
Abstract
Acute and chronic wounds contribute to increased morbidity and mortality in affected people and impose significant financial burdens on healthcare systems. For these challenging wounds, acellular dermal matrices (ADMs) have been used as a biological wound coverage. Unlike engineered dermal matrices, ADMs are prepared through the removal of cells from skin, while preserving the extracellular matrix structure and function. In this study, our primary objective was to develop a detergent-free method for decellularization of the skin to mitigate chemical stress on matrix molecules. Then, we performed a set of in vitro and in vivo experiments to compare this method with nonionic and anionic detergent methods. All decellularization methods satisfactorily removed cells and supported fibroblast growth and migration in vitro. Sulfated glycosaminoglycan content was reduced significantly (p < 0.05) only in the ionic detergent treatment group. In contrast to the detergent-free method, all detergent-based methods significantly reduced scaffold mechanical strength and elastin content (p < 0.05). Three weeks after transplantation, the results showed reepithelialization, angiogenesis, and migration of host cell into scaffolds with no induction of immunogenic reaction in all ADM groups tested. In our study, the detergent-free method showed better preservation of matrix composition and biomechanical properties, but after transplantation, all methods of ADM preparation resulted in equally biofunctional matrices as wound coverage.
Introduction
A
Adequate coverage of the wound is essential for healing regardless of the types of wound. The primary purpose of a wound dressing is to achieve a rapid closure of the lesion and reduce further insult during the wound repair process. Chronic and large surface area wounds share a common characteristic of delayed wound repair, which can be mitigated through application of skin grafts and skin substitutes. Despite the advantages of full-thickness skin grafts (FTSGs) and split-thickness skin grafts (STSGs), issues such as limited supply and morbidity of donor site for FTSG and low vascularity, contracture, decreased elasticity, sensory loss, and undesirable cosmetic results for STSGs remain problematic.3–6
To overcome these drawbacks, tissue-engineered alternatives have been developed. Among those alternatives, biological scaffold materials composed of extracellular matrix (ECM) have been reported to facilitate the constructive remodeling of different tissues in both preclinical animal studies and in human clinical applications. Structural, biomechanical, and biochemical properties of ECM scaffolds are inherently determined by the material properties of original tissue, because it consists of the structural and functional molecules secreted by the resident cells of each tissue and organ. 7 Thus, de novo engineered skin substitutes, using biopolymers, lack the addition of the mosaic heterogeneity of cell-derived molecules that are found in normal skin. To fabricate a skin substitute with molecular heterogeneity and biochemical and mechanical properties of normal skin, a decellularization process is desirable.
Acellular dermal matrices (ADMs) are decellularized allogeneic skin and have been used clinically for different purposes, including treatment of abdominal wall defects, 8 full-thickness burns, 9 prevention of postparotidectomy gustatory sweating, 10 cleft palate repair, 11 resurfacing of intraoral defects, 12 lip augmentation, 13 reconstructive breast surgery, 14 and chronic wound repair. 15
The ideal bioscaffold resulting from decellularization of skin should preserve the original ECM materials with similar biomechanical properties to the skin and not induce an immune response. It should support rapid and directed proliferation of infiltrated cells, be easily revascularized, and exude stability throughout the repair process while being remodeled, over time, just as any other tissue. Practically, the ADM should be easy to apply by the end-user and have a commercially viable shelf-life. 16
A number of different decellularization methods have been reported and used to produce commercially available human- and xenogenic-derived products.17–20 However, decellularization would have a negative effect on the graft function if it altered the biochemical composition, structure, and biomechanical properties of tissue. Conventional decellularization processes require the use of harsh reagents that cause unavoidable adverse effects on the ECM components and molecules contained within (reviewed in Crapo et al. 21 ). It is well known that the ECM is not only a scaffold but also a reservoir of all molecules such as growth factors and cytokines secreted by tissue-resident cells. 22 Any decellularization method that leads to preservation of these molecules and mitigates ECM damage is highly desirable and should therefore ideally improve wound healing.
In this study, the primary objective was to develop a detergent-free method of decellularization to minimize any potential damage on matrix molecules and compare this method with Triton X-100 as a nonionic and N-lauroyl sarcosinate (NLS) as an anionic detergent method of ADM preparation against specific design criteria.
Our criteria comprised indicators and measures of mechanical performance, removal of cellular components, product biocompatibility, and preservation of ECM molecules. Although there are several methods published for decellularization of the skin, those studies have typically focused on in vitro cytocompatibility and scaffold characterization, with minimal in vivo evidence to assess biofunctionality. Consequently, there is relatively little clinically relevant information with which to make decisions regarding the selection of ADM for various applications. Thus, as a secondary objective, we evaluated the wound healing outcome of using these ADMs for transplantation in a full-thickness skin wound in a mouse model. The results of this comparable head-to-head study of decellularization methods demonstrated that a detergent-free process preserves matrix composition and mechanical properties significantly better than detergent-based methods.
Materials and Methods
Ethics statement
All methods and procedures, as well as the use of animals and tissue specimens derived from animals and human, are approved by the Ethics Committee of the University of British Columbia.
Animals and skin preparation
Male C57/B6 mice (Jackson Laboratory) aged 3–4 months were used as skin donors and BALB/c female mice aged 2–3 months were used as recipients of ADM grafts. Donor C57/B6 mice were anesthetized with CO2 and euthanized by cervical dislocation. The mouse dorsum was depilated using a shaver and hair removal cream. Skin was then scrubbed with povidone–iodine solution (Triadine; H&P Industries, Inc., Franklin, WI). Full-thickness donor skin was removed from the mouse dorsum. The removed skin was washed three times in phosphate-buffered saline (PBS), and panniculus carnosus and hypodermis tissues were removed manually using a surgical blade. After washing with PBS, the remaining part of skin tissue was processed for decellularization as described below.
