
Abstract
The 5′-untranslated region (5′-UTR) of mRNA contains structural elements, which are recognized by cell-specific RNA-binding proteins, thereby affecting the translation of the molecule. The activation of an innate immune response upon transfection of mRNA into cells is reduced when the mRNA comprises chemically modified nucleotides, putatively by altering the secondary structure of the molecule. Such alteration in the 5′-UTR in turn may affect the functionality of mRNA. In this study, we report on the impact of seven synthetic minimalistic 5′-UTR sequences on the translation of luciferase-encoding unmodified and different chemically modified mRNAs upon transfection in cell culture and in vivo. One minimalistic 5′-UTR, consisting of 14 nucleotides combining the T7 promoter with a Kozak consensus sequence, yielded similar or even higher expression than a 37 nucleotides human alpha-globin 5′-UTR containing mRNA in HepG2 and A549 cells. Furthermore, also the kind of modified nucleotides used in in vitro transcription, affected mRNA translation when using different translation regulators (Kozak vs. translation initiator of short UTRs). The in vitro data were confirmed by bioluminescence imaging of expression in mouse livers, 6 h postintravenous injection of a lipidoid nanoparticle-formulated RNA in female Balb/c mice. Luciferase measurements from liver and spleen showed that minimal 5′-UTRs (3 and 7) were either equally effective or better than human alpha-globin 5′-UTR. These findings were confirmed with a human erythropoietin (hEPO)-encoding mRNA. Significantly, higher levels of hEPO could be quantified in supernatants from A549 cells transfected with minimal 5′-UTR7 containing RNA when compared to commonly used benchmarks 5′-UTRs. Our results demonstrate the superior potential of synthetic minimalistic 5′-UTRs for use in transcript therapies.
Introduction
Even though the earliest reports of RNA transfection into cells date back to the early 1960s, and even though Wolf et al. 1 demonstrated the impressive potential of intramuscularly injected mRNA for expressing a reporter protein already in 1990, the field of mRNA transcript therapy has progressed to clinical applications only during recent years. Furthest advanced are mRNA-based tumor vaccines,2–4 and mRNA vaccines against viral infections have turned out highly potent in preclinical studies.5,6 Regenerative treatments for heart failure, diabetic wound healing, and vascular diseases 7 have progressed to a first clinical trial, 8 promising developments for bone healing are on the way,9,10 and mRNA-encoded transcription factors are highly efficient for generating induced pluripotent stem cells. 11 Similarly, mRNA-based genome editing tools are not only potent but also offer conceptual and/or safety advantages over protein-based or virus-encoded alternatives. 12 Last but not least, mRNA transcript therapy has been shown to be efficient and even life-saving in preclinical models of human hereditary metabolic disease.13,14
Numerous approaches have been undertaken to enhance the therapeutic potential of mRNA molecules. Incorporation of modified nucleotides has led to a significant reduction of mRNA immunogenicity.15,16 Incorporation of modified nucleotides has been shown to significantly reduce immunogenicity of mRNA in numerous studies in the past decade. 15 To the best of our knowledge, the work from Karikó et al. 17 was the first demonstration of reduced binding of pseudouridine-containing mRNA to TLRs. In a previous report, we demonstrated that replacement of only 25% of uridines and cytidines with 2-thiouridine and 5-methylcytidine synergistically decreased mRNA binding to TLR3, TLR7, TLR8, and RIG-I in human peripheral blood mononuclear cells. 16 In another study, it has been shown that sequence engineering alone without any chemical modifications results in enhanced translation and reduced immunogenicity. 18
Besides nucleotide modifications, structural elements such as untranslated regions (UTRs) have also been shown to affect the translational efficiency of the mRNA molecule. The average length of 5′-UTRs, which are much shorter than 3′-UTRs, ranges between 100 and 200 nucleotides and does not vary greatly among different taxa. 19 Our previous study, 20 comparing five different cellular UTRs for enhanced translation and mRNA stability, showed that human cellular CYBA UTR sequences increase mRNA translation without affecting the half-life of recombinant RNA transcripts. In another study from Holtkamp et al., 21 two copies of the beta globin 3′-UTR supported high expression in dendritic cells. Other studies have used UTRs derived from the human CMV enhancer and human growth hormone, 22 human alpha-globin 5′-UTR, 23 Xenopus beta globin, 24 or natural UTRs of the transgene. 23
The functionality of UTRs, besides being cell-type specific, is highly dependent on the secondary structure. Incorporation of modified nucleotides has been shown to alter the secondary structure of the mRNA molecule, thereby resulting either in complete loss of function 18 or reduced binding to pattern recognition receptors. 16 To overcome such limitations, it would be beneficial to design “minimalistic” UTRs, which are cell-type independent and not drastically affected by the choice of modification.
The major objective of the current study was to combine minimalistic 5′-UTR-based mRNA design with the use of chemically modified nucleotides aimed at improving the translational efficiency. Since majority of the previous studies have been performed using a Kozak element as translational regulator, we also investigated how its function was affected by the choice of modified nucleotides. Elfakees et al. 25 reported a unique translation initiation element (TISU) present in ∼5% of the protein-coding genes with short UTRs. This was also included in the current study as an alternative to Kozak element. The lead short minimal UTR was also benchmarked against some of the naturally occurring 5′-UTR in viruses and cellular genes.22,26,27
To address the issues of cell- and sequence specificity, experiments were performed with two different mRNA sequences (Luciferase and human Erythropoietin [hEPO]) in human lung alveolar epithelial cell line (A549) and human hepatocellular carcinoma cells (HepG2). In vitro results with luciferase were later confirmed in mice after intravenous injection of mRNA-lipid nanoparticles (LNPs). Formulation used for these experiments has been described previously. 28 We show that synthetic sequences, designed on a rational sequence-design approach, outperform some of the most widely used 5′-UTRs, both in vitro and in vivo. Moreover, the functionality of these minimal UTRs was not affected by the choice of modifications and cell types used. To conclude, short synthetic sequences from the current study present themselves as promising 5′-UTRs for the development of mRNA therapeutics.
