
Abstract
Current reconstruction methods of the laryngotracheal segment fail to replace the complex functions of the human larynx. Bioengineering approaches to reconstruction have been limited by the complex tissue compartmentation of the larynx. We attempted to overcome this limitation by bioengineering laryngeal grafts from decellularized canine laryngeal scaffolds recellularized with human primary cells under one uniform culture medium condition. First, we developed laryngeal scaffolds which were generated by detergent perfusion-decellularization over 9 days and preserved their glycosaminoglycan content and biomechanical properties of a native larynx. After subcutaneous implantations in rats for 14 days, the scaffolds did not elicit a CD3 lymphocyte response. We then developed a uniform culture medium that strengthened the endothelial barrier over 5 days after an initial growth phase. Simultaneously, this culture medium supported airway epithelial cell and skeletal myoblast growth while maintaining their full differentiation and maturation potential. We then applied the uniform culture medium composition to whole laryngeal scaffolds seeded with endothelial cells from both carotid arteries and external jugular veins and generated reendothelialized arterial and venous vascular beds. Under the same culture medium, we bioengineered epithelial monolayers onto laryngeal mucosa and repopulated intrinsic laryngeal muscle. We were then able to demonstrate early muscle formation in an intramuscular transplantation model in immunodeficient mice. We supported formation of three humanized laryngeal tissue compartments under one uniform culture condition, possibly a key factor in developing complex, multicellular, ready-to-transplant tissue grafts.
Impact Statement
For patients undergoing laryngectomy, no reconstruction methods are available to restore the complex functions of the human larynx. The first promising preclinical results have been achieved with the use of biological scaffolds fabricated from decellularized tissue. However, the complexity of laryngeal tissue composition remains a hurdle to create functional viable grafts, since previously each cell type requires tailored culture conditions. In this study, we report the de novo formation of three humanized laryngeal tissue compartments under one uniform culture condition, a possible keystone in creating vital composite tissue grafts for laryngeal regeneration.
Introduction
Hemi-
Currently, for patients undergoing laryngectomy there are no reconstruction methods available that restore the complex functions of the human larynx. Transplantation of native laryngeal allografts has been reported in detail, in two cases worldwide.4,5 However, the need for life-long immune suppression makes it a therapeutically irrelevant approach for most patients.
The laryngeal architecture provides the foundation for swallowing, breathing, and phonation, and therefore, laryngeal reconstruction based on the native cartilage framework may be a suitable approach. Generation of acellular laryngeal scaffolds with intact tissue architecture can be achieved by removal of the cellular components, yielding an acellular extracellular matrix (ECM) scaffold of low immunogenicity. 6 To generate functional laryngeal tissue, scaffolds need to be revitalized, ideally with recipient derived cells. However, the complexity of laryngeal tissue composition remains a hurdle to bioengineering approaches, since previously each cell type requires tailored culture conditions.
In this study, we report the ex vivo generation of laryngeal vasculature, epithelial mucosa, and muscle-like tissue from human primary cells based on decellularized canine laryngeal scaffolds, under a single, uniform culture condition.
Materials and Methods
All procedures were performed in accordance with the National Institutes of Health guidelines for care and use of laboratory animals, and with approval of the Institutional Animal Care and Use Committee at the Massachusetts General Hospital.
Isolation and perfusion decellularization of whole larynxes
Harvest of cadaveric larynxes was performed from beagles (24–36 months, n = 11) postmortem. In brief, a midline neck incision was performed, fascia and platysma were split, and the common carotid arteries (CCAs) were located and cannulated with 16G cannulas (McMaster-Carr), followed by ligation of the internal and external carotid arteries.
Next, the proximal external jugular veins (EJs) were cannulated with 16G cannulas. The larynx was separated from surrounding tissue and explanted en bloc with the esophagus attached. We next tied off the esophagus cranially and caudally to the larynx and flushed the vasculature via the CCAs with heparinized saline for blood removal and to identify vascular leaks. Detected leaks were ligated with 3–0 silk suture. Larynxes were then mounted in a custom-built glass chamber and decellularization was initiated by perfusion from both CCAs in a single pass manner. A constant pressure of 80 mmHg was applied using a feedback control unit to regulate flow rate. Sequentially, larynxes were perfused with phosphate-buffered saline (PBS) for 30 min, 1% v/w sodium dodecyl sulfate (SDS) for 5 days, deionized water for 2 h, 1% Triton-X 100 for 6 h, and finally washed with PBS for 4 days to allow the removal of the detergents from the matrix. All solutions were sterile and closely monitored for bacterial and fungal growth. All reagents were purchased from Sigma-Aldrich.