Cell isolation and culture
Mouse dermal fibroblasts were explanted from B6 mice skin as previously described. 23 Human primary fibroblasts were isolated from the discarded foreskins of consenting donors. Skin specimens were briefly washed several times with 1× PBS (pH 7), containing 1% antibiotic, minced into small pieces, and then fixated with fetal bovine serum (FBS) for 4 h on a tissue culture plate. After 4 h, one drop of 1× Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS and 1% antibiotic was added to FBS drops overnight. The next day DMEM was used to cover fixated skin section in the dish. Skin pieces were maintained in culture until fibroblasts reached 60% confluency, after which cells were trypsinized and passaged.
Decellularization methods
In this study, we used three different methods based on effective agents for decellularization that have been reported previously: Triton (nonionic detergent), NLS (nondenaturing anionic detergent), and latrunculin B (LatB) treatment followed by hyper- and hypotonic solutions as a detergent-free method. Triton and NLS have previously been reported for preparation of ADM, whereas LatB, to the best of our knowledge, has only been reported for preparation of decellularized muscle. The LatB method, described herein, was modified for the preparation of ADM.
All donor skin was treated with 2 U/mL dispase II (Invitrogen/Gibco; Cat. # 17105-041) in high-glucose DMEM at 37°C for 90 min to remove the epidermis and disrupt cell attachment to the dermal matrix before preparation of the ADM, using one of the following methods:
Triton method
This was based on a previously described decellularization method by Takami et al. with some modifications. 24 After dispase treatment and washing three times with PBS, the dermal matrix was incubated in 0.5% Triton X-100 (Fisher Scientific, ON, Canada) for 24 h at room temperature with continuous shaking and renewing the solution after the first 12 h. Subsequently, ADM was extensively washed with PBS for 12 h.
NLS method
Samples in this treatment group were subjected to a modified previously described decellularization method that has led to developing a commercial product named Matracell®. 25 Following dispase II treatment and washing three times with PBS, the dermal matrix was incubated in 1% NLS (Sigma-Aldrich) for 24 h at room temperature with continuous shaking and renewing the solution after the first 12 h. Subsequently, ADM was extensively washed with PBS for 12 h.
LatB method
A nondetergent protocol was used according to the method of Gillies et al. with some modifications. 26 Briefly, after dispase II treatment and washing three times with PBS (this step was not in the original protocol), skin samples were incubated in 50 nM LatB (Enzo Life Science; BML-T110) in high-glucose DMEM (Gibco) for 2 h at 37°C to depolymerize actin filaments. Then, samples were washed with distilled water twice for 15 min and subsequently were incubated in 0.6 mol/L potassium chloride (Sigma-Aldrich) for 90 min, followed by 1.0 M potassium iodide (Sigma-Aldrich) for 90 min. Following the high ionic solution incubations, skin samples were washed in distilled water overnight, and then, the potassium chloride and potassium iodide incubations and overnight distilled-water incubations were repeated. Samples were washed with distilled water twice for 15 min between incubation steps. All steps were performed at room temperature with continuous shaking.
At the final step, all ADMs prepared with the three methods were treated with 50 U/mL Benzonase® nuclease (Santa Cruz; sc-202391) for 12 h at 37°C and then washed with PBS for another 12 h. All solutions used for ADM preparation were filter sterilized, and all procedures were performed aseptically. Solutions contained sodium azide (0.02% w/v) at all times, except during the last PBS washing step, to prevent microbial growth.
Histological examination of ADMs
For histological evaluation of the ADM structures, conventional hematoxylin and eosin (H&E) as well as Masson trichrome staining was done on paraffin sections. Paraffin-embedded ADMs were sectioned at a thickness of 5 μm. After removing the paraffin, sections were first rehydrated with a decreasing series of alcohol concentrations to water, and then, standard protocol was followed for H&E staining or Masson trichrome staining. After sealing, samples were examined by light microscopy at a different magnification to inspect the presence of cells (stained by hematoxylin to a bluish-purple color) and collagen fibers (stained by eosin to a pink color or blue with aniline blue in Masson trichrome). Histological analysis was performed on samples from six independent decellularization procedures, three slides for each sample, and three sections in each slide.
DNA quantification and fragment length analysis
To assess the presence of residual cells or debris within the ADMs after decellularization, total DNA content of the ADMs and native skin was measured as described previously. 27 Briefly, samples were freeze-dried, cut into thin strips and small pieces, and then digested with 50 μg/mL protease K in 0.5 mL of lysis buffer. The DNA was extracted with phenol/chloroform method and purified by 2 M NaCl- isopropanol precipitation and 70% ethanol washing. Precipitated DNA samples were dehydrated to remove residual ethanol and then rehydrated in 1× TE buffer. The residual DNA content was measured at 260 nm using a NanoDrop spectrophotometer. To determine DNA fragment size, samples were separated by electrophoresis on a 1% low melting point agarose gel stained with SYBR-safe dye at 80 V for 75 min and visualized with the Gel Doc EZ system (Bio-Rad, Inc.)
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blot analysis
Skin and ADMs were freeze-dried, minced, and homogenized in the presence of a protease inhibitor. Total protein content was measured using the bicinchoninic acid method (Thermo Fisher Scientific; Cat. # 23225). Due to the discrepancy between protein content of skin and ADM, protein amount equal to 1 mg from each sample was loaded into wells of 10% gradient polyacrylamide gels (Bio-Rad, Hercules, CA). Proteins were transferred to polyvinylidene difluoride membranes (Millipore Sigma; IPVH00010) and subsequently blocked in 5% skim milk in PBS with 0.1% Tween-20. The blots were probed with the anti-β-actin mouse monoclonal antibody (Sigma-Aldrich; Cat. # A5441) for 2 h and detected using goat anti-mouse immunoglobulin G (IgG) (H + L) cross-adsorbed secondary antibody, DyLight 800 (Cat. # SA5-10176) at dilutions 1: 30,000 and 1:10,000, respectively. Blots were scanned with LI-COR Odyssey Classic imaging system. Molecular weight was determined using PageRuler Plus Prestained Protein Ladder (Thermo Fisher Scientific; Cat. # 26619).
