
Abstract
Significant bone loss due to disease or traumatic injury requires surgical intervention to modulate the natural healing process of bone. The current bone grafting options, autografts and allografts, can potentially lead to donor site morbidity or mechanical failure over time. The use of tissue engineering is a promising alternative, but the mechanical stability and integrated vasculature in vivo still remains a major challenge. In this work, we introduce a scaffold that mimics the cylindrical structure of native cortical bone and provides biological cues without the addition of growth factors to promote stem cell differentiation along the angiogenic lineage. Biocompatibility of the scaffold was tested with two human endothelial cell types, human microvascular endothelial cells and human umbilical vein endothelial cells, and the angiogenic decellularized scaffold matrix led to a 78% increase in angiogenic protein secretion from human bone marrow-derived stem cells. Histological analysis of the scaffolds implanted subcutaneously in the dorsum of BALB/c mice confirmed vessel development and integration at 4 weeks with a decrease in fibrous capsule thickness up to 8 weeks. Future work will need to be performed to evaluate this novel scaffold as a vascularized tissue-engineered graft in a large animal model.
Impact Statement
Bone loss and skeletal deficiencies due to musculoskeletal diseases, traumatic injury, abnormal development, and cancer are major problems worldwide, frequently requiring surgical intervention. There has been a shift in paradigm to utilize tissue engineering applications. This novel bone technology has the potential to promote bone regeneration for large bone defects without the addition of growth factors and offers a unique architecture for cell attachment, proliferation, and differentiation. This scaffold serves as a tailored therapeutic for bone injuries and defects, leading to an increased quality of life by decreasing the risk of reoccurring surgeries and complications.
Introduction
Cortical bone is the dense bone structure that provides whole bone with its structural integrity. The higher compressive properties of cortical bone are due to the tightly packed arrangement of cylindrical subunits called osteons. Osteons are hollow structures composed of concentric collagen layers with a hollow channel. This channel, known as the Haversian canal, is where blood vessels and nerves are housed. Neovascularization sprouts out from the Haversian canal to the surrounding bone through the Volkmann's canals. 1 This intricate network is vital for bone remodeling and blood and nutrient transport. Research has shown that the development and maturation of bone are coupled with the process of new blood vessel formation, or angiogenesis.2,3 For example, nonhealing or damaged bone consists of large three-dimensional (3D) regions with locally hypoxic environments that lack initial vascularization. 4
Many tissue-engineered grafts that aim to regenerate bone lack the necessary vascularization for long-term graft integration and viability. Recent vascularization approaches in bone regeneration are largely aimed at promoting early vascularization by the delivery of proangiogenic growth factors, stem cells, or prevascularized tissue-engineered constructs. Although the utilization of growth factors in angiogenesis has proven to be successful in vitro and in vivo, there are insufficient data concerning the dose–response dynamics and the related cellular kinetics. 4 Stem cell-based therapies are a promising route for bone regeneration by increasing vascularized bone formation in vivo when cocultured with endothelial cells.4–6 Nonetheless, the timing at which specific cell types need to be added to the system is a sensitive parameter and plays a vital role in vessel development. Furthermore, without a stable structure, vascular networks developed in vivo can collapse or lead to reduced vascular permeability. 4
A scaffold that has the ability to promote differentiation of human bone marrow-derived mesenchymal stem cells (hMSCs) into vascular endothelial cells without the addition of exogenous growth factors would be key in addressing the issues seen with bone vascularization induced by soluble factors and coculture cell therapies. Therefore, the goal of this work is to develop and characterize a fabricated cylindrical osteonal bone structure that will promote bone development and the circumferential growth of endothelial cells into a vascular lumen. We hypothesize the lumen will maintain its proangiogenic properties after decellularization in vitro and in vivo. To achieve this, we evaluated the influence of a 3D scaffold structure and composition on endothelial cell proliferation and morphology. We also assessed the ability of the decellularized angiogenic scaffold matrix with mineral content to induce stem cell differentiation along the vascular endothelial and osteoblastic lineage in vitro and in vivo.
Materials and Methods
Poly-
The base electrospinning solutions were prepared by dissolving PLLA (7% w/v) in DCM and DMF with 10% gelatin and PDLA (22% w/v) in DMF and THF. PEO (10% w/v) solution was prepared by dissolving PEO (10% w/v) in deionized water and 100% EtOH. A 5 mL syringe with an 18-gauge blunt needle attachment was used to electrospin all the solutions. First, 5 mL of the PEO solution was electrospun onto an 8 cm diameter rotating aluminum mandrel (∼2000 rpm) at a working distance of 10 cm, pump rate 5 mL/h, with +15 kV/−11 kV voltages to create a PEO scaffold.
Next, a PLLA scaffold (no gelatin or PDLA) was electrospun onto a separate rotating mandrel with similar parameters. Seven millimeter wide strips of the PEO scaffold and 5 mm wide strips of the PLLA scaffold were then manually twisted together and loaded into a rotating device and rotated clockwise, while 3 mL of PEO was electrospun onto the twist (Fig. 1A). Then, a 3:1 ratio of PLLA and PDLA was electrospun onto the PEO-coated PEO/PLLA twist. All solutions were electrospun at a working distance of 10 cm and extrusion rate of 5 mL/h. The working distance varied between 7 and 15 cm depending on the polymer and environmental conditions. PDLA was used as the sintering polymer to bind the individual cortical scaffolds (Fig. 1B). After electrospinning, the scaffolds were placed into a desiccator overnight to remove any residual solvents. The scaffolds were then soaked in distilled water and the PEO/PLLA twist was removed (Fig. 1C) to fabricate osteon-like structures. The scaffolds were then crosslinked in 25 mM glutaraldehyde under vacuum. Scaffolds (n = 3) were dried overnight and sputter coated with gold and palladium for scanning electron microscopy (SEM) imaging and qualitative assessment.