Materials and Methods
Plasmid preparation
The synthetic 5′-UTR sequences 1–7, as well as reference UTRs, were cloned by a polymerase chain reaction (PCR)-based strategy. The coding sequence for firefly luciferase was amplified from pGL4.10 plasmid (Promega). For each UTR, a specific set of primers were designed. The reaction was initiated by Pfu DNA polymerase (Promega). Correct PCR products (desired UTR with the gene of choice) were cloned into pUC57-Kana vector (GenScript).
Generation of mRNA
To generate in vitro transcribed mRNA, plasmids were linearized by BstBI digestion and purified by chloroform extraction and ethanol precipitation. 29 Purified linear plasmids were used as a template for in vitro transcription. Plasmid templates (0.5 μg/μL) were subjected to in vitro transcription using 3 U/μL T7 RNA polymerase (Thermo Fisher Scientific), with a defined choice of natural and chemically modified ribonucleotides (Jena Biosciences). The modification set 1 was synthesized as described elsewhere.16,30 As for the modification set 2, instead of 5-methylcytidine (25%) and 2-thiouridine (25%), 5-iodouridine (35%), and 5-iodocytidine (7.5%) were used. The complete in vitro transcription-mix was incubated at 37°C for 2 h. Afterward, 0.01 U/μL DNase I (Thermo Fisher) was added for additional 45 min at 37°C to remove the plasmid template.
RNA was precipitated with ammonium acetate at a final concentration of 2.5 mM, followed by two washing steps with 70% ethanol. The pellet was resuspended in aqua ad injectabilia. A C1-m7G cap structure was added enzymatically to the 5′ end of the transcript using Vaccinia Virus Capping Enzyme (NEB) following manufacturer's instructions. The 3′ end of the transcript was subjected to enzymatic polyadenylation of ∼120 nucleotides, using E. coli poly(A) Polymerase (NEB). As all constructs were incubated for equal time, they had comparable poly(A) tails. RNA quality and concentration were measured spectrophotometrically on a NanoDrop2000C (Thermo Fisher Scientific). Its correct size and purity were determined via automated capillary electrophoresis (Fragment Analyzer, Advanced Analytical).
Cell culture
A549 (ACC-107) and HepG2 (ACC-180) cells were purchased from DSMZ. They were cultured in Minimum Essential Media (MEM) with Glutamax (Gibco/Life Technologies) and RPMI 1640 plus GlutaMAX (Gibco/Life Technologies), respectively. Both media were supplemented with 10% heat-inactivated fetal bovine serum (Gibco/Life Technologies) and 1% penicillin/streptomycin (Gibco/Life Technologies). Cells were cultured in a humidified 5% CO2 incubator at 37°C.
In vitro transfection
Both cell lines, A549 and HepG2, were transfected with different doses of mRNA in the range from 3.9, 7.8, 15.625, 31.25, 62.5, and 125 to 250 ng/well. A549 and HepG2 cells were seeded at the density of 2 × 104 cells/well and 4 × 104 cells/well, respectively, in a 96-well plate. Twenty-four hours postseeding, cells were transfected using the commercial transfection reagent Lipofectamine®2000 (Thermo Fischer Scientific). Complexes were prepared at a ratio of 2 μL Lipofectamine2000 per 1 μg mRNA. The mRNA was diluted 1:20 in water and Lipofectamine2000 1:10 separately in a serum-free MEM. mRNA was added to the Lipofectamine2000 solution followed by 20 min incubation time at room temperature (RT). The concentration of the final mRNA/Lipofectamine2000 solution was 25 ng/μL, and a serial dilution 1:2 was performed. Ten microliter of the complex solution was added to the cells and cells were incubated for 24 and 48 h, respectively. For every mRNA construct, replicates of three or six were prepared.
Firefly luciferase assay
For detection of firefly luciferase activity, cells were lysed for 30 min at RT in lysis buffer (25 mM Tris-HCl, 0.1% TritonX-100, pH 7.4). Luciferase assay was performed as described previously.31–33 Photon luminescence emission was measured for 5 s using Tecan Infinite® 200 PRO.
Lipid formulation
For in vivo experiments, mRNA was formulated in LNPs as previously described. 28
In vivo studies
For in vivo studies, Balb/c mice (Charles River Laboratories) at the age of 6–8 weeks were used. All animal experiments were carried out according to the guidelines of the German law of protection of animal life and reviewed by the local ethics committee. Mice were injected intravenously with 20 μg LNP formulated mRNA. In vivo imaging of luciferase expression was performed at 6 h postdelivery using an IVIS Lumina XR Imaging System (Caliper Life Sciences) and organs were harvested and analyzed for ex vivo luciferase measurements as described previously. 28
Enzyme-linked immunosorbent assay analysis
Quantification of hEPO protein was performed using hEPO Quanktikinene IVD ELISA Kit (R&D Systems) following manufacturer's instructions.
Statistical analysis
Each experiment was performed with at least three technical replicates per sample. Results are shown as means ± SD unless otherwise stated. Statistical analysis was performed using GraphPad Prism software (version 6). Data were tested for normal distribution using D'Agostino–Pearson omnibus normality test. Multiple comparisons were conducted by two-way ANOVA, followed by Sidak's test (pairwise comparison) or Dunett's test (many-to-one comparison). A p-value ≤0.05 was considered statistically significant.
