
Abstract
Whole-organ engineering is an innovative field of regenerative medicine with growing translational perspectives. Recent reports suggest the feasibility of decellularization and repopulation of entire human size hearts. However, little is known about the susceptibility of epicardial adipose tissue (EAT) to decellularization. In this study, human size hearts of ovine donors were subjected to perfusion-based decellularization using detergent solutions. Upon basic histological evaluation and total DNA measurement myocardial regions prove largely decellularized while EAT demonstrated cellular remnants, further confirmed by transmission electron microscopy. Western blot analysis showed a significant reduction in lipid-associated and cardiac proteins. However, gas chromatography revealed unchanged proportional composition of fatty acids in EAT of decellularized whole hearts. Finally, cell culture medium conditioned with EAT from decellularized whole hearts had a significant deleterious effect on cardiac fibroblasts. These data suggest that perfusion decellularization of human size whole hearts provides inconsistent efficacy regarding donor material removal from myocardial regions as opposed to EAT.
Introduction
E
Within less than a decade a rapid sequence of scientific cornerstone achievements has boosted the general expectations and the hope for a clinically relevant solution facilitated by means of tissue engineering. 5 The successful decellularization of an entire rodent heart by means of coronary perfusion with detergent solutions has been the ground for further developments involving repopulation of decellularized whole hearts with cardiac interstitial cells and myocytes, as well as endothelial cells.6,7 Meanwhile an upscaling adaptation of the involved methods has been initiated and most advanced reports underline the technical progress toward whole-heart decellularization and repopulation as an alternative approach to generate functional cardiac organs to treat end-stage heart failure. 8
However, there are a number of issues beyond the mere physical size that compose to the interspecies difference of the cardiac organ when rodents are compared to larger mammalians or humans. When decellularization efficacy and feasibility has been addressed in previous reports the focus of the experimental workup has been placed on the myocardial compartment of the heart, the cardiac valves, and the adjacent great vessels.9–12 However, none of previous reports has particularly evaluated the impact of decellularization on the epicardial adipose tissue (EAT) and the potentially associated implications for the future in vivo use of these scaffolds. Therefore, we aimed at evaluating the effects of standard perfusion decellularization of whole hearts using detergent solutions on EAT. Histological and biochemical assays, as well as chromatographic analysis, were conducted, and finally an in vitro viability assay was performed to gain an insight into the evolution of EAT during perfusion decellularization of whole hearts.
Materials and Methods
All experiments involving material from animal source were conducted in accordance to the German law (§6 and §8 of animal welfare act of North-Rhine-Westphalia [TierSchG]), the Guide for the Care and Use of laboratory animals published released by the US National Institutes of Health (NIH Publication 85-23, revised 1996) and approved by the Animal Care Committee of the Heinrich-Heine-University, as well as in compliance with the local governmental authorities in North-Rhine-Westphalia, Germany (LANUV, German state office for nature, environment and consumer protection of NRW).
Whole-heart decellularization and EAT sampling
Ovine hearts were obtained from a local slaughterhouse (Laame, Wuppertal). Particular arrangements were made, and attention was paid to keep the time-to-decellularization as short as possible. Freshly explanted hearts were subjected to coronary perfusion with a heparin solution (50 U/mL) containing streptokinase (10 U/mL) before transportation to the laboratory facility. Coronary ostia were selectively cathetered, and pressure-controlled perfusion decellularization was performed using a custom-made perfusion system as previously described for rodent heart decellularization. 13 In brief, detergent based protocol was used involving a total of three cycles of 10 L solution of sodium dodecyl sulfate (SDS, 0.75%) and deoxycholic acid (DCA, 0.25%). Hearts were rinsed by coronary perfusion involving four consecutive cycles of each 5 L of distilled water for 24 h in total, whereby solutions were exchanged four times. A constant perfusion pressure of >70 mmHg was maintained throughout the perfusion steps.
For standardized sampling of EAT the great cardiac vein and the epicardial coronary arteries were used as anatomic landmarks. In addition to EAT, standardized specimens from the myocardial region were taken from the left ventricular wall.
Protein isolation from EAT and quantification
For protein isolation, 25 μg ovine EAT was homogenized in 180 μL of RIPA buffer containing protease inhibitors on ice. Afterward protein concentration was determined using a commercial BCA assay (Thermo Scientific) according to the manufacturer's instructions and using a standard curve with bovine serum albumin. Protein lysates were diluted 1:6 with laemmli buffer and proteins separated by SDS-PAGE. Western Blot was performed using primary antibodies against myozap (polyclonal; charge 2-A, 11/09; dilution 1:1000), perilipin 1 (monoclonal; Cat. No. 651156; clone 112.17; dilution 1:10), perilipin 4 (S3-12; polyclonal; Cat. No. GP38; charge hNT-A 09/13; dilution 1:1000), and vimentin (polyclonal; Cat. No. GP58; charge hCTB 6/08; dilution 1:1000), all obtained from Progen (Heidelberg, Germany). 14 Primary antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and beta actin (β-actin) were obtained from Cell Signaling Technology (MA) and Sigma-Aldrich (Taufkirchen, Germany), respectively. For signal detection a fluorescent secondary antibody (for perilipin 1) or horseradish peroxidase reaction was used and protein bands visualized using a gel imaging system (Amersham Imager 600; GE).
Transmission electron microscopy
For transmission electron microscopy (TEM), native and decellularized epicardial tissue samples were fixed for several hours in 2.5% gluteraldehyde and 4% paraformaldehyde (PFA) in 0.1 M cacodylate buffer (pH 7.4) at 4°C. Then, samples were incubated in 1% osmium tetroxide in 0.1 M cacodylate buffer for 2 h. Dehydration was achieved using acetone (50%, 70%, 90%, and 100%), and block contrast was applied (1% phosphotungstic acid/0.5% uranyl acetate in 70% acetone). A SPURR Embedding Kit (SERVA, Heidelberg, Germany) was used to embed samples, which were polymerized overnight at 70°C, before cutting into 80 nm sections using an Ultracut EM UC7 (Leica). Images were captured using an H600 TEM (Hitachi, Tokyo, Japan) at 75 kV.
Gas chromatography
Initially 4–5 mg fat, 500 μL C15:0 (1 μg/μL) as an internal standard, and 1 mL methanolic NaOH (c = 1 M) were mixed in a vial, sealed and saponified at 90°C for 60 min. Once the saponified fat sample was cooled down slightly, it was transferred to a test tube. Then, 3 mL of n-hexane was added, followed by a centrifugation step for 5 min at 3000 g. Since the free fatty acids are located in the hexane phase, this was discarded. Subsequently, the fat sample was neutralized with 1 mol of hydrochloric acid. To precipitate any proteins, 0.5 mL of extraction solution was added. After vortexing, 3 mL of n-hexane was added to extract the total fatty acids (TFAs). The sample was vortexed again, centrifuged for 5 min at 6000 g, and ultimately the organic phase transferred into vials. Using nitrogen, the sample was evaporated at 50°C for 5 min. To produce fatty acid methyl ester, hydrochloric acid methanol was used as derivatization reagent. Then, the samples were evaporated again with nitrogen at about 50°C. As a solvent for the fatty acid methyl ester, 1 mL of n-hexane was used and 1 μL of it were injected to the gas chromatograph with the help of an auto sampler.