Scanning electron microscopy
Lyophilized ADMs were cross-sectioned using a surgical blade. Samples were first gold coated and then scanned with the Hitachi S-3000N scanning electron microscope (Hitachi, Tokyo, Japan). Comparisons between ADMs and normal skin were made for morphological changes to collagen fibers in cross sections and surface topography of epidermal and hypodermal sides.
Hydroxyproline content
Lyophilized ADMs or fresh tissues were collected from five independent experiments and digested with proteinase K (in Tris-HCl buffer) (Thermo Scientific) at 54°C while shaking for 5–6 h, and then, homogenates were incubated with an equal volume of 12 N HCl at 105°C overnight to hydrolyze the collagen (1 mg tissue/1 mL HCl 6 N). After evaporation of the acid under a nitrogen flow, the dried samples were suspended in 40 μL of ethanol:dH2O:triethylamine (TEA) (2:2:1) and redried. Derivatization to phenylthiocarbamyl was achieved by adding 40 μL of ethanol:dH2O:TEA:phenylisothiocyanate (PITC) (Edman's reagent; Pierce Biotechnology) (7:1:1:1) to each sample and incubating at room temperature for 20 min before drying. The samples were then resuspended in 1 mL of analysis solution (dH2O:Acetonitril, 7:2) and cleared before high-performance liquid chromatography analysis. In addition, and using the same procedure, 1 mg of purified type I bovine collagen was hydrolyzed and derivatized as a control. For the calibration curve, different amounts of hydroxyproline (1–320 μg) were dried from freshly prepared stock solution of trans-4-hydroxy-
Glycosaminoglycan content
Quantification of sulfated glycosaminoglycans (sGAGs) was performed using the 1,9-dimethylmethylene blue (DMMB) assay adapted from Barbosa et al. 28 Briefly, lyophilized samples were collected from five independent preparations and digested overnight with 100 μg/mL proteinase K (Sigma-Aldrich, St. Louis, MO) at 56°C followed by DNAse treatment. Then, 100 μL of digested mixture was added to 1 mL of the GAG-complexating DMMB solution, which was a 16 μg/mL DMMB (Sigma-Aldrich) in 5% ethanol solution (paper filtered) with a 0.2 M guanidine hydrochloride solution containing 0.2% formic acid and 0.2% sodium formate. The resulting sGAG–DMMB complex was precipitated from solution and then solubilized in the decomplexating solution, which was 50 mM sodium acetate solution buffer (pH 6.8), containing 10% 1-propanol and 4 M guanidine hydrochloride. Absorbance of the decomplexed solution of DMMB and sGAGs was measured at 656 nm. sGAG concentration was calculated by calibrating against a standard curve of chondroitin sulfate-A (Sigma-Aldrich), ranging from 0 to 70 mg/mL (0–7.0 μg/assayed sample). Assays were performed five times and every time in duplicate. Results were expressed in μg of GAG/mg of dried tissue.
Biomechanical assessment
Biomechanical characteristics of native and decellularized skin were evaluated by uniaxial tensile stress testing. Specimens were cut into a dog bone shape using a standardized jig with the neck of the dog bone shape measuring 2 cm in length and 0.4 cm in width. Tensile testing was done using a KES-G1 Micro-Tensile testing set-up (Kato Tech, Kyoto, Japan), with a 5 kg load cell. Two pieces of sandpaper were then used to firmly secure the samples to the specimen holder. Samples were then stretched until breakage at a deformation rate of 0.1 cm/s. Young's modulus in megapascals was derived from the engineering stress/strain curve and calculated using the area density of the ADM. For statistical purposes, the mean and p-value of five samples from each group were evaluated and compared.
Immunofluorescent staining
For the assessment of elastic fibers in ADMs, the presence of proliferative fibroblasts after in vitro culture on ADM, and for characterizing different cell types that migrated into ADMs after transplantation, we used immunofluorescence staining for specific markers. Paraffin-embedded samples were sectioned at a thickness of 5 μm. After deparaffinization and rehydration, antigen retrieval was performed except for elastin staining, and then, nonspecific antibody binding sites were blocked with 5% bovine serum albumin (BSA) in PBS. For vimentin and ki67 staining, cells were permeabilized before incubation with primary antibodies. We used the following primary antibodies: anti-elastin (ab21610), anti-Ki67 (ab16667), anti-vimentin (ab92547) from Abcam, anti-Cd45 (eBioscience; 14-0451-82) 1:100 in 2% BSA in PBS, and secondary antibodies: Rhodamine Red™-X AffiniPure goat anti-rabbit IgG (H + L) (# 111-295-144), Alexa Fluor® 488 AffiniPure goat anti-rat IgG (H + L) (# 112-545-003) (Jackson ImmunoResearch Laboratories, West Grove, PA), and Alexa Fluor 488 goat anti-rabbit IgG (H + L) (Thermo Fisher; R37116), 1:1000 in 2% goat serum in PBS. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) before imaging. Images were collected by systematic uniform random sampling of tissue sections using the 20× dry objective of a Zeiss AxioObserver Z1 confocal microscope fitted with a CSU-X1 spinning disc (Yokogawa Electric) and AxioVision 4.8 (Zeiss). To quantify elastin area fraction, confocal images were analyzed using the program ImageJ 1.50i (National Institutes of Health). Images were converted to an RGB stack format and the scale was adjusted to micrometers, using a scale bar of the images, and then the threshold was set to exclude background and saturated pixel intensities. Immunolabeled areas were automatically detected and the area fraction was calculated for elastin. For final quantification, the area fraction for 15 fields of view per sample was averaged over triplicate independently prepared samples.