Results
Design of minimalistic 5′-UTRs
To create recombinant RNA transcripts with short synthetic 5′-UTRs, the corresponding DNA sequences were cloned into a plasmid vector upstream of the gene of interest (GOI). Table 1 lists the positions of different bases in the mRNA relative to the start codon. T7 promoter (TAATACGACTCACTATA) was combined with the Kozak element consensus sequence (GCCACC) upstream of the start codon (ATG) (minimal UTR1; Table 1). Transcription from T7 promoter begins with the first G after the TATA element. The following six bases after the TATA element (GGGAGA) are needed for high yields and homogenous 5′ mRNA ends during in vitro transcription.34–38 This template-sequence results in an RNA, which has a GGGAGAGCCACC stretch as its 5′-UTR. For comparison purposes a fusion sequence consisting of a T7 promoter and a complete Kozak consensus element (GCCGCCACC) was generated by inserting a C between GGGAGA and GCCACC (minimal UTR2; Table 1), thereby bringing the G at position +5 (base position in mRNA) in alignment with −9 position from ATG (Table 1); this construct was designated as minimal UTR2.
Sequences of Synthetic 5′-Untranslated Region with Annotation of Each Nucleotide Position
Each 5′-UTR sequence comprised T7 promoter (T7), the desired 5′-UTR, and a luciferase coding sequence. Listed are the respective sequences, each starting with a GGG as a transcription-starting site and ending with an ATG, a start codon. Numbers above the sequences indicate coordinates of those nucleotides where transcription starts, as GGG is the first triplet transcribed into mRNA. Numbers underneath the sequence indicate coordinates of nucleotides upstream of a start codon (ATG).
5′-UTR, 5'-untranslated region.
Effect of spacer nucleotides between T7 promoter and Kozak element
We first compared mRNA translation efficiency for two different luciferase-coding transcripts (minimal UTR1 and 2), produced using either unmodified, modification 1 or modification 2 sets of nucleotides. Transfection experiments were performed in both A549 and HepG2 cells. Incorporation of an extra C into the sequence at position 7 significantly increased protein levels posttransfection in a modification and cell type independent manner at both time points investigated (Fig. 1). Therefore, further constructs were designed using minimal UTR2 as a template.
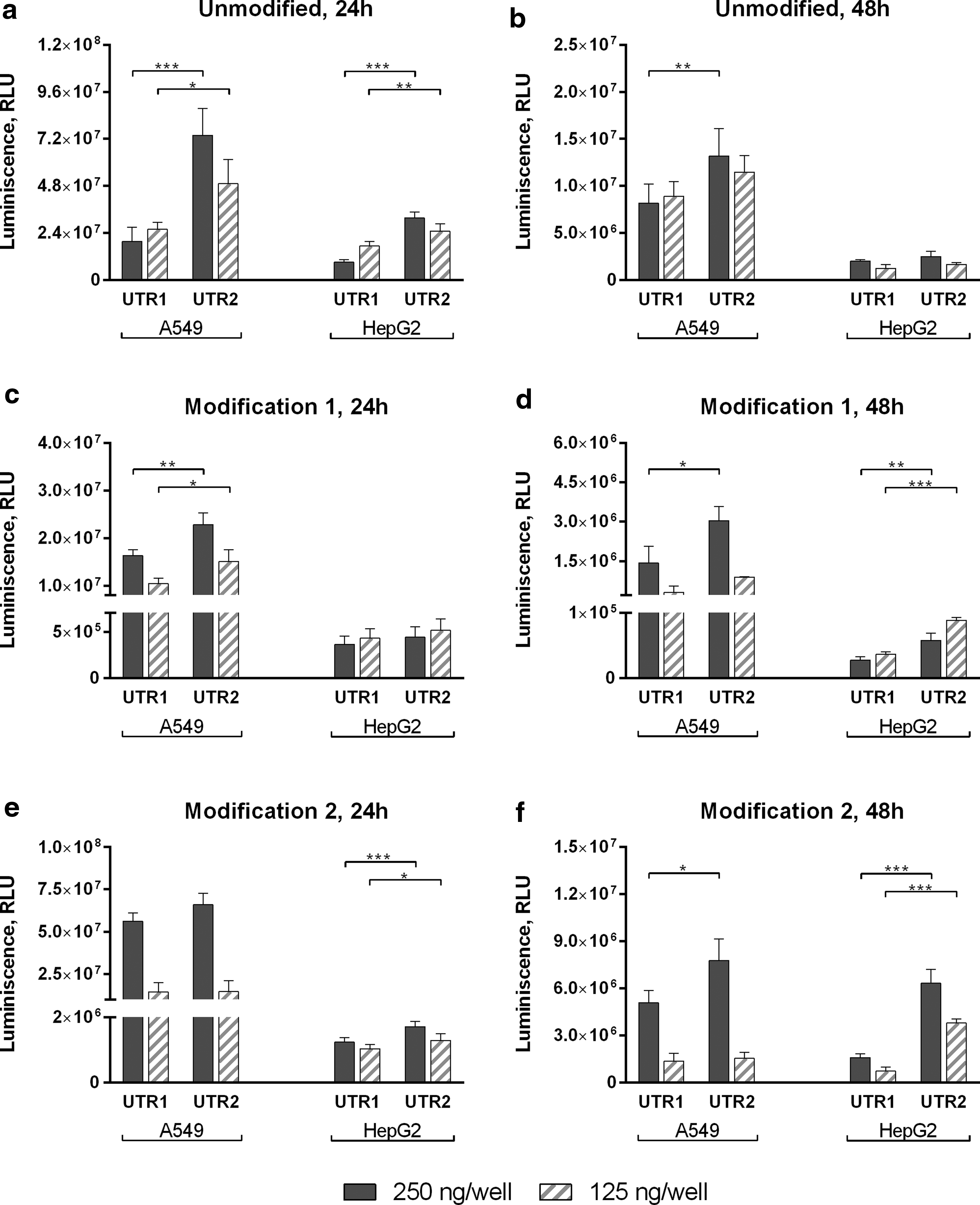
Luciferase activity in protein lysates from A549 and HepG2 cells transfected with minimal UTR1 and minimal UTR2 (Table 1) containing Luc mRNAs. Luciferase activity was measured 24 h
We next investigated the effect of increasing the distance between the T7 promoter and Kozak element by incorporating either T, A, C, or G at the −7 position into the minimal UTR2 construct, thereby generating minimal UTRs 3–6, respectively (Table 1). As a positive control, 5′-UTR from human alpha-globin was inserted at this position (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/tea). 5′-UTR from human alpha-globin was selected as a positive control as it has been shown to result in higher protein amounts in cellular systems. 39 All five constructs were produced with unmodified ribonucleotides, as well as with two different sets of chemically modified ribonucleotides. Transfection experiments were performed in A549 cells. Minimal UTR3 resulted in either comparable (unmodified and modification 1) or higher (modification 2) expression when compared to human alpha-globin 5′-UTR containing mRNA. Similar trends were observed at 24 and 48 h posttransfection (Fig. 2).