DNA isolation and photometric quantification
Genomic DNA was isolated using a commercial kit (QiAamp DNA Mini Kit; Qiagen) according to the manufacturer's recommendations. Proteins were eliminated by addition of proteinase K and precipitated by supplementation of saturated NaCl solution followed by centrifugation. DNA concentration was quantified photometrically at 260 nm. The obtained values were set in relation to the dry weight of the EAT. DNA measurements were performed on decellularized EAT specimens, and results were compared to fresh EAT controls.
Histological DNA visualization
Acridine orange was used for nucleic acid staining as a metachromatic dye. It emits green fluorescence (maximum at 525 nm) upon excitation (excitation maximum at 502 nm) when it binds to dsDNA. Therefore, EAT was shortly washed with PBS, fixated in 3.7% PFA in PBS for 15 min at room temperature and incubated in acridine orange staining solution (0.01%) for 1 h. The slides were washed shortly with distilled water, embedded for fluorescent microscopy and examined under a confocal microscope (Zeiss LSM 700).
Histological analyses
For H&E staining tissue specimens were fixed in PFA (4% in PBS) overnight at room temperature and processed according to standard protocols. In brief, tissue specimens were rinsed in tap water, dehydrated in an ascending series of alcohol solutions, and finally incubated in 100% xylene before transfer into 60°C prewarmed paraffin. After 3 days tissue specimens were embedded in paraffin and processed into tissue sections of 5 μm. For optimal substrate attachment “Knittel Glasses” were used. Sections were rehydrated and stained for 5 min in hematoxylin followed by 5 min incubation in eosin solution, followed by dehydration and mounting using Roti-Histokitt II (Roth, Karlsruhe, Germany).
For Lipid stainings tissue samples were cryoembedded, and cryosections of 7 μm were obtained. Oil Red staining was performed according to previously published protocol. 15 Sudan Black staining was performed according to the standard protocol used at the Institute of Pathology, University Hospital Duesseldorf. In brief, Sudan Black (0.1 g; Roth) was added to 100 mL 70% ethanol and heated to boiling. Staining solution was filtered after cooling. Sections were incubated for 3 h at 60°C for staining. After rinsing in distilled water nuclei were stained in nuclear fast red (Roth) and rinsed under running water before embedding.
Fluorescent imaging of nucleic acids of cryoembedded section was performed after brief fixation in PFA (4% in PBS) by incubation in 4′,6-diamidino-2-phenylindole (DAPI) solution (0.2 μg/mL in PBS; Roth).
Images were obtained using an upright microscope (DM 2000; Leica) equipped with a digital camera (F-view, UC30; Olympus) using light or fluorescent microscopy, where applicable, and applying Leica Application Suite V3.7 software.
In vitro assessment of biocompatibility
To assess the biocompatibility of decellularized EAT a commercial assay was applied (WST-1). In brief, culture medium was conditioned with native and decellularized EAT by incubation for 24 h at 37°C in a 5% CO2 atmosphere as previously published. 16 WST-1 assay was performed according to the manufacturer's manual (Sigma-Aldrich, St. Louis). To that end, primary cardiac fibroblasts of neonatal rats (1–4 days old) were seeded in six-well dishes with a final concentration of 106 cells/well. Cells were incubated in conditioned media for 24 h before assay. Cells kept in standard culture medium served as controls. Absorbance of the samples was measured after 30, 90, and 150 min at 440 nm. Data represent the results of three experiments (n = 3).
Statistics
All values are presented as mean ± SEM. Two-tailed nonparametric Mann–Whitney test was used for comparison of intergroup differences, except for values obtained by WST-1 assay, where Kruskal–Wallis with a Dunn's multiple comparison test was applied. p-Values <0.05 were considered as statistically significant. Statistical tests were performed with GraphPad Prism (version 6.0; La Jolla).
Results
Differential impact of decellularization on the EAT versus myocardial interstitium
After decellularization the epicardial fat tissue shows distinct morphology with an opaque appearance and visible swelling, whereas other regions with epicardial myocardial tissue turn into a transparent mass (Fig. 1). The increase of transparent appearance may be due to extraction of cellular material and also due to certain bleaching effect of the detergent treatment. There was a difference in the macroscopic appearance of myocardial regions versus EAT after decellularization.
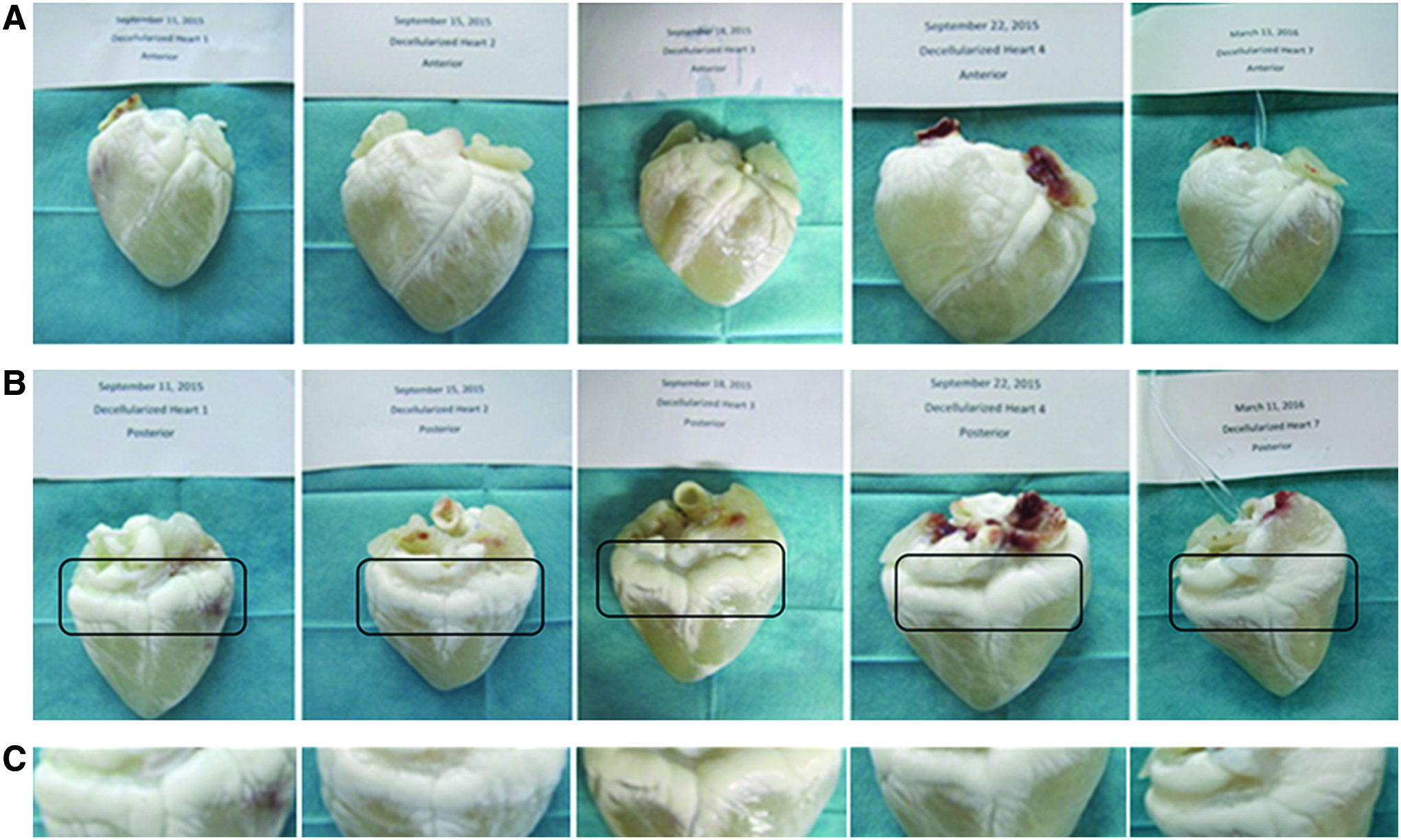
Macroscopic changes in EAT versus myocardial region after decellularization of whole hearts. After decellularization, EAT remained nontransparent in contrast to changes induced in the myocardial wall tissue.