Human microvascular endothelial cell culture
The purpose of this in vitro study was to test the biological response of the endothelial cells to the scaffolding material. Many researchers have reported the positive and negative effects of endothelial cell protein secretion on stem cell mineral deposition, but there are limited data on the effects of insoluble mineral content on endothelial cell activity.2,7–11 The information from this study was crucial in determining the effect of the mineral content and hydroxyapatite (HAp) in the trabecular scaffold on the development of an endothelial lumen in the cortical scaffold. We used immortalized human microvascular endothelial cells (HMECs) derived from human foreskin, and pulmonary and hepatic endothelium (gifted from Dr. Nicole Rylander's Laboratory at Virginia Tech, VA). HMECs are the primary cell type involved in the development of small vessels and undergo rapid morphological differentiation in vitro into capillary-like structures.12–14
The cells (passage 5) were maintained in MCDB 131 Medium (no glutamine) purchased from Life Technologies (Grand Island, NY), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). HMECs were seeded onto two-dimensional (2D) PDLA_PLLA/10%Gel scaffolds at 7000 cells per substrate. The experimental groups (n = 3) were nonmineralized scaffolds (PDLA_PLLA/10%Gel) and scaffolds that were mineralized for 7 days by static mineralization (PDLA_PLLA/10%Gel_Min). The negative control was a PDLA_PLLA/10%Gel scaffold. All substrates were adhered to the well plate with sterile silicone glue and sterilized with 70% ethanol and UV radiation. Substrates were preconditioned with sterile media for 24 h before cell seeding. Total protein was measured over 28 days using a Pierce™ BCA Protein Assay Kit. The scaffolds were also stained for collagen using Masson's trichrome stain and quantitatively assessed. The amount of collagen deposited by the HMECs on the scaffold was compared to determine the effect of mineral content on cellular behavior.
In vitro circumferential endothelial cell growth
The purpose of the study was to evaluate the effect of the scaffold's architecture on endothelial cell morphology. The cylindrical 3D cortical scaffold architecture was expected to promote circumferential cellular growth and lead to the development of an endothelial lumen. To test this hypothesis, human umbilical vein endothelial cells (HUVECs), gifted from the Sofou Laboratory (Rutgers University, NJ), were seeded on 2D electrospun PLLA/PDLA scaffolds. HUVECs are a robust cell line commonly used for in vitro studies as a model for vasculature physiology and vasculature development, and have been shown to have a positive impact on bone regeneration.7,11,15,16 The experimental groups were 2D (PDLA_PLLA/10%Gel_2D) and 3D cortical scaffolds previously mentioned (PDLA_PLLA/10%Gel_3D). The control group was HUVECs seeded directly onto tissue culture polystyrene (TCP) plastic well plates. All substrates were sterilized by 70% EtOH soak and UV radiation. The substrates were also preconditioned with media for 24 h before cell seeding. The HUVECs (passage 5) were maintained in an endothelial growth basal medium supplemented with 10% FBS, 2% human fibroblast growth factor, 0.02% hydrocortisone, 0.05% vascular endothelial growth factor (VEGF), 0.05% recombinant-3 insulin growth factor, 0.05% ascorbic acid, 0.05% human endothelial growth factor, 0.05% GA-100, and 0.05% heparin (media kit purchased from Lonza, Inc., Allendale, NJ). HUVECs were seeded inside the hollow canal of the 3D cortical scaffolds (10,000 cells per substrate) using a sterile 25-gauge needle and syringe.
Phalloidin and 4′,6-diamidino-2-phenylindole (DAPI) staining and fluorescent confocal microscopy were performed to evaluate the endothelial cellular growth over 28 days (n = 3). The scaffolds were also cryotome sectioned into 100 μm cross-sectional slice and imaged for morphological assessment, and then stained for collagen using PicroSirius Red Stain Kit (Polysciences, Inc.). The kit also stains yellow and green for collagen types I and III.
Decellularized proangiogenic cortical scaffold
Decellularized cortical scaffolds were created to promote the differentiation of hMSCs along the vascular endothelial lineage. The rationale for this study was to form an angiogenic endothelial matrix within the osteonic structure by allowing endothelial cells to secrete proangiogenic factors into their developed extracellular matrix (ECM), and then decellularize the matrix to remove the cells and intercellular content. Two-dimensional PDLA_PLLA/10%Gel scaffolds were used for this study. The cells (passage 5) were maintained in Ham's F-12K (Kaighn's) medium (purchased from Fisher Scientific Co., Suwanee, GA), supplemented with 10% FBS, 2% P/S, 100 μg/mL heparin, and 30 μg/mL endothelial cell growth supplement (all purchased from Sigma Aldrich). The scaffolds were attached to well plates with sterile silicone glue, sterilized by UV radiation and preconditioned with media for 24 h before cell seeding. The HUVECs were seeding on the scaffolds at 25,000 cells per scaffold.