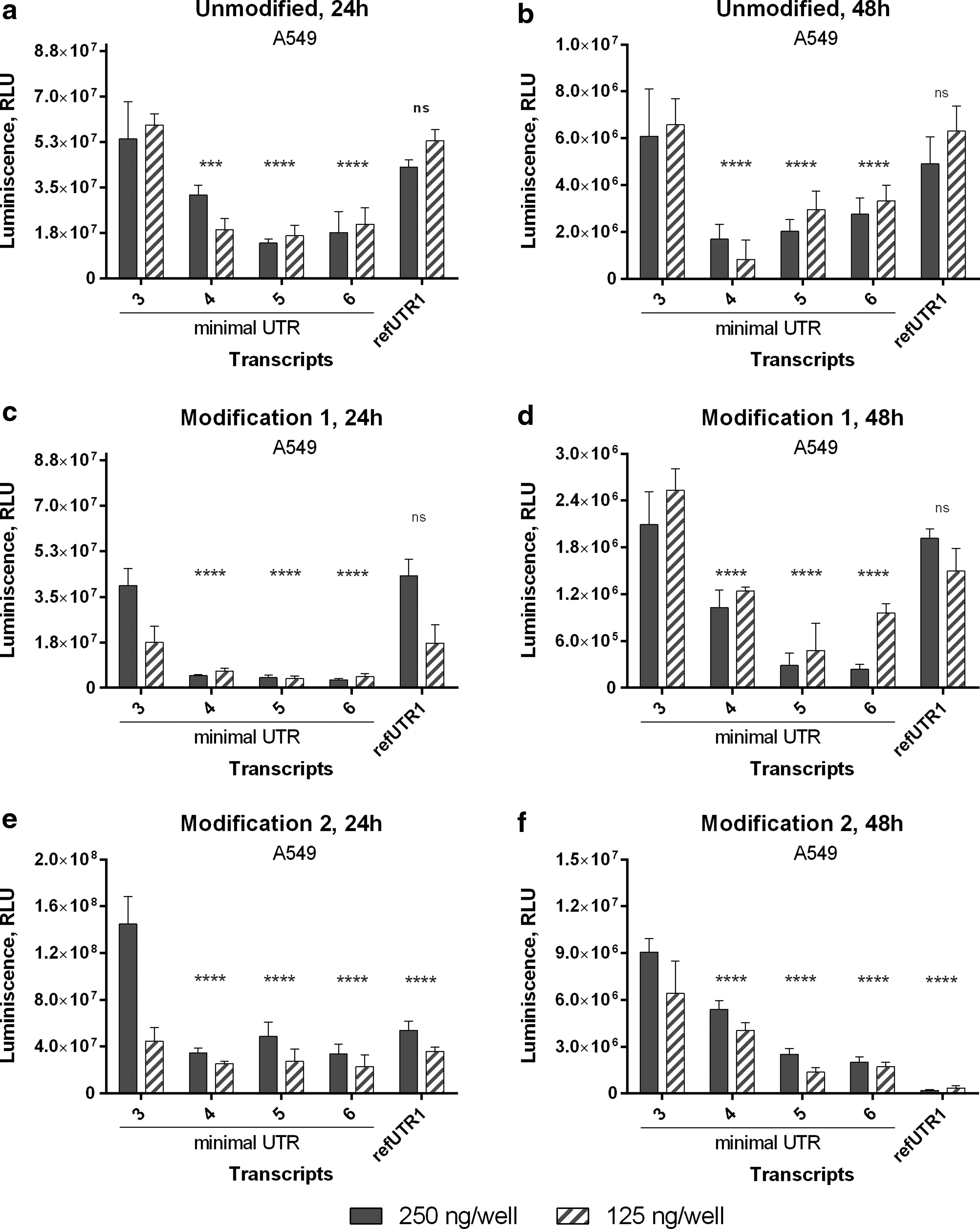
Luciferase activity in protein lysates from A549 cells transfected with different UTRs containing Luc mRNA. Luciferase activity was measured 24 h
Effect of translation regulator
We next investigated the role of the translational regulator on transgene expression and how its functionality is affected by the type of nucleotide modification. Previous studies have shown that certain coding sequences have very short 5′-UTRs harboring a consensus sequence, namely the TISU-element. 40 Minimal UTR3 was appropriately changed to replace the Kozak element by a TISU-element to generate minimal UTR7. Transfection experiments with luciferase mRNA were performed in A549 and HepG2 cells. In both cell lines, the functionality of translational regulator was affected by the kind of modified nucleotides incorporated in the mRNA during in vitro transcription. When using unmodified mRNA, comparable expression levels were observed independent from the regulatory sequence investigated. Modification 1 in combination with the TISU element resulted in a significantly higher expression compared to values observed with Kozak element. An opposite trend was observed when modification 2 was used (Fig. 3). Similar results were observed at both time points.