Cellular composition is altered after decellularization
For a gross evaluation of the decellularization efficacy histological sections of native and decellularized myocardial tissue were analyzed (n = 3 for each group). Hematoxylin and eosin staining revealed a marked difference in the analyzed groups (Fig. 2). In contrast to the native tissue, in decellularized samples neither entire cell nuclei nor gross cell debris were visible. However, upon higher magnification typical areas of reduced acellularity (e.g., vascular walls) were observed with retained hematoxylin staining, indicating a residual DNA smear in this region (Fig. 2).

Perfusion decellularization leads to a marked washout of cellular components and marked changes in the histological appearance of myocardial tissue. After perfusion decellularization of whole hearts using detergents the histological appearance is changed with removal of the cellular mass predominantly stained by eosin
Histological impact of decellularization on EAT
Perfusion decellularization leads to remarkable structural changes of EAT on the histological level (Fig. 3). Cellular organization of EAT is largely destroyed, and nuclei cannot be identified by light microscopy. However, using methods of lipid staining (Oil Red, Sudan Black), large amounts of lipids are detected in EAT after perfusion decellularization (Fig. 3A–H). Fluorescent staining of nucleic acids by DAPI confirms the disruption of nuclei, while occasionally slight signals within the residual tissue mass suggest a smear in deliberated nucleic acids being retained after decellularization.
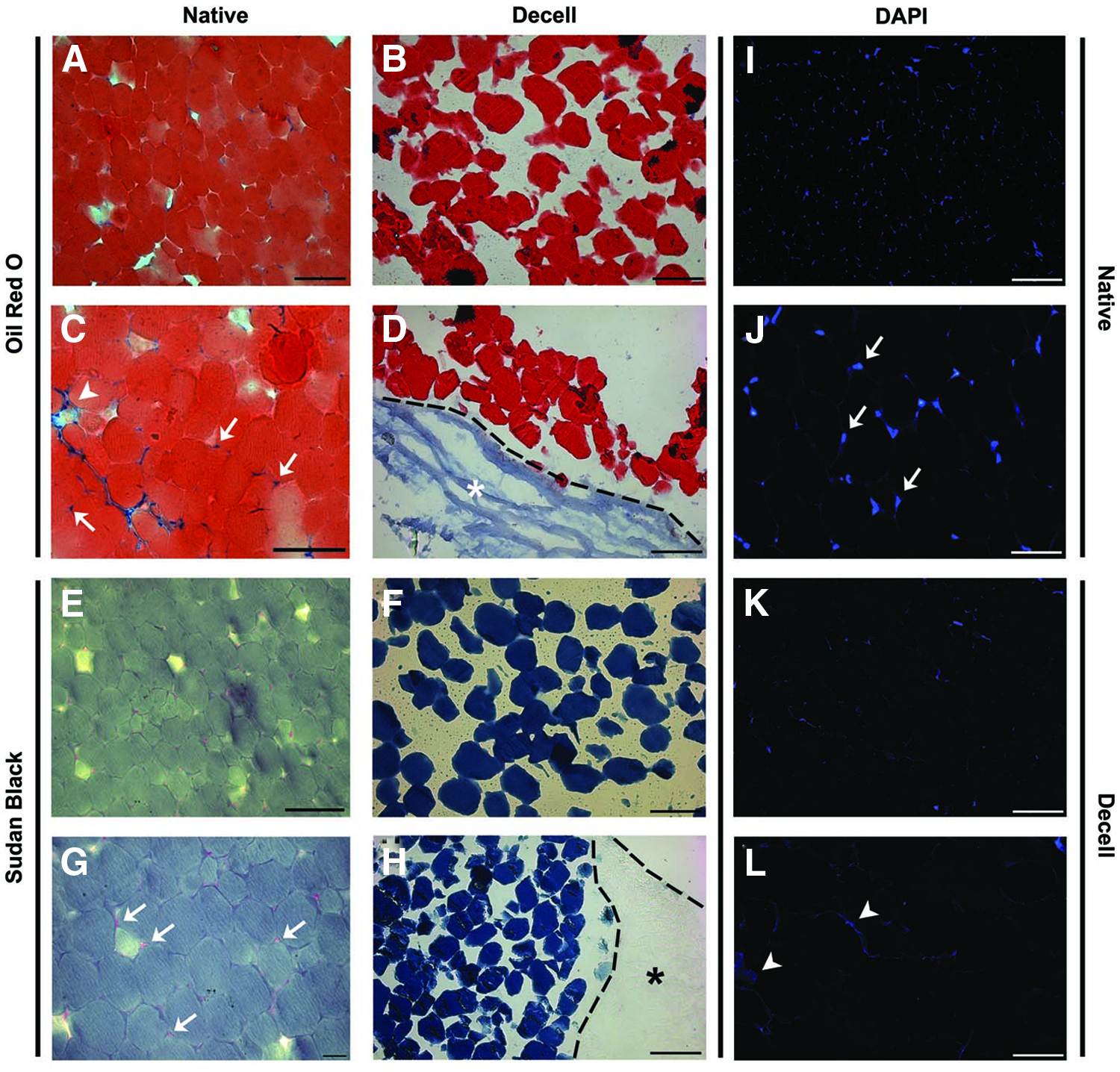
Histological changes of EAT induced by decellularization. Two different fat specific staining methods, that is, Oil Red
Total protein content in EAT is strongly reduced by perfusion based detergent decellularization
Quantitative analysis revealed a strong reduction of total protein content in the EAT of decellularized hearts compared to native control hearts. Within our native samples, we could measure values from 0.999 to 4.263 μg/μL protein/wet weight, whereas in decellularized EAT, total protein amounts were reduced by ∼90% (Fig. 4A). A gross overview of protein content of native and decellularized EAT by nonspecific protein staining (coomassie) revealed a clearly distinguishable profile in decellularized versus native EAT, which is characterized by a substantial loss of protein mass across all size classes (Fig. 4B).