Cytocompatibility
Cell adherence to the surface of ADMs was determined qualitatively through staining actin filaments with cytopainter Phalloidin 647-iFluor reagent (Abcam; ab176759), and viability was assessed using a live/dead toxicity assay (Molecular Probes®; Invitrogen, Carlsbad, CA). Cells used in this study include mouse dermal fibroblast and human foreskin dermal fibroblast. The high-glucose DMEM culture medium supplemented with 10% FBS was added to each ADM and incubated in a 37°C, CO2-regulated incubator for 24 h before cell seeding. For cell attachment and viability tests, 15 × 103 and 10 × 103 cells were seeded onto the top surface of each ADM, respectively, and were maintained in culture. Cell culture was stopped at 24 h postseeding, and phalloidin staining or live/dead staining was performed. For the cell attachment test, after 24 h of culture, the ECM scaffolds were fixed with 4% paraformaldehyde, stained with cytopainter Phalloidin 647-iFluor reagent (Abcam; ab176759) for f-actin to visualize cells, and counterstained with the nuclear dye DAPI. For the viability test, after 24 h, the scaffolds containing cells were washed three times with 1× PBS, pH 7.0, and incubated with a mixture of ethidium homodimer and calcein-AM according to the manufacturer's instructions. After 30 min, the scaffolds were washed three times with 1× PBS and visualized using a Zeiss AxioObserver Z1 confocal microscope fitted with a CSU-X1 spinning disc (Yokogawa Electric) and AxioVision 4.8 (Zeiss), and images were analyzed by Zen software.
Subcutaneous implantation of ADMs and biocompatibility study
Four female BALB/c mice per treatment group were anesthetized using isoflurane and aseptically prepared for surgical subcutaneous engraftment of ADMs. Briefly, bilateral 1 cm incisions were created on the mouse dorsum followed by minimal dissections under the panniculus carnosus. ADMs (8 mm in diameter) or freshly excised C57/B6 mouse skin was placed onto dorsal pockets. Each had two implants from the same treatment group, one on the left and the other on right dorsal side; C57/B6 fresh skin sample acted as a control and three decellularized dermal matrices as treatment groups. All the skin incisions were closed with 5/0 vicryl sutures. They were implanted for 1, 2, 4, and 8 weeks into each mouse. All mice remained healthy, with no overt signs of inflammation over the experimental period. At proper time points, recipient mice were euthanized and the implants along with upper skin and surrounding connective tissue were removed (Fig. 4). The size of the implanted ADM was measured using a ruler and then processed for routine H&E histologic examination.
Recall antigen and T cell proliferation assays
Spleens from ADM-matched BALB/c mice were harvested and prepared by removing fat and mesentery. Splenocytes were isolated using a 40-μm cell strainer (Fisherbrand; Fisher Scientific, United Kingdom) and the back end of a plunger from a 10-mL syringe as previously described. 29 In all cases, ≥95% of cells remained viable (data not shown). Splenocytes were also isolated from C57/B6 mice spleens for the two-way mixed lymphocyte reaction (MLR) controls. After isolation of splenocytes from presensitized mice and labeling them with carboxy-fluorescein diacetate succinimidyl ester (CFSE), each group was cocultured with 8 mm punch biopsies of native or decellularized skin from a corresponding group for 4 days.
To ensure that only viable cells were evaluated, 7-amino-actinomycin D (7-AAD) (live/dead) exclusion dye was used as a selection marker. Only 7AAD negative cells were gated for final analysis. Any adherent cells were detached by gently washing the tissue with medium. Cells were incubated with monoclonal antibodies for anti-mouse CD3 APC (eBioscience), anti-mouse CD4 PerCP-Cyanine5.5 (BD Biosciences), anti-mouse CD8a PE (eBioscience), or an equivalent isotype-matched negative control antibody. Samples were processed (minimum 30,000 live-events per sample) using a BD FACS machine (BD Biosciences). Data were acquired and analyzed using BD CellQuest Pro software (version 4.0.1 for Mac; BD Biosciences). The gating strategy excluded debris to ensure positive gating of a lymphocyte population on the forward scatter–side scatter dot plot. Of the live cells, CD3+, CD4+, and CD8+ cells were shown in a dot plot against CFSE, and the percentage of proliferated cells was measured on a histogram, expressed here as the proliferation index (i.e., the proportion of cells that have proliferated in response to antigen).
Transplantation procedure
BALB/c mice were anesthetized and hair from their backs was removed using clippers and a depilatory cream. Full-thickness wounds, including the panniculus carnosus, were created on the back of the mice using a 6 mm in diameter punch device in two different sites on the back of the recipient mice (Fig. 6A). Four female BALB/c mice per treatment group were used, and each mouse received either two ADMs (prepared using the same method) or control skin grafts (fresh allogeneic skin). Allogeneic skin was harvested from donor C57/B6 mice. Skin grafts and ADMs (8 mm diameter) were engrafted on the 6 mm wound bed and sutured at 90° intervals using 7-0 Prolene sutures (12, 3, 6, and 9 o'clock). The intervening gaps were then addressed using simple interrupted sutures with 8-0 nylon sutures (6–8 sutures) (Fig. 6A). Immediately following surgery, OPSITE dressing (Smith & Nephew) was sprayed onto the graft site and covered by Vaseline-impregnated gauze. Tegaderm film was applied over the graft site and then secured by a 2 cm width Co-flex bandage.
Statistical analysis
Data are mean ± standard error of the mean of three or more independent sets of experiments. The statistical differences of mean values among ADMs and control normal skin were tested with one-way analysis of variance. Posthoc comparisons were done using Bonferroni correction for multiple comparisons. p-Values <0.05 were considered statistically significant.
Results
Assessment of decellularization
We compared freshly isolated mouse dorsal skin with site-matched decellularized skin matrix variants, namely, Triton, NLS, and LatB. Histological examinations are shown in Figure 1A. Successful decellularization was defined as the absence of nuclei in H&E staining, with relative preservation of dermis structure. To qualitatively assess the extent of preservation of collagen fibers during decellularization, we used Masson trichrome staining (Fig. 1B). Both staining methods revealed that dermal and epidermal cells (nuclei included) were completely removed by all decellularization treatments, while the basic dermal architecture of collagen bundles was preserved.

Assessment of decellularization.
