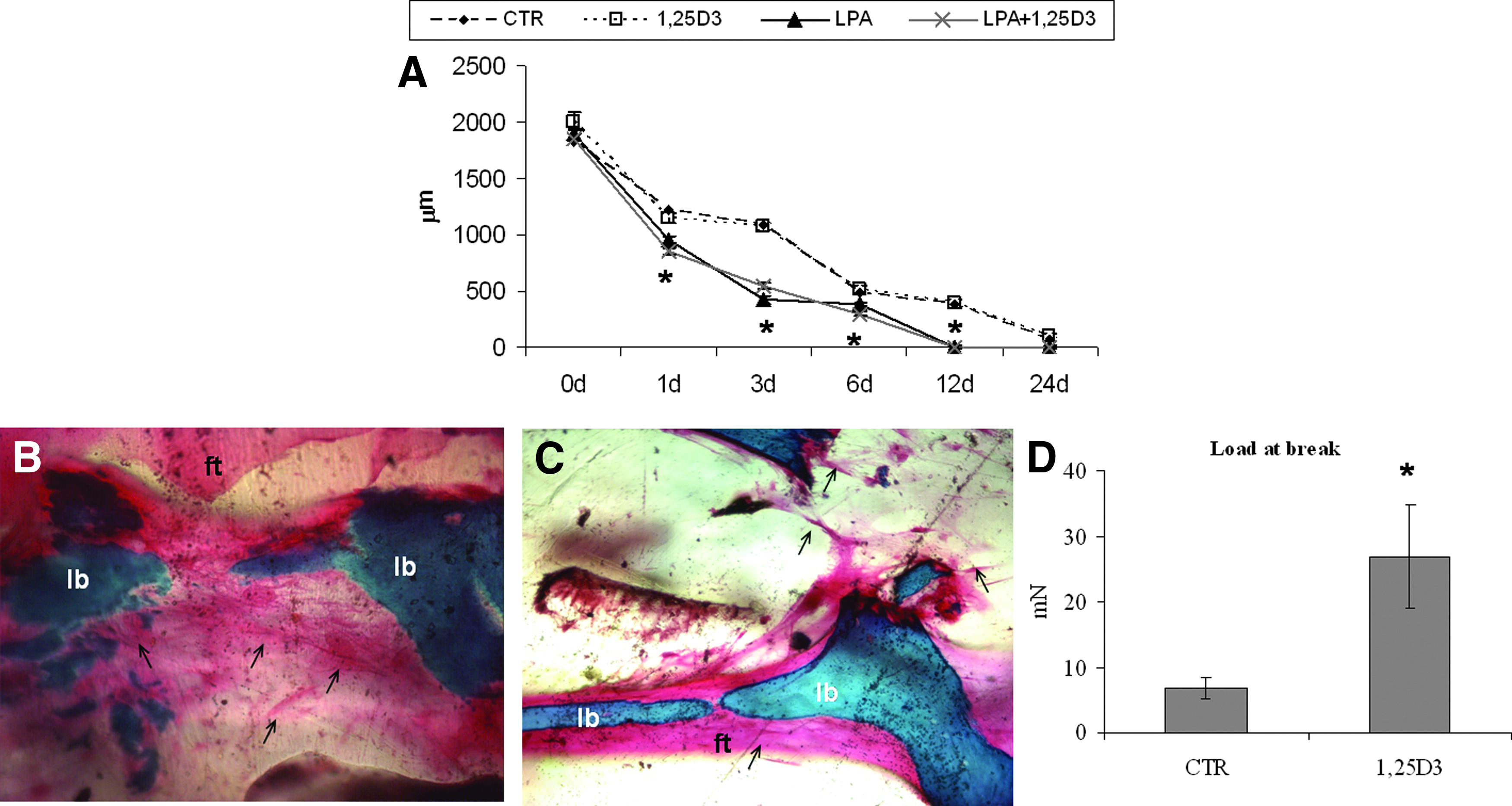
LPA expedite bone fragments approach, whereas 1,25D3 promotes ECM synthesis.
Discussion
Tissue regeneration by endogenous cells recruited from the defect area “cell homing,” without the necessity of cell transplantation, is a promising strategy in various fields of regenerative medicine. 23 With regard to bone, the migration of cells from the site of defect could be important to faster bone healing and speed up bone regeneration. We used a 3D collagen gel, generally used to demonstrate the mechanisms by which cells contract, but only recently considered a potentially useful biological tool for 3D bone tissue engineering 24 to develop a potentially injectable bioactive scaffold, which can speed up bone tissue formation in sinus lift augmentation, in bony void, and in fracture repair.