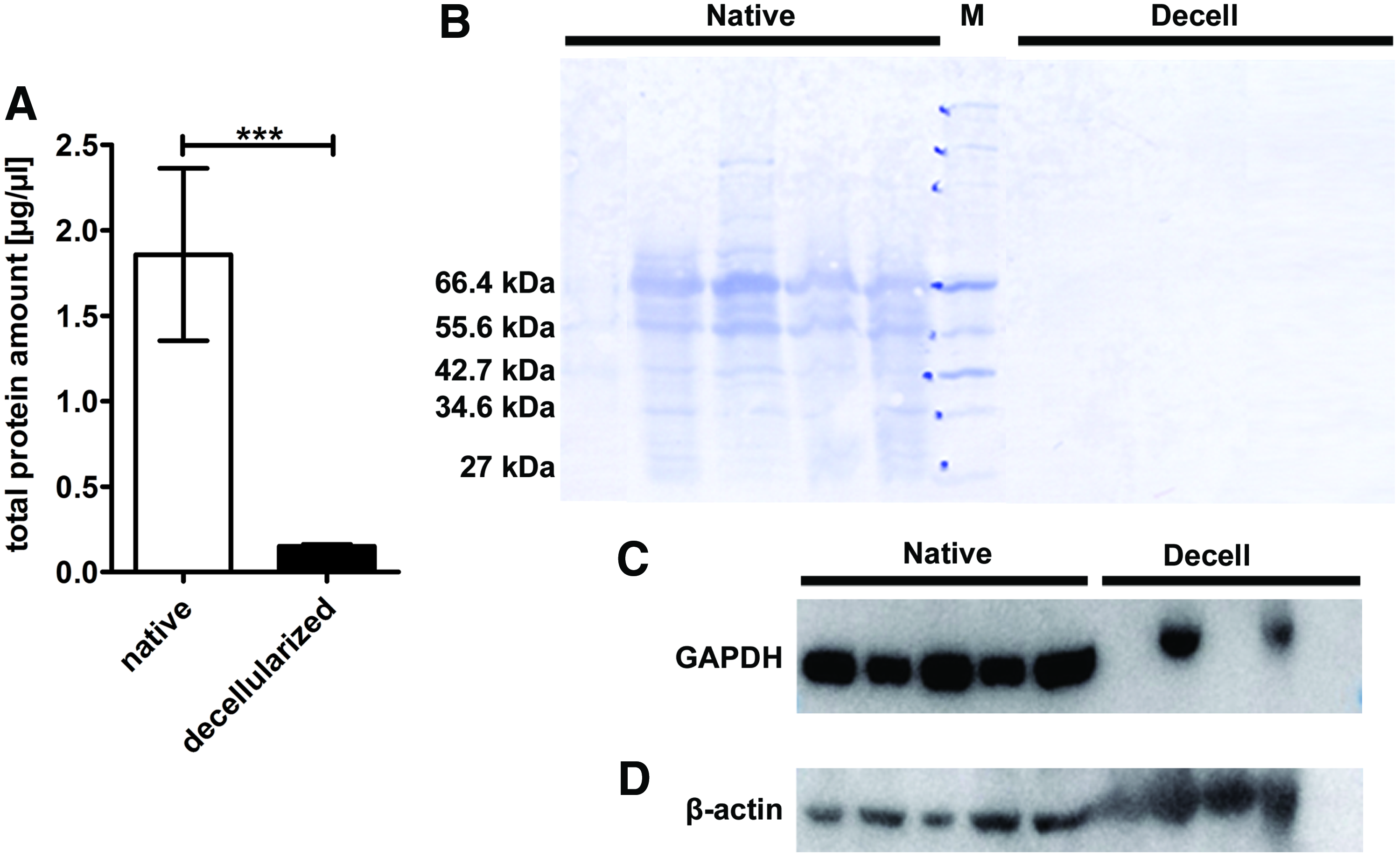
Total protein content and major cellular proteins are remarkably altered in EAT after decellularization.
A more specific analysis by Western blot indicates that in case of GAPDH in four out of five hearts the applied decellularization protocol has resulted in complete or substantial elimination of this cytosolic protein, whereas the removal of the cytoskeletal protein β-actin proved to be ineffective in four out of five hearts when perfusion decellularization was applied (Fig. 4C).
Western blot analysis confirmed the removal of cellular proteins with almost complete discharge of specific lipid-associated proteins
To characterize the susceptibility of specific protein types to extinction from the tissue by decellularization we studied the protein levels for representative specific lipid-associated proteins, two candidates from the perilipin (PLIN) family, as well as for two further cellular proteins not being specific for EAT (vimentin and myozap). Our analysis revealed that after decellularization perilipin 1, which is eponymous for the PLIN gene family, is strongly diminished (0.77 ± 0.47 AU in decellularized EAT vs. 86.16 ± 6.94 AU in native EAT; p = 0.0079) (Fig. 5A). In addition, perilipin 4 (S3-12), which is also a lipid-associated protein and member of the PLIN gene family, seems to be completely removed, as it was not detectable in any of the decellularized specimens by the methods applied in this study. In contrast, all native specimens demonstrated a significant amount of perilipin 4 (38.76 ± 3.62 AU) (Fig. 5B). To broaden the picture of protein extinction by decellularization we evaluated the protein content of the filamentary protein vimentin by Western blot. Decellularization led to a significant reduction of vimentin from 93.34 ± 17.68 AU in native EAT to 1.30 ± 0.91 AU in decellularized samples (p = 0.0079) (Fig. 5C). Finally, a similar marked extinction of the intercalated disc protein myozap was observed (0.35 ± 0.23 AU in decellularized EAT vs. 27.33 ± 8.27 AU in native controls; p = 0.0079) (Fig. 5D). Concluding, Western blot analysis confirmed the efficacy of perfusion-based decellularization using detergents for removal of cellular proteins with almost complete discharge of lipid-associated proteins.