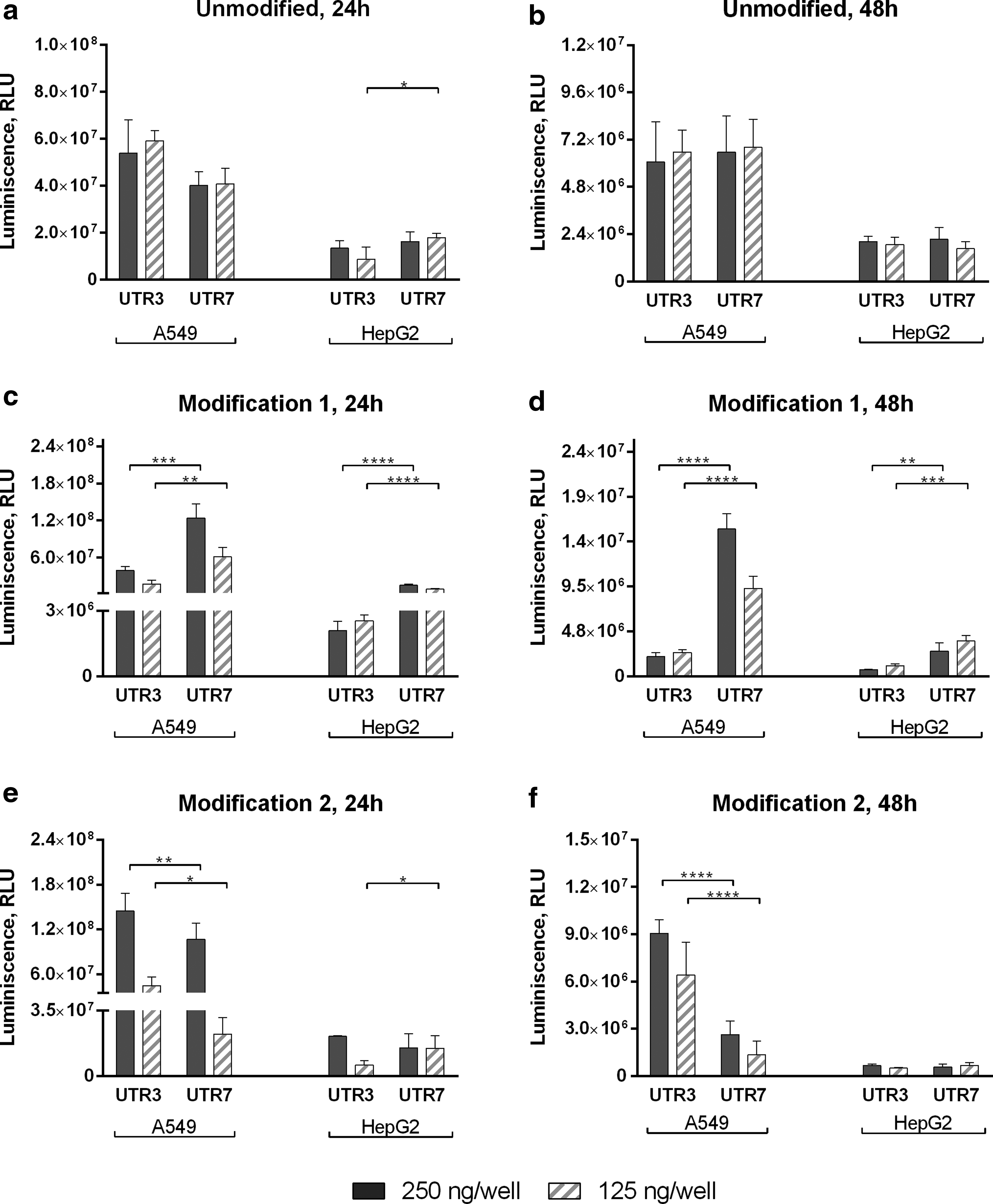
Luciferase activity in protein lysates from A549 and HepG2 cells transfected with either Kozak (UTR3) or TISU (UTR7) element within Luc mRNA. Luciferase activity was measured 24 h
In vivo comparison of different Luciferase coding mRNA constructs
Because of the encouraging results achieved with the minimal UTR3 (expression rate comparable to those from the human alpha-globin UTR) and UTR7 (even better than UTR3 when modification 1 was used), we aimed to test these UTRs in vivo. Experiments were performed in female Balb/c mice (6–8 weeks old). Besides human alpha-globin 5′-UTR (refUTR1), an additional luciferase construct was made containing 5′-UTR from the human CMV enhancer and 3′-UTR from the human growth hormone (refUTR2, Supplementary Table S1) to better benchmark our minimal UTRs UTR3 and UTR7. Both reference UTRs have been reported to enhance protein amounts from the corresponding mRNAs. 22 The respective RNAs, all of them were containing modification 1, were formulated in our previously described lipid formulation 28 and injected intravenously into mice (20 μg/mouse). In vivo imaging was performed 6 h postinjection and luciferase expression was quantified as photons/sec/cm2/sr (Fig. 4). Moreover, spleen and liver were isolated at 6 h postinjection and luciferase activity as also measured ex vivo in these organs.

Luciferase expression in mice injected with 20 μg of different luciferase coding RNAs produced using modification 1. Luciferase expression was measured at 6 h post-i.v. injection using In Vivo Imager
Similar to our in vitro results, minimal UTR3 was as effective as human alpha-globin 5′-UTR (refUTR1), and minimalUTR7 resulted in significantly higher expression compared to its Kozak element containing counterpart (minimal UTR3). Surprisingly, the reference UTR proposed by Guild et al. 22 (refUTR2, Supplementary Table S1) was significantly inefficient compared to minimal UTR7. In vivo imaging results were confirmed by ex vivo luciferase measurements from spleen and liver.
To further validate the translational efficiency of minimal UTR7, it was compared to additional previously published short translation regulators, namely refUTR3-5 (Supplementary Table S1). A549 cells were transfected with mRNA containing UTR7 or refUTR3-5, harboring unmodified and chemically modified transcripts, respectively. Comparable expression was observed when compared to reference UTR3, independent from the modification used. Minimal UTR7 resulted in higher expression when compared to reference UTR4 (unmodified, modification 2) and reference UTR5 (modification 2) after 24 h, but the effect diminished after 48 h (Fig. 5).