Representative scanning electron micrographs of the surfaces of both sides of ADMs and their cross sections are shown in Figure 1C. Scanning electron microscopy (SEM) micrographs showed morphological and structural differences between the hypodermal and epidermal sides of ADMs. In brief, a network of collagen, reticular fibers, and connective tissue with varying diameters were observed on the hypodermal sides. Even though all three ADMs showed a relatively smooth epidermal surface, resembling sheets of tightly compacted materials, they exhibited different surface topography. Cross-sectional micrographs of the materials displayed fibrous architecture with variable porosity, and interestingly, the cross section of the NLS–ADM has a distinct structural pattern resembling that of normal skin (Fig. 1C). Figure 1D and E shows the residual DNA contents before and after decellularization with three methods. The residual DNA content, which represents the remaining cell debris, is not detectable in ADMs. In the last step of our decellularization methods, we used Benzonase treatment and these results showed that our methods were adequate for eliminating the residual DNA cell content. Similar findings were observed with western blot for detection of actin protein as an abundant intracellular protein. The amount of actin in ADMs was undetectable compared with that of untreated skin (Fig. 1F).
Collagen and GAG content
The collagen and sGAG content of normal skin and decellularized scaffolds was assessed quantitatively. Collagen content, indicated by quantification of hydroxyproline, was equally preserved in all three methods of decellularization (Fig. 2A). In contrast, decellularization with NLS significantly reduced sGAG, while other decellularization methods did not significantly change the GAG content (Fig. 2B).
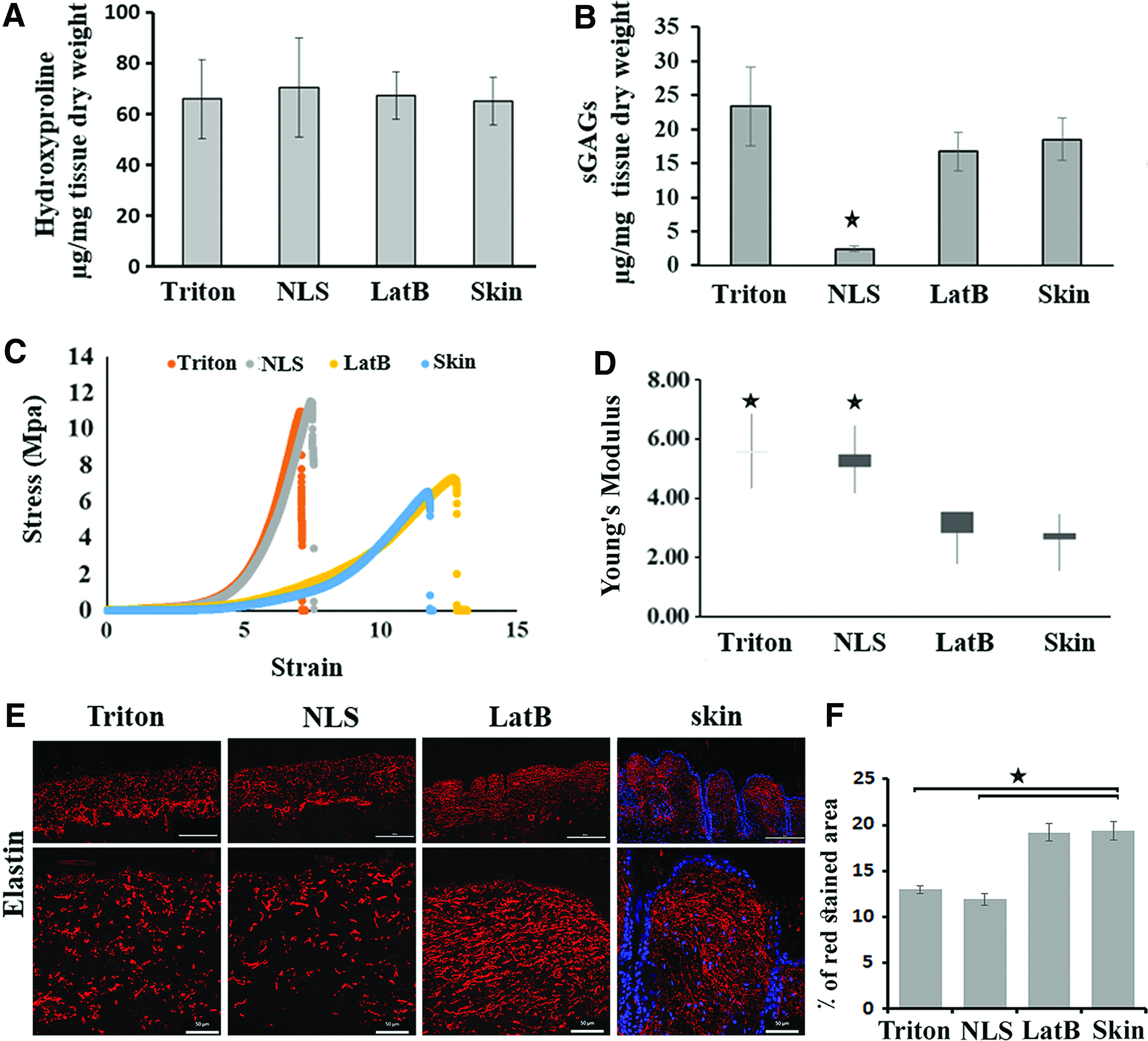
Characterization of ADMs. Samples were collected from five independent preparations for following experiments.
Tensile strength
Tensile strength, as a measure of engineered stress, was determined for all ADMs as well as mouse skin as a control. Performance-wise, LatB decellularized skin was most comparable with normal skin (Fig. 2C). Furthermore, the tensile modulus of NLS and Triton ADMs was significantly higher than that of both LatB–ADM and normal skin (p < 0.05) (Fig. 2D).