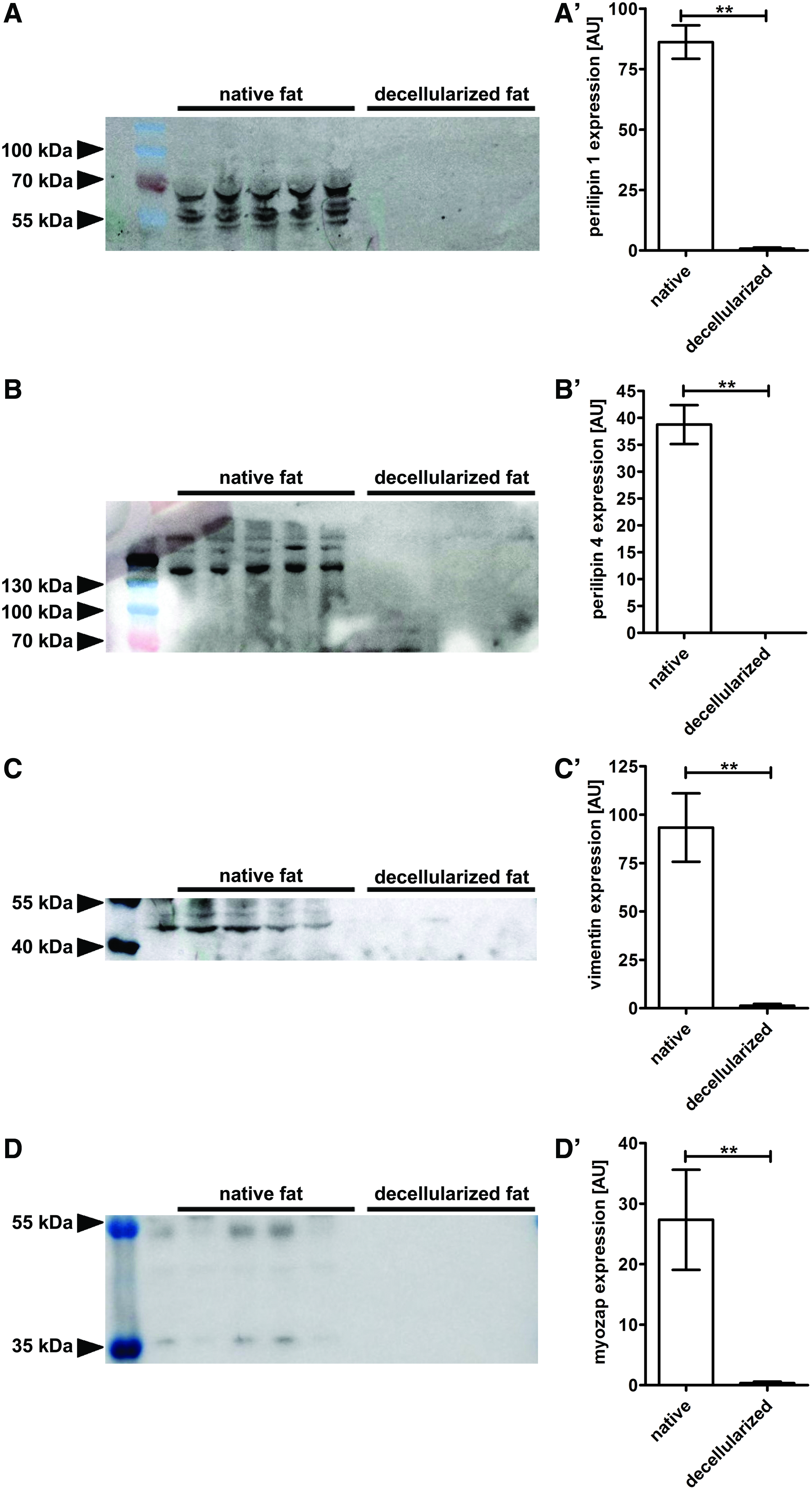
Western blot analysis confirms a significant reduction of cellular components with almost complete discharge of lipid-associated proteins. The protein level of perilipin 1
Decellularized samples show only sporadic cell components, but abundant collagen fibrils
Semi-blue staining was performed to demonstrate the penetration depth of osmium tetroxide as an indicator for the susceptibility of the EAT to diffusion based treatments. Subsequent TEM revealed blue staining in areas that were reached by the applied solution. Images taken in different resolutions were used to compare decellularized EAT versus native control (Fig. 6). Surprisingly, penetration depth of osmium tetroxide was higher in native controls than in decellularized EAT, although the initial impression of both representative images might be vice versa. However, as the final analysis is depending on the total tissue depth as a reference point, mass transport and swelling phenomena as observed during decellularization may represent a bias in this analysis, where the final cross-sectional area is different in the two herein analyzed conditions.

Semi-blue staining of decellularized EAT versus native control. Representative images of native
Ultrastructural changes in EAT after decellularization
In native EAT typical cell borders were observed with clearly delineated fat vacuoles (Fig. 7A). However, occasionally the ultrastructure of cell organelles was not entirely preserved, possibly as a consequence of incomplete tissue preservation in native samples due to the delay associated with the tissue transport from the abattoir to the laboratory (Fig. 7B).

TEM images of EAT prior and after decellularization. TEM was used for an in-depth analysis of the structural changes in EAT. Representative images of native
After decellularization only sporadic cell components were observed, but abundant collagen fibrils were found within the tissue (Fig. 7C, D). Dark spots, for example, at top of the Figure 7C, represent artifacts as a consequence of uranyl acetate agglomeration at nonplanar areas. In conclusion, abundant collagen fibrils can be observed in both samples, whereas clear cell borders and cellular components can only be observed in native samples of EAT.
Fatty acids are largely unaffected by perfusion based decellularization of human-size whole hearts
For a focused analysis of EAT after decellularization, fatty acids were additionally analyzed by gas chromatography. As depicted in Figure 8, neither was the relative amount of fatty acids indexed to the dry bio mass significantly changed nor was their proportional composition altered. In particular, there were no differences in the TFA content between native and decellularized whole hearts. In this study, the average TFA amount of EAT of the native heart amounts to 914.40 ± 23.29 mg/g dry mass, whereas the respective values for the decellularized hearts ranged around 886.88 ± 31.21 mg/g dry mass.
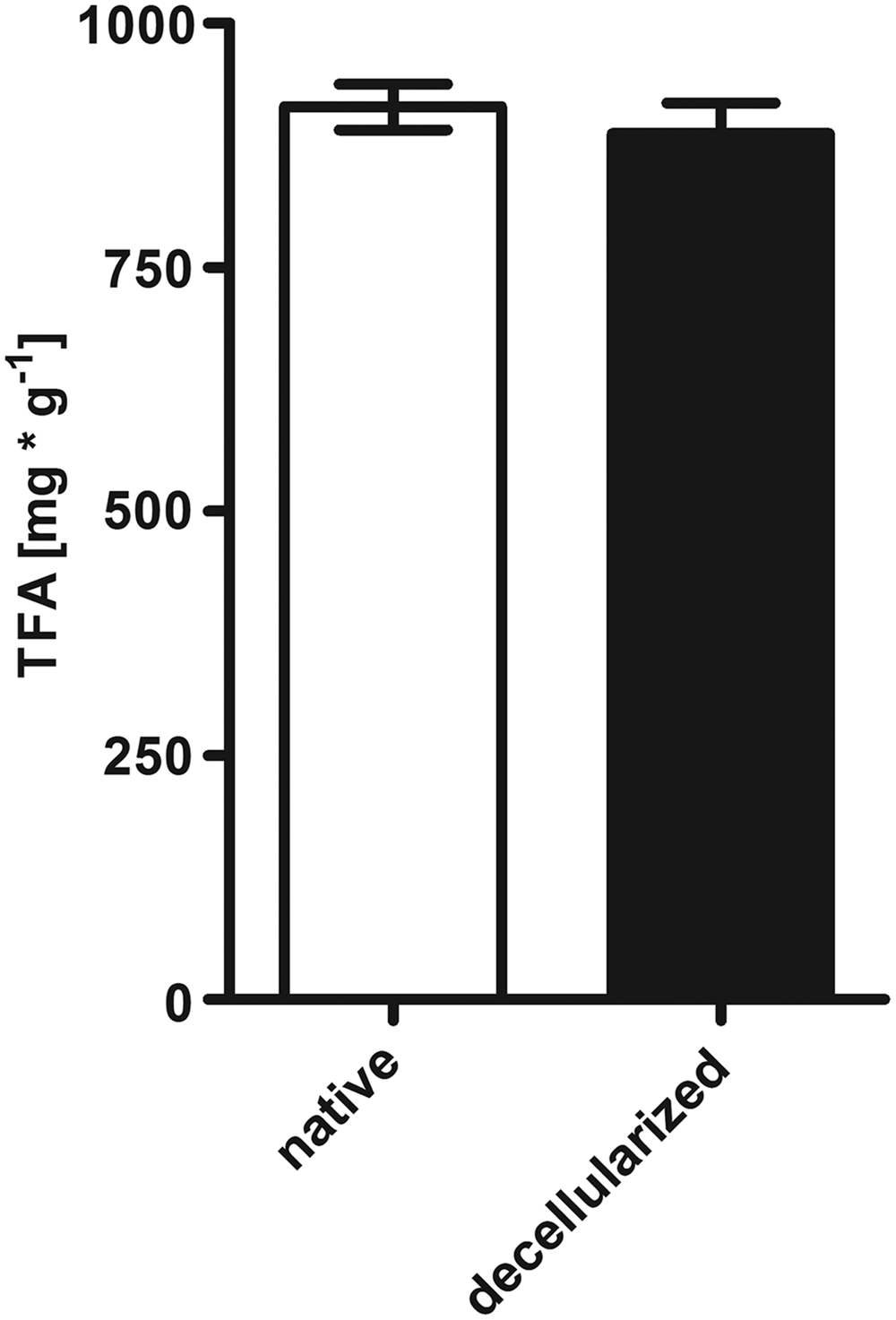
Gas chromatographic analysis of TFA amount. There was no difference in the TFA content of EAT between native and decellularized whole hearts (p = 0.5556). Results are presented as mean ± SEM (n = 4–5 animals per group). TFA, total fatty acid.