PrestoBlue® and Chondrex assays were performed on day 14 for the indirect measure of cellular viability and to measure the presence of collagen/noncollagenous proteins, respectively (n = 3). The positive controls were HUVECs seeded directly onto plastic well plates and the negative controls were the PDLA_PLLA/10%Gel scaffold without cells. The substrates were then decellularized using an FDA-approved freeze-thaw method that has shown success in similar studies.17,18 Decellularized substrates (n = 3) were analyzed quantitatively for cellular viability and collagen deposition, and fluorescently stained for qualitative analysis. The remaining decellularized scaffolds were preconditioned in media overnight. hMSCs (passage 8) were maintained and expanded in alpha-modified minimum essential media (α
Scaffold preparation for in vivo study
The scaffold groups for the subcutaneous in vivo study are outlined in Table 1. The groups included nonmineralized scaffolds with and without a prevascularized lumen, NonMin − Lumen and Min + Lumen, and mineralized scaffolds with a prevascularized lumen, Min + Lumen (n = 6 per group per time point). The rationale for these groups was to assess the biocompatibility, osteogenic and angiogenic potential of the complete scaffold in vivo (NonMin − Lumen vs. NonMin + Lumen and Min + Lumen), evaluate the effect of mineralization and HAp on vasculature development in vivo (NonMin + Lumen vs. Min + Lumen), and investigate the effect of the prevascularized lumen on vasculature integration (NonMin − Lumen vs. NonMin + Lumen) at 4 and 8 weeks. Porous electrospun PDLA_PLLA/10%Gel scaffolds were wrapped around a single fabricated osteon as shown in Figure 1D. The entire assembly was wrapped with a nanofibrous PDLA_PLLA/10%Gel scaffold and sintered at 54°C for 45 min to yield a stable scaffold 5 mm diameter by 10 mm in length. The scaffolds in the Min_HAp groups had 10% HAp in the trabecular scaffold and were mineralized by static mineralization for 3 h at 5 V in simulated body fluid, yielding ∼20% mineralization.
21
The scaffolds (n = 6 per time points) were attached to well plates with sterile silicone glue, sterilized with 70% EtOH and UV light exposure, and conditioned with endothelial media for 24 h. HUVECs were seeded inside the cortical scaffold at 25,000 cells per scaffold. The cells (passage 5) were maintained in Ham's F-12K medium (purchased from Fisher Scientific Co.), supplemented with 10% FBS, 2% P/S, 100 μg/mL heparin, and 30 μg/mL endothelial cell growth supplement (all purchased from Sigma Aldrich). On day 14, the scaffolds were decellularized by the freeze-thaw procedure as previously discussed. Before scaffold implantation, hMSCs at passage 1 (obtained from Texas A&M Health Science Center) were seeded throughout the 3D scaffolds at 100,000 cells per scaffold. The scaffolds with hMSCs were maintained for 7 days in media (α
Outline of Scaffold Groups for Subcutaneous Scaffold Implantation Study
The scaffold groups were nonmineralized scaffolds with and without a prevascularized lumen, NonMin − Lumen and Min+Lumen, and mineralized scaffolds with a prevascularized lumen, Min + Lumen. N = 12 per group.
hMSC, human bone marrow-derived mesenchymal stem cell; HAp, hydroxyapatite.
Subcutaneous scaffold implantation
All surgeries were performed in compliance with Rutgers Institutional Animal Care and Use of Committee. The goal of the in vivo subcutaneous study was to characterize the osteoblastic and vascular phenotype expression in response to the 3D prevascularized osteoinductive scaffold implanted into the dorsum of BALB/c mice.
Female BALB/c mice (6 weeks and ∼18–21 g) were used for this study based on their common use for scaffold implantation compatibility studies.22–26 On the day of surgery, the mice were anesthetized by mask induction of isoflurane and oxygen 1–5% or to effect on a heating pad at 37°C and a small area on the dorsum was shaved. Local anesthetic was administered subcutaneously at the incision site and the surgical area was sterilized. The scaffolds were implanted in the dorsum of the mice using a poke incision method. To minimize the number of mice used for study, one NonMin + Lumen scaffold and one Min + Lumen scaffold were implanted in two separate subcutaneous pockets of the same mouse (Table 1). The control scaffold, NonMin − Lumen, was implanted one per mouse (Table 1). The skin around the incision was then pulled together and closed with sterile adhesive tissue glue. Animals were removed from anesthesia and placed into their recovery cage after surgery.
Mice (six per group time point) were sacrificed at 4 and 8 weeks postoperatively by CO2 inhalation. The scaffolds were extracted and placed in 70% EtOH for up to 24 h followed by formalin. Scaffolds were sliced horizontally to yield circular cross-sections, and were then prepared for histology at the Digital Imaging and Histology Core laboratory at the Rutgers-NJMS Cancer Center in Newark, NJ. The sections were stained with hematoxylin and eosin (H&E) to qualitatively assess cellular infiltration and tissue development. Masson's trichrome and Alizarin red stains were also applied to visualize collagen and mineral content. The samples extracted at 8 weeks were also stained with PicroSirius Red stain for collagen types I and III.
The scaffolds were mechanically tested under compression with an Instron 5869 at a crosshead speed of 10%/min. All samples were vacuum soaked in phosphate-buffered saline for 30 min before testing and the fibrous capsule was removed. Compressive moduli, yield stresses, and ultimate compressive stresses were computed and compared between groups to determine change in scaffold properties due to degradation and/or tissue integration. Toughness was measured using the trapezoidal method to measure the area under the stress–strain curve. The thickness of the fibrous capsules at varying location was assessed macroscopically with light microscopy and ImageJ analysis, and validated using calipers.
Statistical analysis
Statistical analysis was performed using Kaleida Graph software. All data collected from the in vitro studies were subjected to either a t-test or ANOVA with posthoc analysis (Tukey test) to determine the statistical significance of differences between groups, p < 0.05. For the in vivo studies, statistical significance was determined at a p-value of 0.05, a standard deviation of 10%, and a sample size (n = 6) that yields a power of 0.98.
Results
In vitro study I results: biocompatibility testing using HMECs
A Pierce bicinchoninic acid protein assay was performed on the HMECs seeded on the 2D PDLA_PLLA/10%Gel mineralized and nonmineralized scaffolds (Fig. 2) to assess total protein. HMECs seeded on the nonmineralized and mineralized scaffold expressed higher levels of total protein than the HMECs seeded directly onto plastic well plates up to day 21. On day 21, the cells seeded on the scaffolds had significantly greater total protein level than the cells seeded directly onto the plastic well plates. On day 28, there was a decrease in total protein observed with all the groups.