Immunofluorescent staining of elastic fibers
To evaluate noncollagenous proteins of ECM in decellularized matrix, elastin staining was performed. The elastic network, elastin, is, in part, responsible for natural skin tension and elasticity. The elastic fibers are much finer than collagen fibers. The role of elastin fibers is to restore the collagen network to its normal condition after deformation. 30 As shown in Figure 2E, elastin fibers appeared fragmented and significantly less abundant in the Triton- and NLS-ADMs (12.98 ± 0.42 and 11.89 ± 0.66) when compared to those of the LatB method and normal skin (19.20 ± 0.93 and 19.35 ± 1.00), respectively (p < 0.05).
Cytocompatibility of ADMs
To investigate the cellular response to these scaffolds in vitro, ADMs were seeded with mouse dermal fibroblasts and human fibroblasts. All cell types cultured for 24 h on ADM surfaces prepared with three different decellularization methods were viable as indicated by green (live cells) staining (Fig. 3A). There was no difference between the three materials with respect to human dermal fibroblast adherence after 24 h in culture (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/tea). The morphologic appearance of the fibroblasts and their density on the surface of ADMs are indicative of fibroblast adherence to the surface of matrix. Differences in fibroblast morphology correspond with the observation of differences in surface topology made evident by SEM (Fig. 1C). To investigate the infiltration of fibroblast into the dermal matrices, fibroblasts were kept in culture on matrices for 1 month. As shown in Figure 3B, all three types of ADMs were able to foster the in vitro culture and infiltration of fibroblasts into the ECMs after 1 month of being in culture. Some of those fibroblasts were positive for the staining of proliferation marker Ki67, indicating ongoing proliferation of cultured and infiltrated fibroblasts (Fig. 3C).
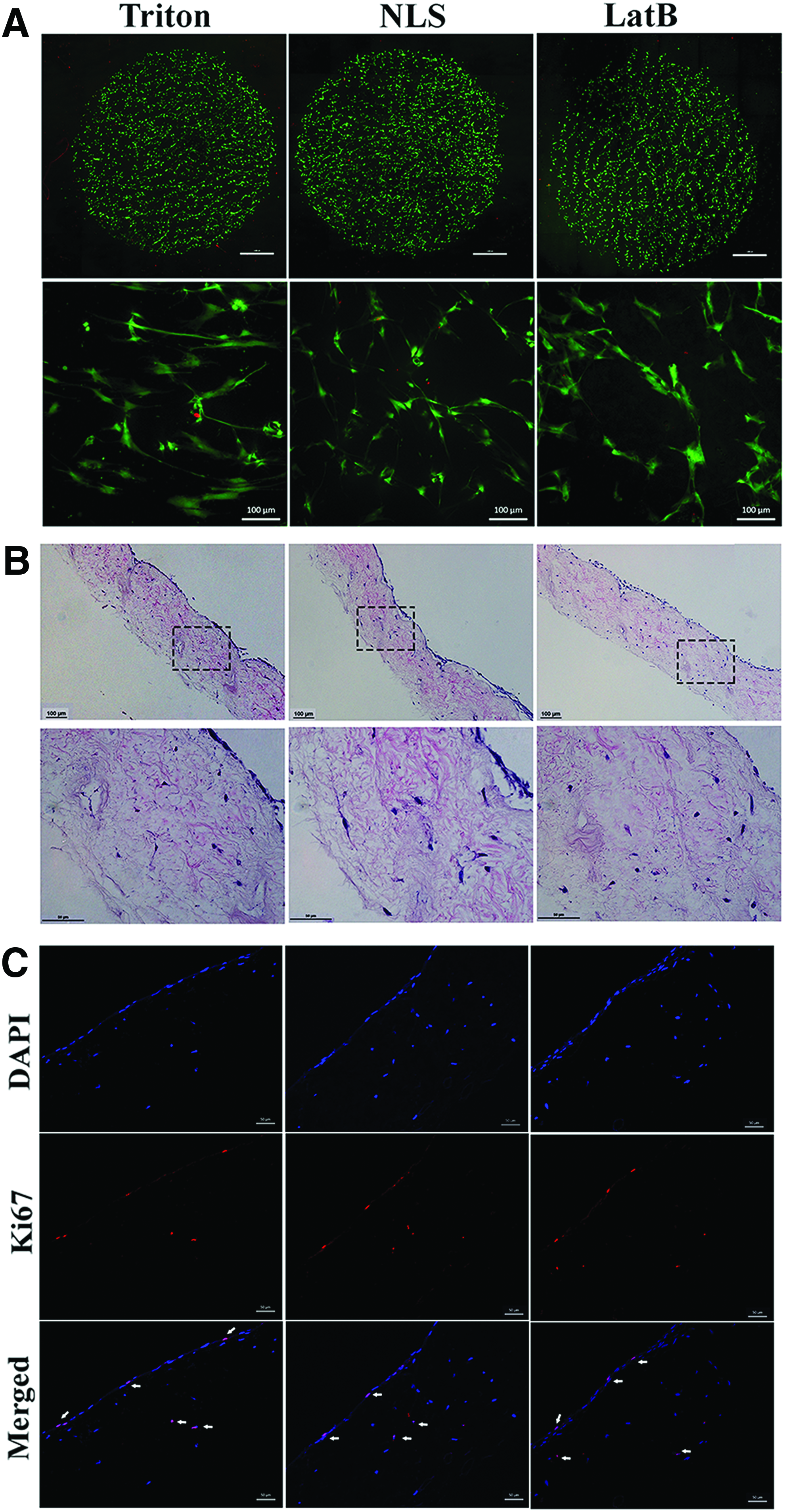
Cytocompatibility of ADMs.
Biocompatibility study
Both fresh and decellularized dermal samples derived from C57/B6 mice were implanted subcutaneously on the dorsum of BALB/c mice to evaluate both immunogenicity and cell infiltration in a secondary in vivo model (Fig. 4). After 1, 2, 4, and 8 weeks, recipient mice were euthanized, the implants were removed, along with upper skin and surrounding connective tissue, and processed for routine H&E histologic examination (Fig. 4A, B). As expected, allogeneic skin presented signs of immune cell infiltrate, indicative of acute rejection by the host, as early as 1 week (Fig. 4A[h]), with notable degradation and infiltrating immune cells within the implants at week 8 (Fig. 4B[l]). In contrast, none of the ADMs (regardless of the decellularization method) prompted any significant immune response. In fact, there were no noticeable differences in gross morphology of ADM implants at any given time. Histology revealed that host cells migrated into all ADMs after the first week of implantation (Fig. 4A[e–g]). Over the 8-week follow-up, there was no evidence of ADM rejection by the recipient mouse (Fig. 4B).