A more detailed analysis of the individual fatty acids and their relative proportional composition in native versus decellularized heart revealed oleic acid to be the major constituent of the fatty acids with a share of 33.77% of the TFA content in native EAT. After decellularization, oleic acid counted for 34.55% of all fatty acids. Next to oleic acid, stearic acid with 31.35% and 32.46%, as well as palmitic acid with 24.07% and 22.19% represented major fractions of TFA before and after decellularization, respectively. A number of other fatty acids were also detected; however, with a minor quantitative proportion: myristic acid (5.01% vs. 4.51%), linoleic acid (2.93% vs. 2.67%), palmitoleic acid (1.86% vs. 2.43%), and linolenic acid (1.00% vs. 1.19%) (native vs. decellularized epicardial tissue of whole hearts, respectively). Using gas chromatography, we were not able to detect arachidonic acid in the EAT of native or decellularized hearts. In conclusion, perfusion based decellularization using detergents is not capable of changing the relative amount or the compositional share of fatty acids in EAT of human-sized whole hearts (Fig. 9).

Specific detection of individual fatty acids by gas chromatographic analysis of EAT before and after perfusion decellularization. Gas chromatographic analysis of fatty acids shows no differences in the composition of the TFA amount between native and decellularized whole hearts for myristic acid
Perfusion decellularization significantly reduces total DNA content in EAT of whole hearts
DNA quantification of EAT revealed a significant reduction of DNA content by about 45% after decellularization (Fig. 10). In this study, the average total DNA content of EAT amounts to 121.69 ± 9.7 ng and 67.36 ± 9.15 ng/mg dry mass of native and decellularized whole hearts, respectively (p = 0.0286). Upon histological evaluation using nucleic acid staining by acridine orange and confocal microscopy nuclear structures were largely removed from EAT. The structural appearance of fatty vacuoles was loosened up and occasionally remnants of fat cells were visible. However, nucleic acid signals indicating a noticeable DNA smear pervading the adipose tissue were detected (Fig. 11).
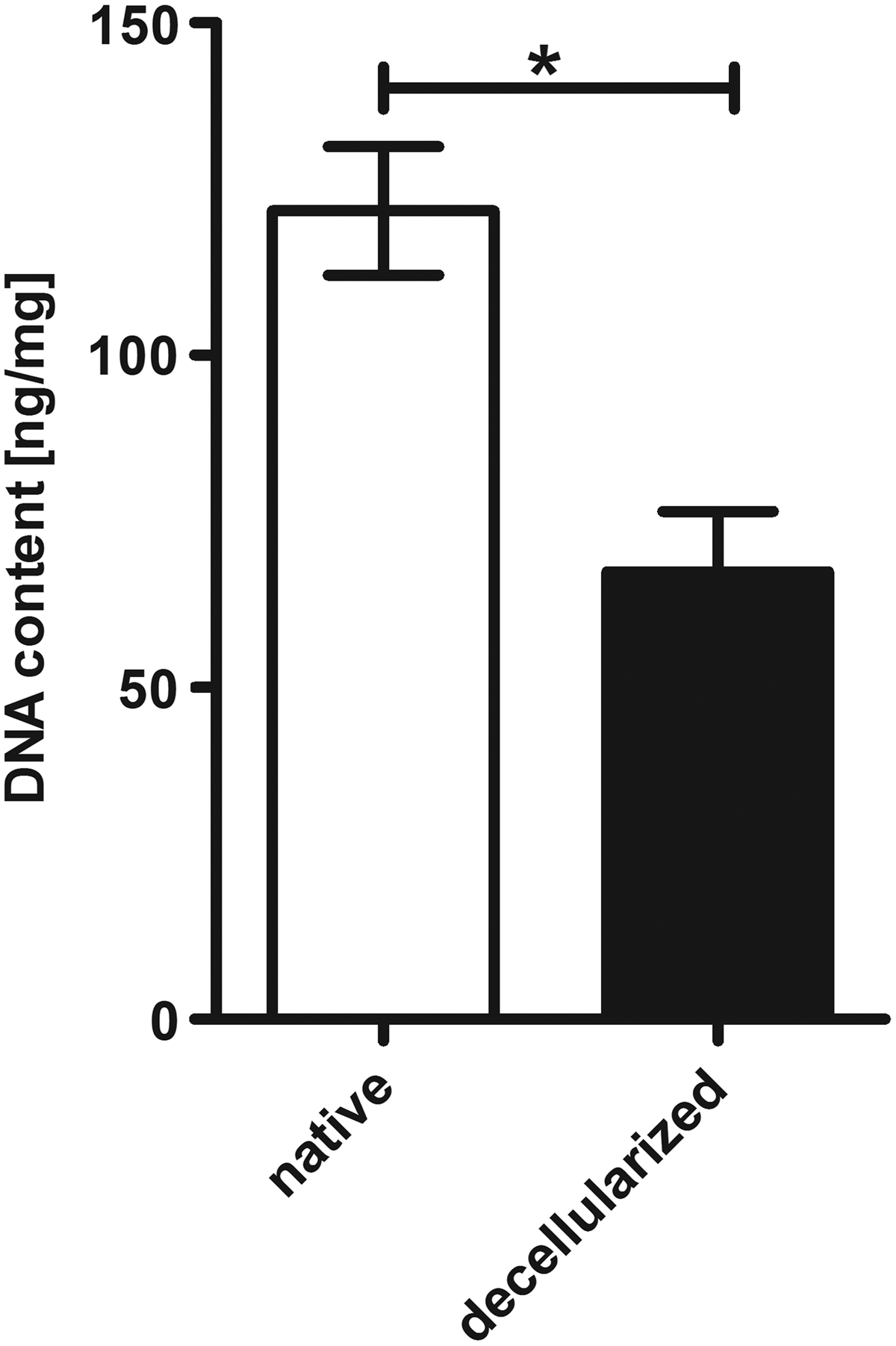
DNA quantification of EAT in native versus perfusion decellularized whole hearts. Perfusion decellularization of human size whole hearts led to a significant reduction of the total DNA content of EAT by 45%. Results are presented as mean ± SEM (n = 4 animals per group). *p < 0.05.