Total protein analysis of HMECs on the control (HMECs_plastic well plate), nonmineralized (PDLA_PLLA/10%Gel) and mineralized (PDLA_PLLA/10%Gel_Min) scaffolds. Statistical analysis: “#” denotes ANOVA Tukey Test (posthoc) p < 0.05 in comparison to the control scaffold on day 21. ANOVA, analysis of variance; HMEC, human microvascular endothelial cell; PDLA, poly-
In vitro study II results: developing a circumferential endothelial lumen
Endothelial growth in response to scaffold architecture and fiber orientation was evaluated using HUVECs. Figure 3 is a collection of the actin/DAPI fluorescent confocal z-stack images of the HUVECs seeded directly onto the plastic well plates and on a 2D scaffold (PDLA_PLLA/10%Gel_2D). Figure 3A and C are the HUVECs on plastic well plates on day 7 and 14. The HUVECs attached to the plastic well plates and elongated in random directions. Figure 3B and D are the HUVECs seeded on the 2D scaffold. The HUVEC cytoskeleton morphology was similar to the cells seeded on the plastic well plates with random directional growth. Cross-sectional slices of the 3D scaffold stained with phalloidin and DAPI also demonstrated the ability of the cells to attach to the inner lumen (Fig. 4A). On day 14, the PDLA_PLLA/10%Gel cortical scaffold (osteon) was also imaged using SEM (Fig. 4B). The rough texture of the scaffolds suggests the development of an endothelial lumen within the cortical structure. The lumen was further characterized using PicroSirius Red stain for collagen types I and III. Collagenous structures result in a bright red stain, whereas collagen type I and type III will stain yellow and green, respectively. An orange color indicates collagen type I or a mixture of different collagen types outside of collagen type I and III. An overlap in the collagen type I (red) and collagen type III (green) stains results in a gray color. The orange color of the scaffold (Fig. 4C) is a mix of the denatured collagen in the scaffold due to the addition of the 10% bovine gelatin (red) and collagen type I (yellow).
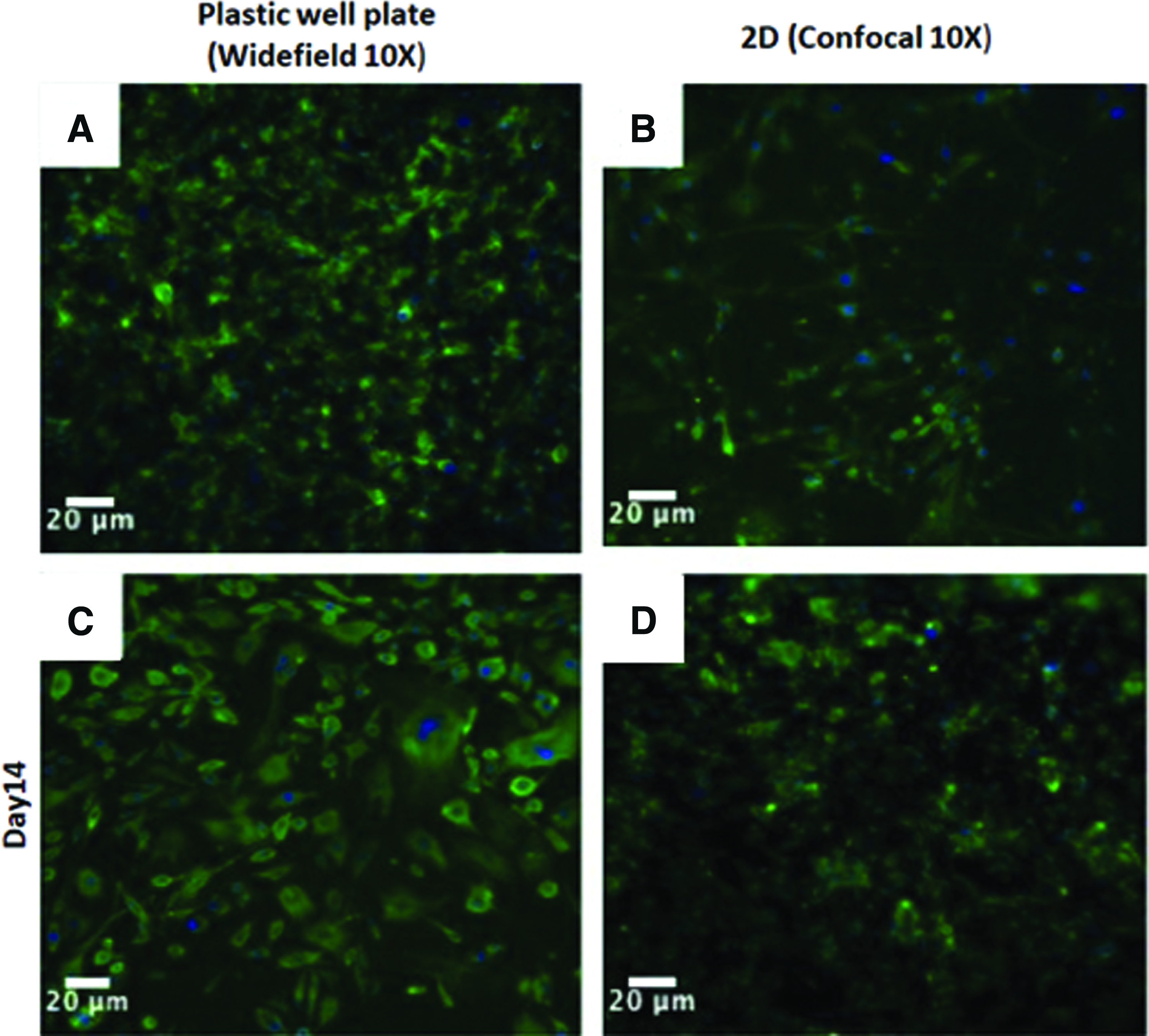
Fluorescent confocal actin/DAPI images of HUVECs on the plastic well plate

In vitro study III: developing and characterizing a decellularized cortical scaffold
In this study, PDLA_PLLA/10%Gel scaffolds were seeded with HUVECs and then decellularized using a freeze-thaw method on day 14. Figure 4 shows the presence of HUVECs nuclei stained with DAPI before (Fig. 4D) and after (Fig. 4E) decellularization. Qualitatively, there is a significant decrease in cellular viability after decellularization. To further validate the freeze-thaw decellularization method, metabolic activity and collagen deposition of the HUVECs were evaluated before and after decellularization using a PrestoBlue® and Chondrex assay. There is an 86% decrease in cellular viability due to the decellularization method (Fig. 5, left). Even though the cellular viability decreased, 96% of the collagenous/noncollagenous matrix was maintained postdecellularization (Fig. 5, right). The preservation of this protein matrix is essential for promoting the differentiation of the hMSCs into vascular endothelial cells. These data support the efficacy of our decellularization technique.