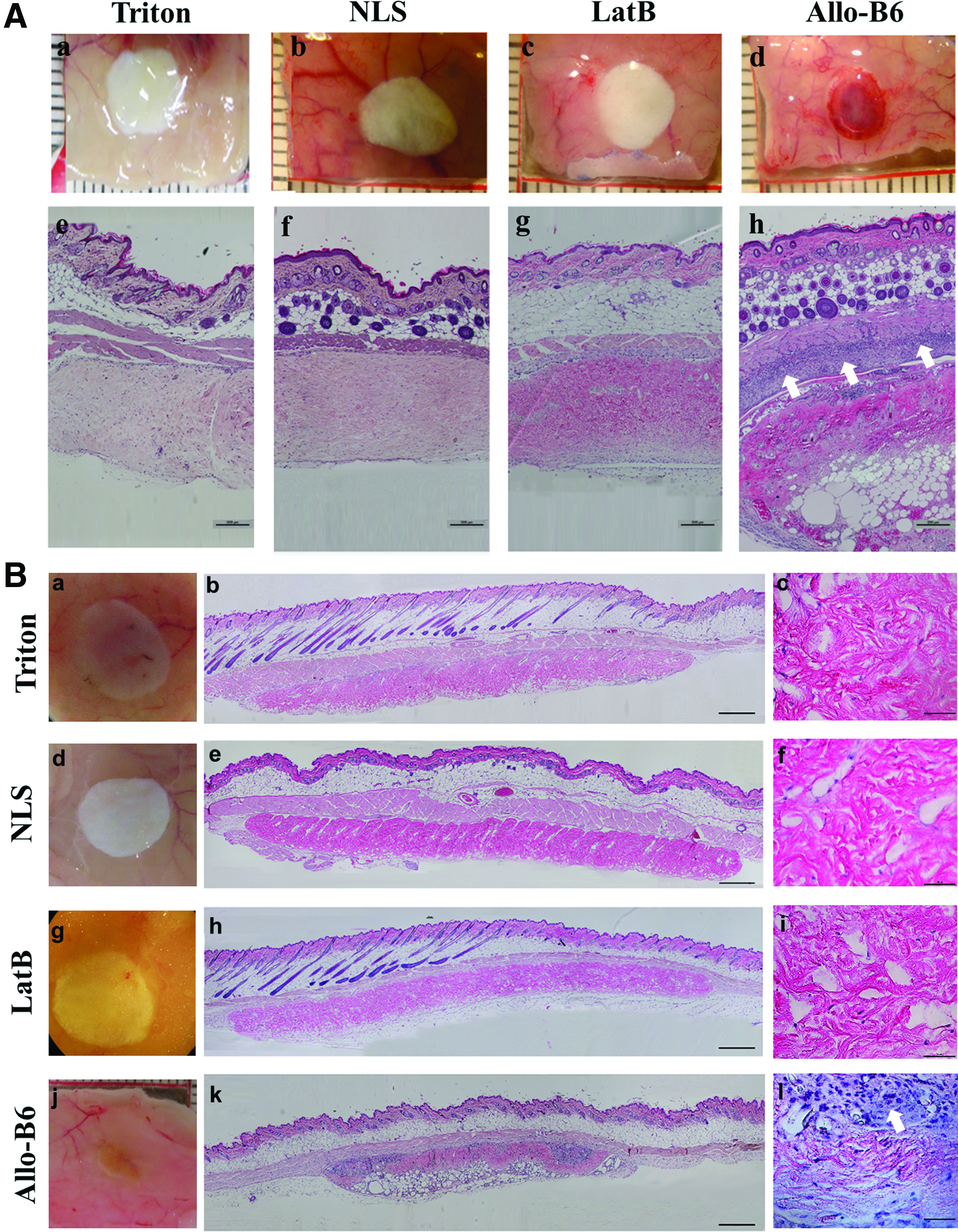
Biocompatibility study of both fresh allo-skin and decellularized dermal samples derived from C57/B6 mice implanted subcutaneously on the dorsum of BALB/c mice after
Assessment of T cell proliferative response to decellularized scaffolds in vitro
When presensitized BALB/c splenocytes were stained with CFSE and cocultured with any type of B6 decellularized dermal scaffold (acting as recall antigen), BALB/c T cell proliferation rate was not significantly different from that of the unstimulated BALB/c splenocytes (data not shown). Stimulation of BALB/c splenocytes by coculturing with allogeneic fresh B6 skin tissue or B6 splenocytes (MLR), however, resulted in a robust BALB/c CD3+ T cell proliferation (8.1 ± 1.33% for B6 skin coculture and 25.4 ± 1.94% for MLR, respectively. p < 0.05). Further analysis revealed that both CD4+ and CD8+ populations of CD3+ cells showed increased proliferation in response to fresh B6 skin tissue or B6 splenocytes and not to ADMs (Fig. 5).

Recall antigenic T cell (CD3, CD4, and CD8) proliferation assay by flow cytometry analysis using CFSE dye.
Biofunctionality of ADM transplantation
One week after transplantation, ADMs appeared to be engrafted in surrounding host tissue, and migration of host cells into implanted ADM was detectable. Epithelialization was observed at the wound edges of the grafted ADM, macroscopically (Fig. 6B) and as evidenced by histological results (Supplementary Fig. S2). Two weeks after implanting, vascularization was detectable in the margin of implants as its color changed to pinkish skin color (Fig. 6B).

Assessment of biofunctionality of ADM in full-thickness skin wound transplantation.