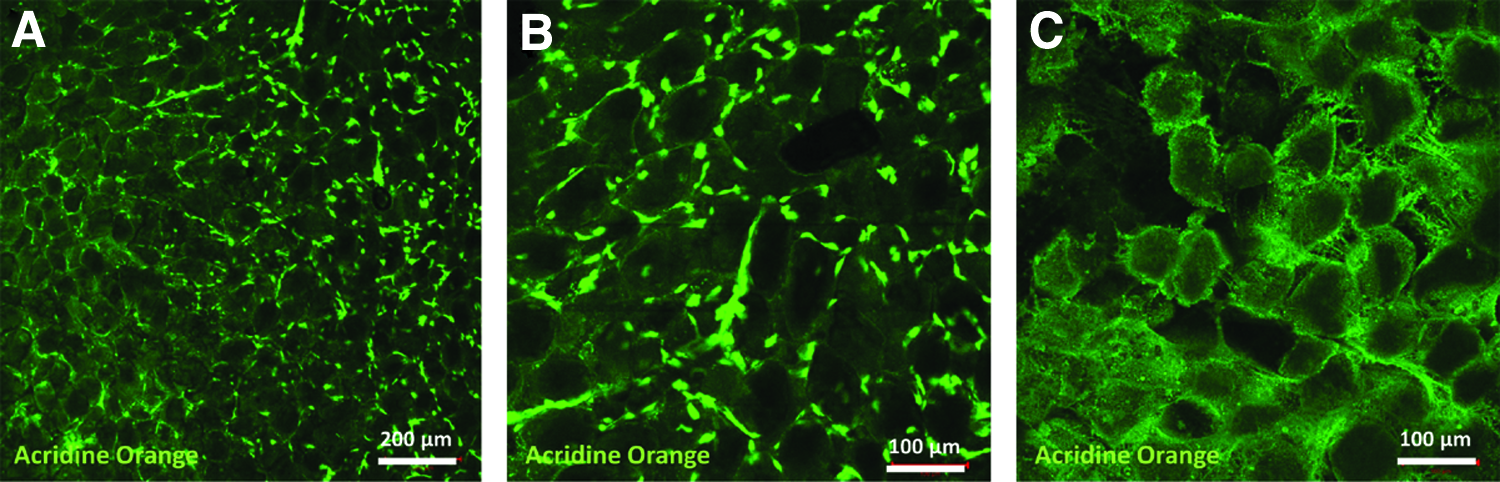
Confocal microscopic images of acridine orange stained EAT. Native EAT consists of clearly organized fat vacuoles and interspersed adipose cells with dense nucleic structures
Perfusion decellularized EAT exerts cytotoxic effects in vitro
The previous results suggested an incomplete removal of donor material (i.e., fatty acids, DNA) from the EAT, suggesting an increased donor material burden in this region of otherwise well decellularized whole hearts. Therefore, we aimed at evaluation of the functional relevance of these findings by conducting a simple in vitro viability assay. Using commercially available WST-1 assay and employing cardiac fibroblasts we observed a significantly reduced viability when cells were incubated with decellularized EAT as opposed to native EAT or unconditioned culture medium (p = 0.0219 at 90 min for decellularized vs. native EAT; p < 0.001 for other time points when decellularized group was compared to native group or control medium) (Fig. 12).
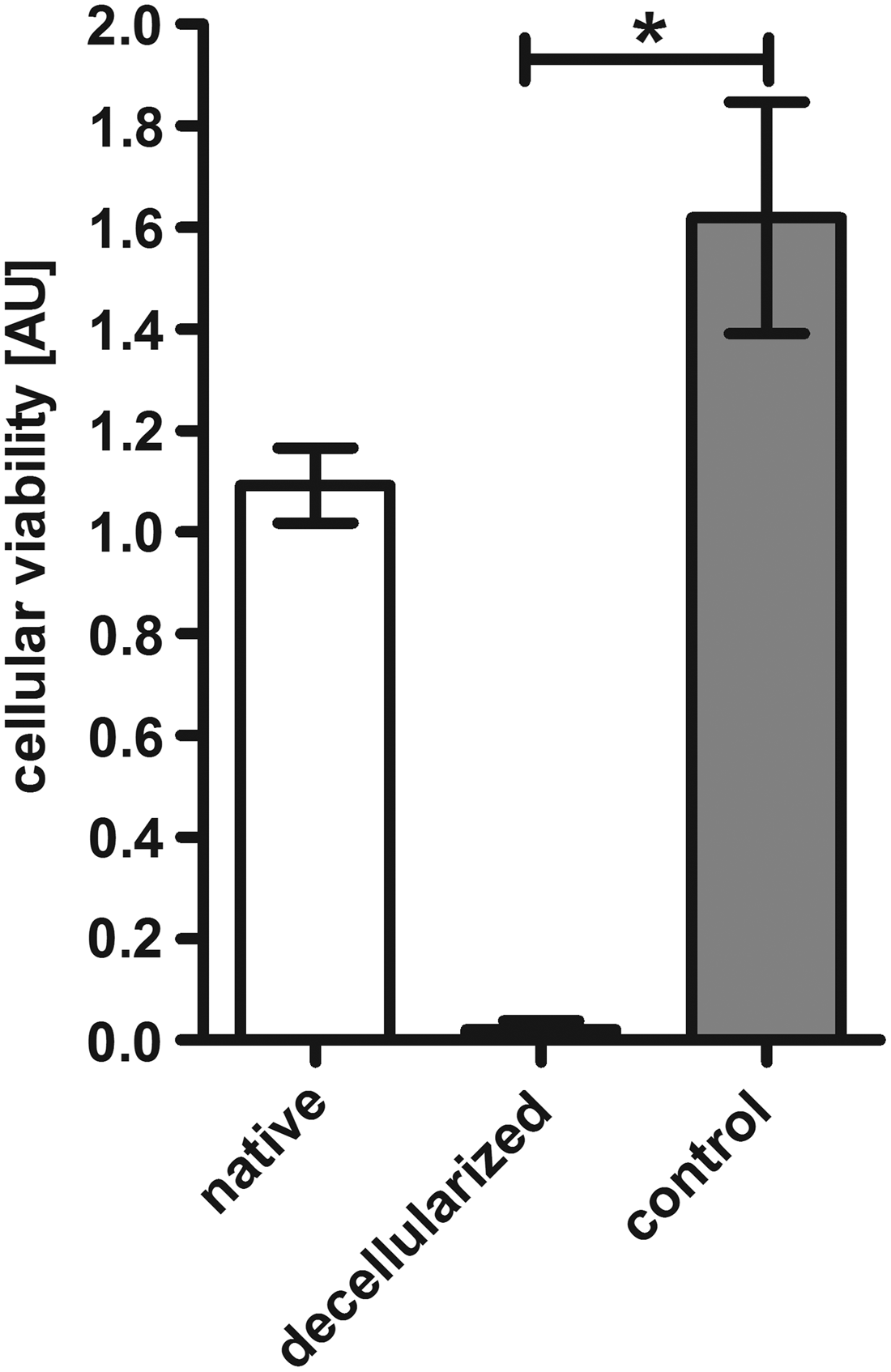
Perfusion decellularized EAT exerts cytotoxic effects in vitro. Primary cardiac fibroblasts were incubated with cell culture media conditioned with fresh native EAT (native) or decellularized EAT (decellularized). Cells incubated with standard cell culture medium without prior conditioning served as controls (control). Decellularized EAT led to highly significant reduction of viability in cardiac fibroblasts. The displayed figure shows the results after 90 min of incubation. *p < 0.05.