(Left) Metabolic activity (indirect measurement of cellular viability) of HUVECs on PDLA_PLLA/10%Gel scaffolds decreased by 86% after decellularization. (Right) Collagenous/noncollagenous protein matrix secreted by HUVECs was maintained by 96% after decellularization. Statistical analysis: # denotes one-way t-test, p < 0.05.
Next, we evaluated the cellular response of hMSCs to the preserved decellularized PDLA_PLLA/10%Gel scaffold on day 7. The hMSCs seeded on the decellularized scaffold had a statistically significant higher level of metabolic activity on day 7 in comparison to the hMSCs seeded on the control scaffold (Fig. 6, left). The control scaffold was a scaffold that was not previously seeded with cells. These data suggest that the hMSCs preferred the maintained endothelial matrix deposited by the HUVECs. Figure 6 (right) also shows that the hMSCs secreted similar levels of collagenous/noncollagenous proteins in response to the material as the hMSCs on the control scaffold, suggesting the matrix did not inhibit the hMSCs innate function.

(Left) Metabolic activity of hMSCs on the decellularized PDLA_PLLA/10%Gel scaffolds was significantly higher than on the control nondecellularized scaffold on day 7. (Right) Collagenous/noncollagenous protein matrix secreted by the hMSCs on the decellularized PDLA_PLLA/10%Gel scaffold was similar to the hMSCs on the control nondecellularized scaffold on day 7. Statistical analysis: # denotes one-way t-test, p < 0.05. hMSC, human bone marrow-derived mesenchymal stem cell.
The protein expression of VEGF was evaluated using IHC (Fig. 7). The green fluorescence stain indicates the presence of intercellular VEGF in the ECM. The scaffolds were counterstained with DAPI to visualize the hMSC nuclei. The negative control, decellularized scaffold without cells, was also imaged and compared to confirm the VEGF expression was from the hMSCs and not residual VEGF on the scaffold (Fig. 7B). This image qualitatively confirms the ability of the decellularized cortical scaffold to promote stem cell differentiation into vascular endothelial cells without the addition of any growth factors. The protein secretion was also measured quantitatively by ELISA for human VEGF (Fig. 7C). In this study, HUVECs seeded directly onto plastic well plates were added as a comparison group. There was a significantly higher concentration of VEGF secreted from the hMSCs seeded on the decellularized scaffold (hMSCs_PDLA_PLLA/10%Gel_Decell) than the hMSCs seeded on the control scaffold, (hMSCs_PDLA_PLLA/10%Gel). As expected, the secretion of VEGF from the HUVECs was significantly greater than the hMSCs seeded on the decellularized scaffold. It is important to note this was the amount of VEGF measured in the cell supernatant on day 7. An increase in secreted VEGF as the hMSCs continued to differentiate in response to the proangiogenic matrix would be expected at later time points.