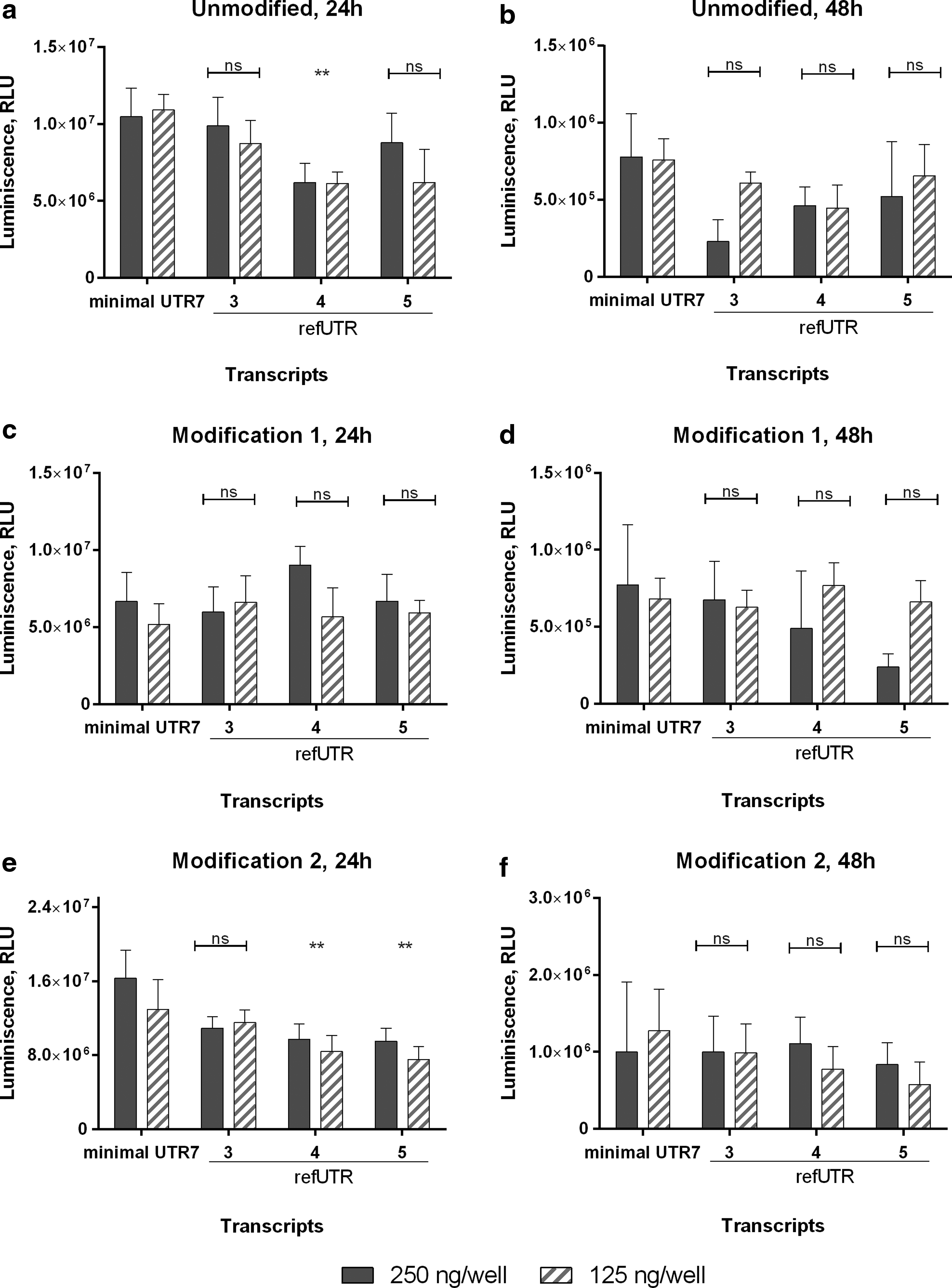
Luciferase activity in protein lysates from A549 cells transfected with different short transcription regulator elements. Luciferase activity was measured 24 h
Sequence-independent function of minimal UTR7
To find out whether or not UTR7 performs superior independently from the subsequent coding sequence, we generated EPO expressing mRNAs containing either refUTR2 (Supplementary Table S1) or our minimal UTR7. mRNA was produced using modification 1 and transfection experiments were performed in A549 and HepG2 cells. Expression of human EPO was quantified via ELISA at 24 h posttransfection. In comparison to refUTR2, significantly higher levels of hEPO could be quantified in supernatants from A549 cells transfected with minimal UTR7 containing mRNA. In HepG2 cells, the construct with minimal UTR7 resulted in higher and reproducible expression (Fig. 6), although the difference was not significant (due to one high EPO measurement point with refUTR2).