We have demonstrated that hOB cultured in a 3D collagen matrix showed a global matrix remodeling in a manner similar to what is observed in fibroblasts, resulting in collagen contraction that was dependent on the activation of Rho kinase in hOB. Encouraged by these findings, we incorporated LPA, a Rho activator, to the collagen gel, demonstrating that this medicated gel could be used as an active scaffold that due to its cell-mediated contraction will favor the closure of the gap between bone fragments. The cytoskeletal changes to osteoblasts seen in LPA-enriched gel could explain the gel contraction results as occurring through an inhibition of cell protrusion, thus leading to contractility. These results are supported by previous studies that have shown that fibroblasts when treated with LPA exhibited large bundles of actin microfilaments and stress fibers, and that the subsequent addition of Y-27632, a Rho-kinase inhibitor, disrupted these fibers. 25 Moreover, in cells where Rho function was blocked, both focal adhesion and stress fiber assembly induced by LPA were completely inhibited. 26 The mechanism of contraction of these cells, therefore, very likely involves the action of myosin bound to actin along stress fibers 27 as phosphorylation of MLCs lead to conformational changes in myosin, enabling it to assemble into filaments and promoting its productive interaction with actin. 28
We chose LPA as an activator of the collagen scaffold for three distinct reasons. First, LPA's receptors couple to Rho and Rac to elicit changes in cytoskeletal organization, 11 thereby regulating cell migration, chemotaxis, and growth 8 in many cells, including osteoblasts through interactions with the LPA1 receptor. 11 Second, LPA is believed to be a factor involved in the regulation of osteogenesis and induction of bone remodeling by inducing in vitro osteoblasts' synthesis of some osteoinductive cytokines12,29 and committing MSCs to osteoblasts. 10 Third, recent in vitro and in vivo studies have shown that LPA is present at elevated levels at sites of tissue injury and inflammation as product of activated platelets, 6 and is also produced by bone cells, thus identifying its direct effect on various cellular activities that could involve control of bone mass and/or composition.9,10 Our results have demonstrated that actin cytoskeletal modification of hOB and MLC phosphorylation are attributes of LPA-induced gel contraction, and due to LPAs effect on hOB proliferation migration and maturation, it can be considered a regenerative factor with potential role in bone healing and repair.
LPA together with EGF and TGFβ are the only three growth factors known to synergistically cooperate with 1,25D3 in promoting hOB maturation 30 ; so its combination with 1,25D3 in a controlled release format or as coatings of metal and ceramic devices could have great potentials in a bone regenerative context like fracture nonunions and bone biomaterial integration.
Active vitamin D3 metabolites have direct effects on human osteoblast function and matrix calcification, providing mechanically robust mineralized bone collagen matrix as vitamin D receptor (VDR)-mediated events. Recently, we have shown that the loading of 1,25(OH)2D3 positively influences the late events in osteoblast maturation and the induction of osteogenic markers, and in accordance with other studies, 31 we found a positive effect on in vitro osteogenesis using calcitriol within a 3D scaffold. 14 Of additional significance are studies reporting cross-talk between 1,25D3 and growth factors known to be active on hOB growth, matrix synthesis, and mineralization.
Toward the aim to modulate gel contraction rate and bone regeneration, our results represent an improvement on data obtained with LPA alone or mixed with 1,25D3, which has a known synergistic role with LPA in osteoblast maturation.11,32
This research presents an important first step in realizing the development of lipid-functionalized injectable material (LPA-enriched collagen gel) that can fasten bone fragments, leading to their apposition by cell-mediated collagen contraction. Furthermore, presence of LPA led to an enhanced hOB homing in its 3D structure and a synergistic effect with calcitriol to promote human osteoblast maturation, resulting in an ex vivo bone-like mineralized collagen matrix, which was mechanically sound.
Footnotes
Acknowledgments
This work was supported by grants from the Italian Ministry of Education, University and Research (MIUR) PRIN 201288JKYY/2012. The study sponsors had no involvement in the study design, in the collection analysis, and interpretation of data, in the writing of the article, and in the decision to submit the article for publication.
Disclosure Statement
No competing financial interests exist.