In vivo scaffold implantation
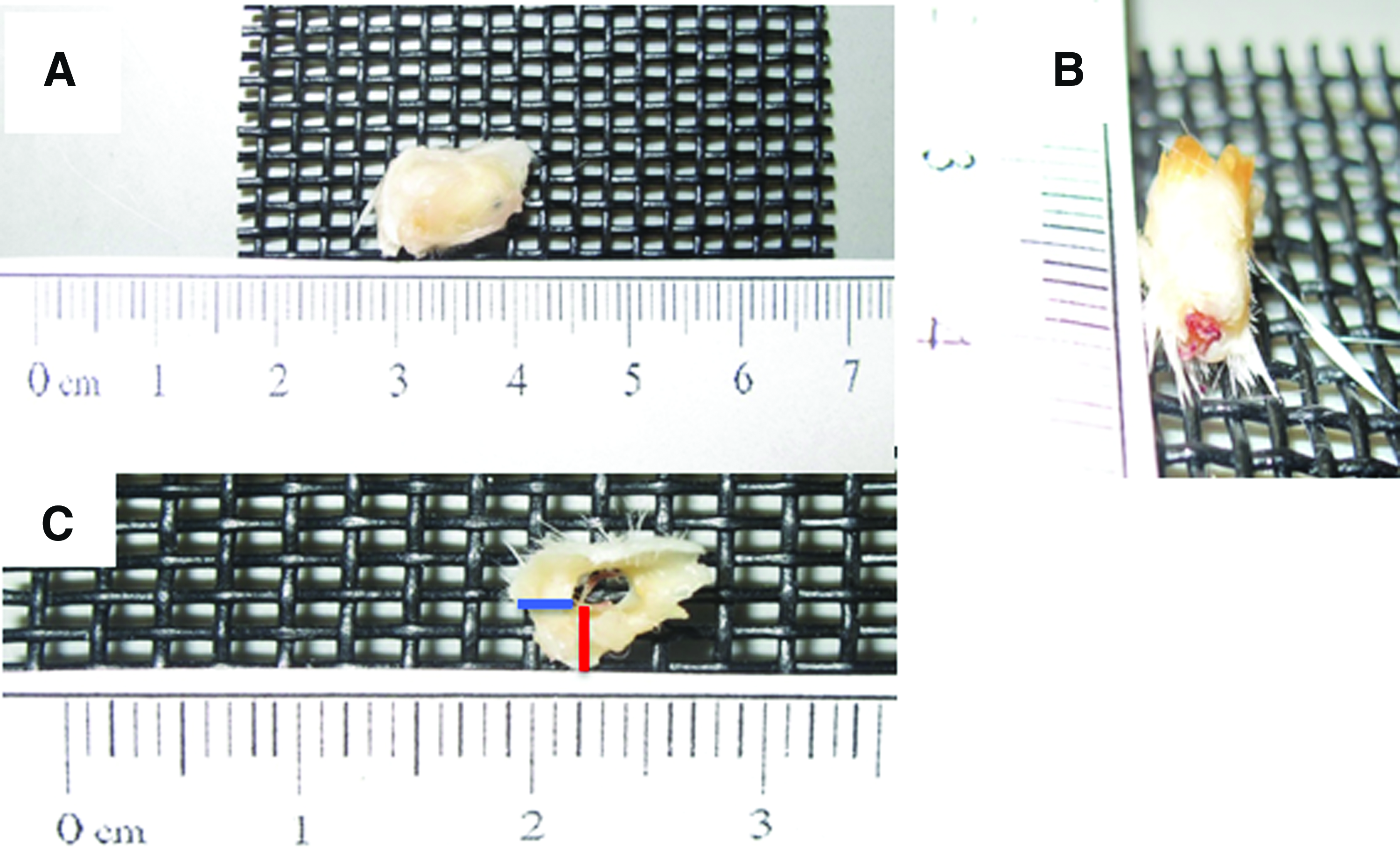
The scaffolds were extracted 4 and 8 weeks after implantation and qualitatively assessed. Macroscopically, all the scaffolds were surrounded by a fibrous capsule at 4 weeks, as shown in Figure 8A. Furthermore, the Min + Lumen scaffolds appeared to be integrated with new vasculature (Fig. 8B). Fibrous capsule thickness (FCT) was measured at the bottom (inferior side) and at the middle (Fig. 8C). The FCT at the bottom was thicker than at the middle at 4 weeks for all the scaffold groups (Fig. 9). The FCT at the bottom decreased at 8 weeks for all the scaffold groups in comparison to the 4 weeks. In addition, the fibrous capsule at the bottom was significantly thicker for the NonMin − Lumen scaffolds than the scaffold with the lumen. Histology was performed at 4 and 8 weeks to qualitatively assess cellular infiltration, scaffold integration, and vasculature within or surrounding the scaffold (Figs. 10 and 11).

FCT measurements (units = μm) taken at the middle and bottom of the scaffold over 8 weeks demonstrated an increase in FCT at the bottom of the scaffold. The values are shown as mean ± standard. The FCT also decreased at the bottom over 8 weeks. FCTs decreased in the thickness over 8 weeks at the bottom location. The thickness of the fibrous capsules surrounding the NonMin − Lumen scaffold was significantly greater than the scaffold groups with the lumen. Statistical analysis: * denotes ANOVA Tukey Test (posthoc) p < 0.05. FCT, fibrous capsule thickness.
As seen in all the figures, there was infiltration of a heterogeneous population of cells within the scaffolds (“S”). Structured vasculature was also evident in some sections of the scaffolds as seen in Figure 10D–F at 20 × , as indicated by “BV” for blood vessels filled with erythrocytes. The structure of the blood vessel is stable at 8 weeks, but appears to be acellular (Fig. 11F). Integration of the scaffold with the surrounding tissue is evident in Figures 10D–F and 11D–G. Masson's trichrome analysis of the excised scaffold at 4 weeks further confirmed the presence of scaffold integration and vasculature (Fig. 10H). The circular layers of the scaffold are apparent in Figure 10G with naive collagen (dark pink) between the layers. The separation of the scaffold layers may have occurred during the sectioning process for histology. Infiltration of collagen and stable blood vessels within a scaffold section is seen in Figure 10H. Mineral content was detected in the Min + Lumen-excised scaffolds at 4 weeks and at 8 weeks (Figs. 10K and 11B). This indicates that the mineralization process was successful at mineralizing the full thickness and that the mineral remains within the scaffold structure in vivo. The mineral content from mineralization and the addition of HAp was maintained up to 8 weeks as seen in the slices stained with Alizarin Red (Fig. 11A–C). The presence of mineral content detected in the nonmineralized samples can be contributed to the HAp in the scaffolds (Figs. 10L and 11C). Collagen was evaluated at 8 weeks by PicroSirius Red stain (Fig. 11H–J). The red stain, indicating collagen type I, is present surrounding the scaffold and between the scaffold layers.

Cross-sectional histological images of the scaffolds, “S”, excised at 4 weeks show the presence of blood vessels (BV), cellular infiltration, and scaffold integration in all the scaffold groups.