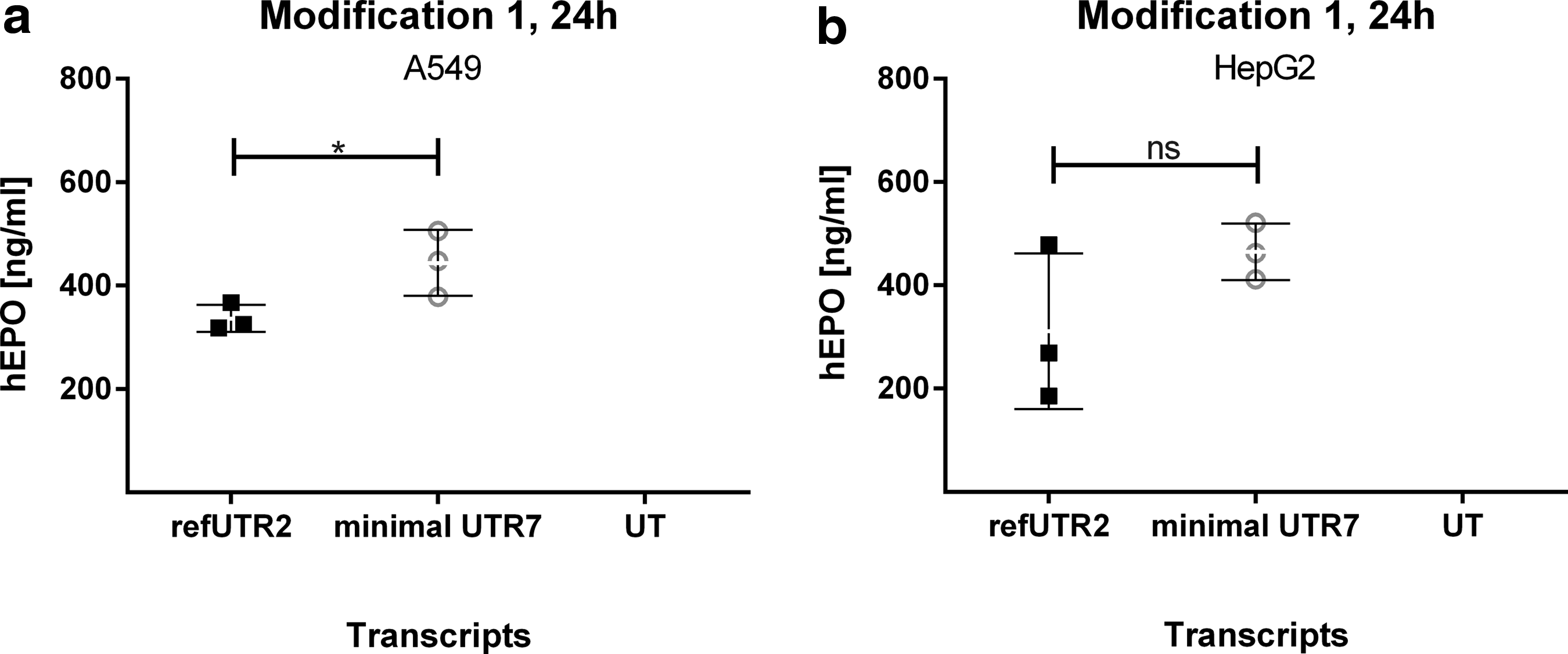
Quantification of secreted human erythropoietin protein levels as measured via ELISA in A549
Discussion
In this study, seven minimalistic synthetic 5′-UTR sequences (minimal UTR 1–7) were investigated with respect to their effect on protein amounts post-mRNA delivery. The goal of this study was to design minimalistic 5′-UTRs, comprising only elements necessary for efficient in vitro transcription and cellular translation and to test their functionality in comparison with commonly used naturally occurring 5′-UTRs. In this regard, we designed synthetic RNA molecules consisting of slightly different minimalistic 5′-UTRs upstream of the GOI, and a 3′ poly(A) tail. For each 5′-UTR, three kinds of mRNAs (unmodified, modification 1, 16 and modification 2) were produced by in vitro RNA synthesis.
Earlier studies37,38 have reported that T7 promoter is composed of two functional domains, an upstream binding region from −17 to −5 and an initiation region from −4 to +6. Base substitutions in the initiation region significantly affected the transition of enzyme from bound to initiation state. Imburgio et al. 36 performed studies of promoter recognition and start site selection by T7 RNA polymerase using a comprehensive collection of promoter variants. In their study, substituting the bases at position +1 to +6, from the wild-type sequence (GGGAGA) to any other base negatively affected the promotor strength. For this reason, short synthetic UTRs designed in this study have been based on wild-type T7 promoter sequence elements.
If we consider wider Kozak consensus, it is not surprising that adding an extra C into the very minimal sequence (minimal UTR1) significantly increased protein translation post-mRNA delivery. The idea behind this is that by inserting an additional C nucleotide at position −7 (seven nucleotides upstream from the ATG codon), a G residue previously placed at the −8 position (in minimal UTR1) is shifted to the −9 position (in minimal UTR2), with respect to the start codon. In an earlier study, Kozak compiled the 5′-noncoding sequences from 699 vertebrate messenger RNAs, 33% contained a G at the −9 position and 37% a C at −7 position. 41 With this in mind, we actually created a favorable wider Kozak sequence (GCCGCCACC) 41 within our fused minimal sequence (minimal UTR2). Our results are in accordance with the hypothesis that having an additional C nucleotide at position −7 and shifting the G nucleotide to position −9 is beneficial for translation efficiency.
In the subsequent experiment, we found that having an additional T at the position +8 (base position in the mRNA) in the minimalistic 5′-UTR, independent whether unmodified or modified, greatly enhanced protein expression compared to the addition of any other nucleotide at the same position. After demonstrating the advantage of using minimal UTR3, we compared it to the 5′-UTR from human alpha-globin gene. We have chosen human alpha-globin as a reference, as it has been reported to have the greatest translation efficiency, followed by the beta-globin. 39
In our previous study using human angiotensin-converting enzyme 2 (hACE2) sequence, incorporation of the 5′-UTR from the human alpha-globin gene resulted in a higher mRNA stability and protein translation rate compared to the natural hACE2 5′-UTRs or the CYBA UTRs.20,23 The full nucleotide sequence of human alpha-globin 5′-UTR consists of 66 nucleotides, 42 while the transcription-starting site is at position −30. Thus, bases 1–29, although present in the gene, are not transcribed into human alpha-globin mRNA in the cell. Therefore, positions 31–60 from NCBI Reference sequence (NM_000517.4) was used as human alpha-globin 5′-UTR (refUTR1). The remaining nucleotides (31–36) from human alpha-globin were replaced by the Kozak element in our constructs.