Discussion
This report analyzes the effect of detergent-based perfusion decellularization of whole hearts on the quantitative presence and qualitative composition of EAT in the final organ matrix. To the best of the authors' knowledge this is the first dedicated study focusing on EAT as a relevant paracrine component of the heart and its modification during the process of whole-heart decellularization.
Whole-organ engineering has attracted considerable scientific attention as an innovative approach to respond to irreversible end-stage organ failure in times of donor organ shortage. As one of the most prominent organs in focus of clinical demand, whole-heart engineering has been recently introduced with allogeneic or xenogeneic donor hearts serving as the base for generation of a decellularized whole-heart matrix.8,11 However, human-size donor hearts demonstrate a remarkably different anatomic composition compared to the rodent heart, which has been in focus of reports involving whole-organ decellularization for several years and thus subject to an in-depth characterization.6,7 Porcine, ovine, and human donor hearts of adult age demonstrate a considerable amount of EAT, which is in contrast to the typical anatomical composition of rat hearts. 17 Moreover, there is no generally accepted protocol for the decellularization of adipose tissue, 18 while scientific literature provides several protocols primarily aiming at adipose tissue as the main target. 19 At the same time, a growing body of evidence underlines the relevance of the EAT as an endocrine organ system playing a major role in the initiation and propagation of adverse cardiovascular remodeling. 20 With the identification of adipokines and their versatile effects on the tissue inflammatory status and cardiomyocyte biology, including contractile function, EAT is no longer a simple anatomic adjunction, but a true modulator of cardiac anatomy and function.16,21 Interestingly, EAT has been clearly present in decellularized human size whole hearts previously reported, as far as macroscopic images have been provided by the authors.8,12,22 By simple macroscopic comparison of pre- and postdecellularization appearance of human size whole hearts, it may be anticipated that different regions of the heart are differentially susceptible to the process of perfusion decellularization: while the myocardial region remarkably changes from the typical muscle tissue to a translucent pale mass, regions at the base of the heart covered with epicardial mass hardly change in their macroscopic appearance. The latter observation holds true for the herein evaluated ovine hearts, and yet it is in line with previously published images of porcine and human donor hearts.8,12,22 Hence, we hypothesize that there may be a difference in decellularization efficacy depending on the different tissue qualities within the same organ. This may be a phenomenon not restricted to a certain species but rather a matter of incapability of current perfusion decellularization techniques to remove donor EAT. However, in contrast, fat is known to be responsible for the opaqueness of tissues, thus, even when cellular compounds are removed, residual fatty components would lead to a less translucent appearance of the treated tissue.
Striving for a deeper understating of the efficacy of decellularization we performed Western blot analysis to evaluate the process of protein extraction. In this study, decellularization proves as largely efficient; as both analyzed lipid-associated proteins, perilipin 1 and 4 were significantly removed by perfusion decellularization. To the same degree vimentin and myozap were almost completely discharged from the remaining tissue. In contrast, DNA measurements demonstrated a low level of efficacy with more than one third of the donor DNA remaining in EAT after decellularization. This is in clear contrast to our own results (not shown) and reported data of other groups on the myocardial regions of the heart obtained with perfusion decellularization using detergent solutions (SDS and DCA).8,12 Free nucleic acids have been shown to elicit pro-inflammatory and deleterious effects on cardiac myocytes and fibroblasts. 23 Thus, persistence of almost one third of donor DNA in decellularized EAT accompanied by dissolved nuclear structures could constitute an environment that may mediate toxic effects to cells that are seeded into the extracellular matrix of decellularized hearts. However, it is difficult to prove the latter hypothesis because a myocardial ECM that is 100% free of donor DNA is currently difficult to achieve to serve as a valid control. As a further explanation, SDS and DCA, both detergents known for their deleterious effects on cells may have led to the observed impaired cell viability. In the experiments performed in this study, cytotox/WST-1 assay clearly showed negative effects of decellularized EAT on cardiac fibroblast viability. Further in-depth studies may elucidate the impact of donor nuclear acids, residual detergents, or possibly further components of EAT on cell viability after cellular repopulation.
In addition to protein and DNA levels, we performed gas chromatographic analysis to study the lipid acids in EAT. Several lipid acids were detected in native EAT. However, after the decellularization process fatty composition remained similar, suggesting the inability of the applied decellularization process to largely discharge fatty acids from EAT and supporting the gross macroscopic impression of the resulting decellularized hearts.
Considering the results of Western blot analyses of individual lipid-associated or cell-specific proteins, as well as the gas chromatographic evaluation of fatty acids, and further considering the histological data concerning lipid content of EAT the herein presented results collectively underline the finding that perfusion decellularization of human size hearts leads to inconsistent results regarding the different regions of the heart with EAT experiencing an unexpectedly poor decellularization.
We interpret these data as a call for reevaluation and improvement of the applied decellularization methods. Our data suggest that further refinement is necessary for an adequate adaptation of the basic principles of decellularization to the complex composition of human size whole hearts. Notably, culture medium preconditioned with specimens of decellularized EAT had a deleterious effect on cardiac fibroblasts, whereas native EAT did not lead to such a negative impact on cell viability. The viability test applied in this study certainly represents a preliminary approach to the complex of biocompatibility and warrants further support by more sophisticated experimental setups. Moreover, some of the negative effects may be associated with residual detergents that have been described in decellularized tissue. 24 However, the missing anatomic separation of EAT from underlying myocardial compartment by fascia and their close anatomic vicinity are features unique to the cardiac anatomy and their implications for a translational use of decellularized whole hearts are multiple. Conditions supportive for in vitro attachment, growth, and differentiation of progenitor or stem cells are not only necessary in the myocardial compartment but also they are most likely needed to the same degree in the adjacent region of EAT. Furthermore, although not in focus of this work, immunological aspects that are related to the donor-recipient compatibility of the EAT need to be addressed using dedicated experimental approaches.
This work is limited by a number of issues. Ovine hearts have been investigated in this work as an exemplary model of human size whole heart. Our results indicate a clear difference between rodent hearts and large mammalian hearts when subjected to perfusion based decellularization. However, with an increasing level of understanding relevant differences in the susceptibility to perfusion decellularization may become visible for ovine, porcine, and human donor hearts, respectively. Due to practical reasons, a single decellularization protocol was applied in this study. Numerous modifications, involving different exposure times and the addition of further chemicals and enzymes, are possible and may result in different outcomes. However, the basic principles outlined by our results are likely to be maintained even when gradual changes to the decellularization protocol are made.
Conclusions
Perfusion decellularization of human size whole hearts results in consistent decellularization effect regarding donor material removal from myocardial regions as opposed to regions of EAT. With a translational ambition spreading through the cardiac tissue engineering community, substantial improvement related to EAT of donor hearts is needed.
Footnotes
Acknowledgments
The authors acknowledge Susanne Bunnenberg-Stiftung at the Düsseldorf Heart Center, as well as a stipend provided by the Schmeil-Stiftung (Heidelberg, Germany) to J.H. The authors are grateful for the support by Mareike Barth (Research Group for Experimental Surgery, Department of Cardiovascular Surgery, Medical Faculty, Heinrich Heine University Duesseldorf, Germany) regarding staining and imaging, as well as Petra Reinecke (Institute of Pathology, Medical Faculty, Heinrich Heine University Duesseldorf, Germany) for protocols and support regarding Sudan Black and Oil Red staining of EAT.
Disclosure Statement
No competing financial interests exist.