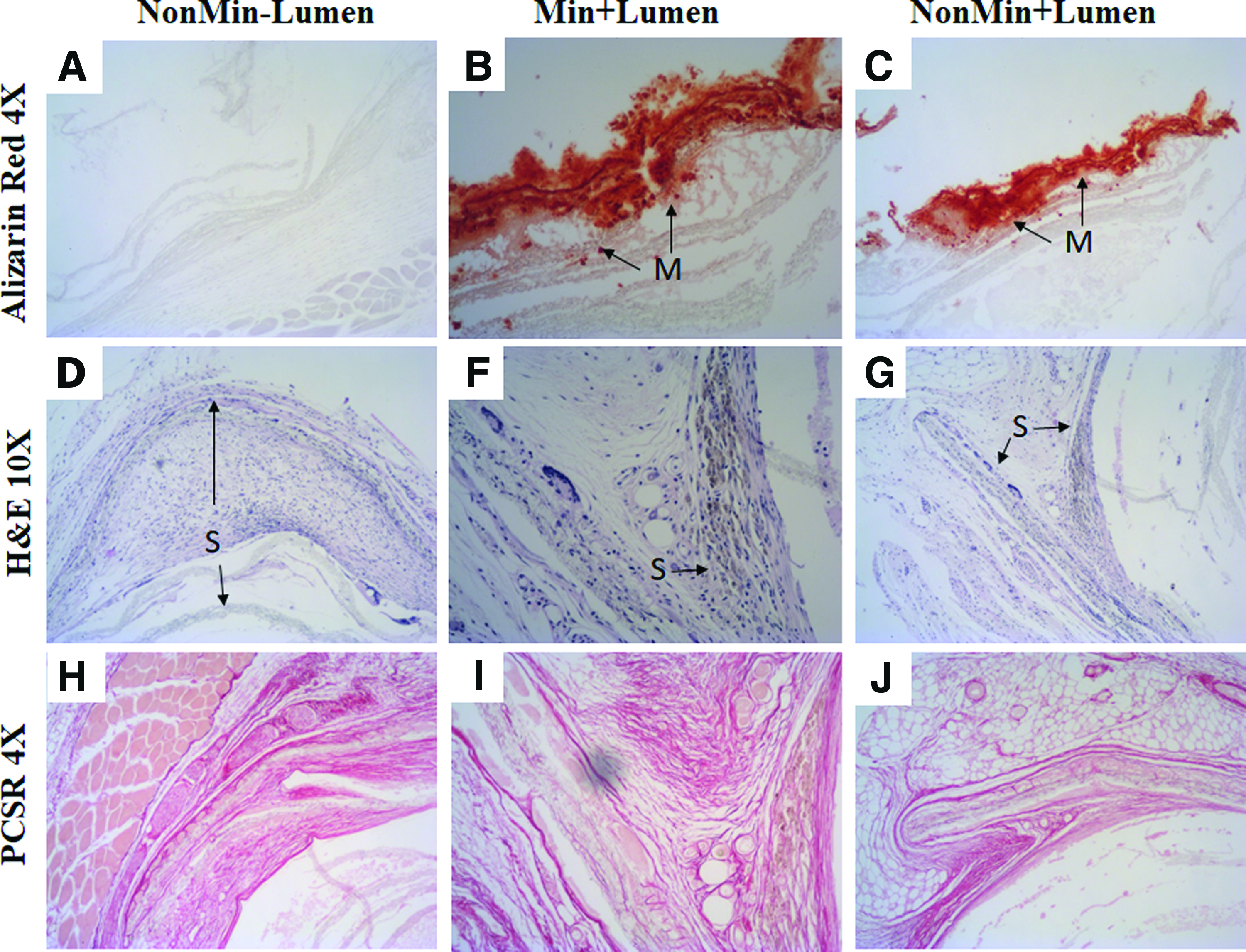
Histological images of NonMin − Lumen, Min + Lumen, and NonMin + Lumen scaffolds excised at 8 weeks.
Mechanical testing of excised scaffolds
The excised scaffolds were subjected to compressive testing at 1 mm/min using an Instron 5869. Scaffolds that were not implanted were also tested as negative controls (NegControl_Min and NegControl_NonMin). The calculated yield strengths, ultimate compressive strengths, Young's moduli, and toughness values are shown in Figure 12. There were no significant differences in the ultimate compressive strength, yield strength, and toughness of the excised experimental scaffolds, 4wks_NonMin_NoHAp and 4wks_Min_HAp, in comparison to their corresponding negative controls, NegControl_Min and NegControl_NonMin, respectively (data not shown). Furthermore, the mechanical properties of the experimental scaffolds seeded with hMSCs before implantation had significantly increased in strength and toughness in comparison to the unseeded positive control scaffolds, 4wks_PosControl. The mechanical properties of the excised scaffolds were also analyzed at 8 weeks and compared to the mechanics of the scaffold excised at 4 weeks. All of the scaffolds extracted at 8 weeks have a significantly greater Young's modulus, ultimate compressive strength, and increased toughness compared to the 4-week explants within the same group (Fig. 12). There was no statistical significance difference in yield strength between the 4- and 8-week Min + Lumen scaffolds or in toughness between the 4- and 8-week NonMin − Lumen scaffolds. The mechanical properties of all the groups were also assessed at each time point. Min + Lumen has statistically greater ultimate compressive strength and toughness than the NonMin − Lumen and NonMin + Lumen scaffolds at 4 weeks and there was no statistically significant difference found between the groups at 8 weeks.

Mechanical properties of scaffold excised at 8 weeks postoperation compared to the scaffolds excised at 4 weeks. There was a statistically significant increase in Young's modulus for all the groups from 4 to 8 weeks. Statistical analysis: * denotes t-test comparing the 4 and 8 week samples within each group, p < 0.05, # denotes ANOVA posthoc comparing the groups at each time point, p < 0.05.