In addition, the type of nucleotide modification significantly affected the performance of UTRs against the benchmark human alpha-globin 5′-UTR (refUTR1). While no significant difference in luciferase expressions was observed when using either refUTR1 or minimal UTR3 mRNA (unmodified or with modification 1), the use of modification 2 negatively affected the functionality of refUTR1 at 48 h. These results highlight the susceptibility of natural UTR sequences to the kind of nucleotide modification(s). An explanation for this observation may be an alteration of the mRNA secondary structure by incorporating certain chemically modified ribonucleotides, which may result in loss of UTR function or alteration of translation.18,20
In a previous study, 20 we demonstrated that the incorporation of CYBA UTR sequences at the 5′ end of the mRNA results in increased translational efficiencies without increasing the stability of mRNA. These effects were sequence- and cell-type dependent. When using Metridia luciferase as a marker gene, the combination of 5′ + 3′-UTR from CYBA gene resulted in highest protein levels in A549 and NIH3T3 cells. The same combination of UTRs, when combined with human BMP2 coding sequence, was least efficient (compared to the other UTR combinations) in C2C12 cells. 20
Comparing protein production from minimal UTR3 and UTR7 revealed a significant difference between modification 1 version of mRNAs. Translation efficiency from minimal UTR7 mRNA performed 4-fold better 24 and 48 h posttransfection. Since both constructs comprise nearly identical 5′-UTRs but are regulated by different translation initiators (differ only with respect to the use of either Kozak or TISU-element as translation initiator), the observed difference can be attributed to the differences in the translation regulatory element. The vast majority of all previous studies has been performed using a Kozak element as translational regulator9,10,16,18,20,23,30,43 without investigating how its function is affected by the kind of nucleotide modifications.
Previous studies have shown that a minimal length of 32 nucleotides at 5′ end is needed to ensure translation initiation at the start codon which is located directly after the Kozak element; if less than 32 nucleotides are used, translation is initiated at a downstream ATG instead of the intended start codon.44,45 Since our synthetic UTRs are less than 15 nucleotides, besides Kozak element, we additionally tested a TISU element as translation regulator. The TISU element was originally discovered in the noncoding region of genes with extremely short 5′-UTRs. 40 It serves as a strong TISU (AAGAUGG), distinguished from the Kozak consensus (RCCAUGG, where R is purine). 40
Modifications 1 and 2 contain modified C nucleotides (5-methylcytidine and 5-iodocytidine, respectively), which can be incorporated at the locations of the two “C”s preceding the start codon when a Kozak element is used, while modifications at these positions in a TISU element are not possible, because it lacks cytidine residues. The opposite trend, observed at the expression level of minimal UTR3 and minimal UTR7, highlights how specific modifications (5-methylcytidine in modification 1 and 5-iodocytidine in modification 2) can significantly affect the performance of regulatory elements (both UTRs and translation regulators).
The superior in vitro performance of minimal UTRs 3 and 7 was also confirmed in mice. For in vivo experiments, a luciferase expressing construct containing refUTR2 was used as a reference. 22 Similar to our in vitro data, minimal UTR7 outperformed minimal UTR3 and the reference benchmarks in spleen and liver, thereby indicating cell-type independence of its functionality. Exchanging the luciferase coding sequence by erythropoietin resulted in a similar translation efficiency in A549 and in HepG2 cells. Inferiority of the reference UTR (refUTR2), 22 compared to minimal UTR7 proves the advantageous concept of minimalistic UTRs in comparison to their longer counterparts (5′-UTR in refUTR2 is 113 nucleotides longer than minimal UTR7). Similar results were reported earlier, where the incorporation of minimal UTR2 was beneficial for mRNA stability and protein translation when compared to CYBA 5′-UTR 20 or natural 5′-UTR from hACE2. 23
Promising in vitro and in vivo results prompted us to benchmark our minimal UTR7 to additional short 5′-UTRs found in natural mRNAs. Orthopoxviruses contain a short homopolymeric Poly(A) tract at the 5′ end, which enables them to repress RNA decay 26 (refUTR3). A metagenomic study of the human genome revealed a 37-nucleotide conserved sequence (refUTR4) that promotes rapid expression in a Vaccinia virus (VACV)-based cytoplasmic expression system. 27 Since the first 26 nucleotides of the VACV element function as a promoter, and only the remaining 11 nucleotides function as an actual translation enhancer, we designed 2 more constructs: the first construct comprises all 37 VACV nucleotides (refUTR4), whereas the other 1 contains only the translation enhancer (11 nucleotides). Minimal UTR7 was either comparable or better than these two reference UTRs.
Based on the in vitro results obtained in A549 and HepG2 cells and the outcome of the in vivo experiments in mice, we propose minimal UTR3 and minimal UTR7 as the most promising candidates for use as 5′-UTRs for transcript therapy. Both minimal UTR sequences have no negative effects on RNA yield during in vitro transcription (data not shown) and the resulting protein expression is equal or even higher compared to naturally occurring 5′-UTRs. The latter function in a cell- and sequence-specific manner and also tend to be much longer than our synthetic short UTRs. Their small size makes them less susceptible to the kind of nucleotide modification, target sequence- and cell-type specific effects.
Although the mechanism for improved performance for these minimal synthetic 5′-UTRs has not been investigated, the lack of miRNA target sites in synthetic sequences may be one of the underlying factors resulting in reduced translational repression. Such mechanistic studies, although beyond the scope of the current study, are central to improve our essential understanding of rational mRNA design. Such artificial, minimalistic but highly efficient 5′-UTRs will reduce the mRNA amount needed to achieve the desired therapeutic effect, while reducing the costs of mRNA therapy.
Footnotes
Acknowledgments
Forschungsgemeinschaft via the Excellence Cluster “Nanosysyems Initiative Munich” is gratefully acknowledged.
Disclosure Statement
All authors except Z.T. are employees and C.P. and C.R. are shareholders of Ethris GmbH, a biotech company developing mRNA therapeutics.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.