Three weeks after transplantation (Fig. 6C), histology results showed complete recellularization and re-epithelialization. Newly formed vessels originating from wound bed penetrated upward through the ADM. The collagenous structure of the ADMs was preserved. To determine which type of cells infiltrated into the graft region, we performed immunofluorescent staining for vimentin as a fibroblast/mesenchymal marker and CD45 as a pan-leukocyte marker. Results revealed that the majority of these cells were fibroblasts/mesenchymal cells and very few CD45+ cells were detectable (Supplementary Fig. S3). At week 4, re-epithelialization effectively protected the ADM implant such that neither bacterial infection nor desiccation of the implanted ADMs was seen (Supplementary Fig. S4). There were no signs of rejection in all ADMs after transplantation at any time points tested.
Discussion
In this study, we have introduced a detergent-free method for preparing ADMs and compared it with Triton X-100 as a nonionic and NLS as an anionic detergent, two detergent-based decellularization methods of ADM preparation. Triton X-100 is one of the most widely used nonionic detergents for decellularization of tissues.24,31,32 NLS, also known as sarkosyl, is a nondenaturing anionic detergent derived from sarcosine. This surfactant is amphiphilic and is believed to be milder than other ionic detergents such as sodium dodecyl sulfate and has been used for decellularization of skin and artery tissues.25,33
This comparison was based on effectiveness of decellularization while maintaining the structure and functional composition of ECM components. Recalling that the advantage of an ADM over an engineered matrix in the preparation is the inclusion of cell-secreted molecules that are resident in normal skin, the best decellularization processes should thus prevent their damage and removal. Despite the variety of reported decellularization methods, there is lack of evidence to support the use of one method over another in consideration of design characteristics. In our study, we sought to explore and evaluate the biofunctionality of three decellularization methods, in parallel, both in vitro and in vivo. In the interest of clinical applicability, we sought to further compare differently decellularized ADMs as skin substitutes in the treatment of full-thickness wounds.
The detergent-free method that we used, namely the LatB method, was a modified version of the method that has been reported before for decellularization of skeletal muscle, and it is based on decellularization without using detergent and proteolytic enzymes. 26 However, in this study, we used dispase II, a neutral protease, to remove the epidermis and improve the efficiency of the decellularization process, as our preliminary results showed inefficient decellularization without using this enzyme. This method utilizes actin filament disruption by treatment with LatB, cell lysis by osmotic shock by exposure to high ionic strength salt solution, and benzonase treatment to remove residual DNA from skin.
Based on H&E and Mason trichrome staining, there was no detectable nucleus staining in ADMs prepared with the three methods. Measurement of DNA content and western blot for detecting intracellular protein, actin, confirmed complete decellularization as no DNA and actin protein were detectable in ADM samples. Also, the overall structure of the ECM was maintained in each ADM when compared with normal skin (Fig. 1). As evidenced by hydroxyproline assay, these methods did not remove collagen—the most abundant ECM structural component (Fig. 2A). Studies using Triton X-100 have shown mixed results from complete to inefficient cell removal and from safe to harsh on ECM molecules. 21 This discrepancy was mainly related to the tissue being decellularized and to details of protocol such as Triton concentration, incubation time, agitation, and the other methods with which it was combined.
Our results showed that the decellularization method using NLS significantly removed sGAGs from skin, while Triton and LatB methods did not show any reduction when compared with normal skin (Fig. 2B). Previous studies have shown that most detergents cause at least some degree of removal of GAG from the scaffolds. 34 GAGs are negatively charged hydrophilic molecules, so they are capable of absorbing and retaining a large amount of water within the matrix. This feature affects the mechanical and viscoelastic behavior of the tissue matrix. 35 Retention of sGAG content in ADM thus has a significant impact on its function.
Biomechanical analysis of the ADMs confirmed the mechanical integrity of the ADM prepared with LatB method, since there was no difference between the normal skin and ADM, while detergent-based methods showed different stress–strain patterns and tensile modulus (Fig. 2C, D).
Furthermore, elastin staining showed that the LatB method has a similar staining pattern to normal skin compared with methods that use Triton or NLS (Fig. 2E). Elastin is a durable and insoluble protein with minimal turnover. It gives recoil and resistance to tissue such as blood vessels, lung, and skin and induces cell activities such as cell migration and proliferation, matrix synthesis, and protease production.36–38 The biomechanical properties of elastin allow skin to bear tensile stress, especially at the mobile parts of the body such as joints. Even though at the beginning of wound healing and on injury, elastin fragments are generated and released into the ECM to induce biological responses in cells as active ligands (elastokines), 39 later on the deposition of elastin in the dermis is aberrant and has only been detected a few months after the initial wound healing stages. 40 Moreover, disrupted elastic fibers lead to reduced elasticity of the mature scar. Also, the presence of elastin in a skin substitute has been shown to reduce wound contraction and improve dermal regeneration. 41 Regarding the mechanical and signaling properties of elastin during wound healing, it is obvious that the restoration of an intact elastic fiber is critical to regain functional skin after injury. 36
In this study, we provided a comparison of ADMs prepared in parallel with different methods of decellularization. Each method was associated with distinct changes to structure and composition of the resulting material. Although our assessment of post-transplanted scaffolds showed overall acceptable results for all ADMs and no single method greatly outperformed the functionality of another, our findings indicated that a detergent-free LatB method was minimally disruptive to native ECM and was the only method that achieved effective decellularization of skin while preserving the sGAG and elastin content of the tissue as well as its biomechanical property, when compared with detergent methods.
A limitation of the present study is that a full-thickness skin wound model was created on the backs of mice and that location is not the proper place to show the importance of having elastic skin substitute because it is not the best body site in terms of enduring tensile stress. It would be appropriate to create skin wounds in the areas such as joints, which bear tensile stress. However, this is not feasible in a small rodent model.
Overall, our results emphasize the importance of decellularization methods for producing ADMs and the design criteria for evaluating efficacy of these methods. We strongly recommend using controlled, parallel preparation and analysis of ADMs in vitro and in vivo to ascertain biofunctional outcome.
Footnotes
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.