Discussion
The results presented validate the development of a decellularized cortical bone scaffold that promotes endothelial growth. SEM images of the fabricated osteon confirmed that the scaffold mimics the hollow cylindrical architecture of native osteons. The total protein data suggest that the PDLA_PLLA/10%Gel nonmineralized and mineralized cortical scaffolds are a favorable environment for endothelial cellular attachment and proliferation, and ECM deposition. This was confirmed with two human endothelial cell lines, HMECs and HUVECs. There was a decrease in total protein measured on day 28 with all the groups. Future studies are necessary to elucidate this confounding result because, although decrease in cellular proliferation is common on substrate at later time points due to cellular contact inhibition, this should not have a direct effect on total protein content.
The concentric electrospun fibers also guided cellular growth circumferentially and led to organized collagen type I formation within the canal of the cortical scaffold. Collagen type I is an ECM protein that is essential for many ECM-dependent cell functions, such as cell migration and proliferation. Moreover, collagen I is a major protein present in the endothelial wall of blood vessels.27–29
The freeze-thaw method used to decellularize the scaffold was validated by an 86% decrease in cellular viability. The HUVECs deposited a collagenous/noncollagenous protein matrix that was maintained, up to 96%, postdecellularization. Stem cells seeded on the collagenous matrix exhibited cellular morphology indicative of endothelial lumens in 2D. Furthermore, the maintained proangiogenic collagenous matrix promoted differentiation of hMSCs, which was confirmed by the presence of VEGF, an early marker for angiogenesis. VEGF has been shown to enhance angiogenesis and bone regeneration, and have a direct effect on osteogenesis by recruiting and promoting osteoblast and osteoclast activity.4,30,31 The prevascularized cortical scaffold combines the advantages of cell-based therapies, proangiogenic factors, and a stable biomimetic construct to promote early vascularization in vitro.
For in vivo evaluation, scaffolds with and without the prevascularized lumen or mineral content were implanted into the dorsum of BALB/c mice and assessed for cellular infiltration and tissue development at 4 and 8 weeks. Qualitatively, the scaffolds were encapsulated in fibrous tissue ∼50 μm thick, and the hollow lumen was infiltrated with vascularized tissue. There was a decrease in the FCT and cellular infiltration from 4 to 8 weeks after implantation (Fig. 9). H&E also confirmed significant cellular infiltration and the presence of blood vessels with erythrocytes at 4 weeks. There appeared to be a direct relationship between the fibrous capsule measured at the bottom of the scaffold and presence of blood vessels. Fibrous encapsulation is indicative of the early wound healing response elicited by the normal foreign body reaction at the tissue-implant interface.32–34 Our findings were contrary to other studies that confirmed an inverse relationship between FCT and blood vessel ingrowth due to the blocking of vascularization and integration.35,36 During the foreign body response, monocytes and macrophages are activated, which leads to collagen matrix deposition and angiogenesis.35–39 Similar studies have concluded that when macrophages are activated by a foreign material surface, they produce a greater number of vessels and angiogenic factors such as MMP-9, TIMP-1, and TIMP-2.38–41 Moreover, cell infiltration is a critical process in promoting tissue integration between implanted material and host tissue.42–44 The presence of a thick fibrous capsule could potentially be inhibiting these processes that allow for successful implant and host integration.
Mechanical testing (1 mm/min) was performed to determine the impact of in vivo conditions on the structural integrity of the scaffolds. The results show no significant structural changes due to rapid degradation in vivo. The scaffolds seeded with hMSCs exhibited significantly greater strength and toughness than the control scaffolds, which could be due to early tissue development and integration initiated by the hMSCs in the scaffolds. The ability to maintain the mineral content and strength in vivo was demonstrated by the statistically significant increase in compressive strength, yield strength, and toughness of the mineralized scaffolds with HAp in comparison to the nonmineralized scaffolds without HAp. Furthermore, there was an increase in mechanical properties exhibited by the 8-week explants in comparison to the scaffolds excised at 4 weeks. This increase in toughness could be the result of matrix integration or tissue formation throughout the porous scaffold. All scaffold groups had similar geometric areas with fibrous capsulation. Therefore, the increase in mechanical properties was not due to differences in area, but a difference in the forces the scaffolds were able to withstand. The scaffolds exhibited ductile behavior and there was no single absolute point of failure for majority of the scaffolds. This slow barreling effect causes a change in area as the load is applied, leading to variations in the computed stresses and strains of samples within the same group. 45
A limitation of this study is that the fabricated osteons are about 10 times greater in diameter than native human osteons. Therefore, future work is necessary to develop and evaluate smaller fabricated osteons with the same diameter to length ratio as demonstrated in this study.
Future studies will also elucidate the bone healing potential of this prevascularized scaffold combined with previous load-bearing scaffolds in a functional long bone defect animal model.
Conclusion
The findings presented highlight the material and biological characterization of a novel prevascularized scaffold for large bone defects. The features of this scaffold address the current issues seen with bioengineered bone replacements by mimicking the architecture of native osteons and promoting stem cell differentiation without the use of growth factors. The data confirm the scaffold's biocompatibility, bioactivity, and durability in vitro and in vivo over time. Future studies will evaluate the regenerative effectiveness of this cortical scaffold combined with our trabecular scaffold in a functional in vivo large animal model.
Footnotes
Acknowledgment
B.T. was supported by the National Institutes of Health under Ruth L. Kirchstein National Research Service Award T32 GM8339 (Biotechnology training grant) from the NIGMS.
Disclosure Statement
No competing financial interests exist.