Computer tomography angiography
Computer tomography scans were acquired through a dual-source CT scanner (SOMATOM Definition Flash; Siemens Health care). The decellularized whole larynx was mounted in a customized polytetrafluoroethylene chamber and perfused with equal parts Visipaque contrast agent and 0.9% saline at a rate of 5 mL/min from both CCAs. Scanning was performed using a multiphase volume dynamic protocol, and a three dimensional (3D) image was rendered from still images using MediCapture (MediCapture, Inc.).
Biochemical assays
Biochemical analysis of native and decellularized canine larynxes was performed to determine the presence of glycosaminoglycan (GAG) and double-stranded DNA (dsDNA). Samples were divided in three groups: cartilage (n = 3), muscle (n = 3), and mucosa/submucosa (n = 3). GAGs analysis was performed using the Blyscan Assay Kit (Biocolor, UK). Dry samples were homogenized in 1 mL of papain buffer containing 7 μL/mL of papain, digested at 65°C, and incubated with the dye reagent. After washing, the samples were read in a 96-well plate, at 656 nm and concentrations were calculated in reference to the relative standard curve.
For dsDNA quantification, tissue samples were digested overnight at 55°C in 310 μL of a solution containing 10 mM Tris (pH = 8.0), 5 mM ethylenediaminetetraacetic acid (EDTA), 0.1 M NaCl, 1% SDS, and 650 μg/mL Proteinase-K (Life Technologies, Carlsbad, CA). dsDNA was then isolated using a phenol/chloroform extraction and alcohol precipitation, and quantified using the Quant-iT PicoGreen dsDNA Kit (Life Technologies) following the manufacturer's protocols. Fluorescence was read at 480/520 nm (excitation/emission) and concentrations were calculated in relationship to the standard curve. All biochemical data are presented as the total mass of the component extracted normalized to tissue sample wet weight and normalized to dry weight (dWT) (lyophilized tissue).
Biomechanical testing
For biomechanical testing, we compared the first tracheal ring of decellularized larynxes to native tracheal rings. Rings were cut into 1 cm long strips for uniaxial tensile testing, as previously described. Trachea strips were loaded into the grips of the Instron 5544 Uniaxial Tensile Tester Machine (Instron, Inc., Norwood, MA) and were pulled to failure at a strain rate of 1.00 mm/min. During the tensile testing, trachea displacement was visualized using a Fastec HiSpec4 high-speed camera (Fastec Imaging, Inc., San Diego, CA), with videos taken at 120 frames per second. Videos acquired were used to generate strain maps of the trachea. Average strain over the area was calculated using the High-Density Mapping software algorithm. 7 High-Density Mapping measures subpixel displacement based upon a random light intensity object. The average strain was calculated and then visualized into a heat map showing the localization of strain during the tensile testing.
Histological stains
Tissue samples were fixed overnight in 4% paraformaldehyde (PFA; Sigma-Aldrich), paraffin-embedded, and sectioned at 5–7 μm thickness. Histological assessment was performed using standard protocols for the following stains: hematoxylin and eosin (H&E) (Sigma-Aldrich), Mason's Trichrome (American Master Tech.), Mason Pentachrome (American Master Tech.), Safranin-O/Fast green stain, and Toluidine blue stain. For immunofluorescent analysis, we performed antigen retrieval on deparaffinized sections using a citrate-based antigen unmasking solution (H-3300; Vector Laboratories).
The sections were blocked with 1% bovine serum albumin (BSA) (Sigma-Aldrich) and incubated overnight at 4°C with the following primary antibodies: Rabbit (rbt) anti-LamAC (ab108595; Abcam), rat anti-KI67 (14-5698; eBioscience), rbt anti-Collagen IV (ab6586; Abcam), ms anti-CD3 (ab16669; Abcam), ms anti-CD68 (MCA341GA; AbD Serotec), rbt anti-CD163 (bs-2527R; Bioss, Inc.), rbt anti-CD80 (bs-2211R; Bioss, Inc.), goat anti-E-Cadherin (AF648SP; R&D Systems), rbt anti-CK5 (ab52635; Abcam), ms anti-P63 (CM163A; Biocare Medical), ms anti-acetyl K40 (ab24610; Abcam), rbt anti-CCSP (ab40873; Abcam), ms anti-MU5AC (ab3649; Abcam), ms anti-Fox J1 (14-9965-82; eBioscience), ms anti-CD31 (M082301-2; DAKO), rbt anti-VE-CAD, ms anti-myosin heavy chain (MYH) (MF20; DSHB), and ms anti-embryonic MYH (F1.652; DSHB).
After primary antibody binding, the sections were washed and incubated for 40 min at room temperature with the appropriate donkey-raised secondary antibodies, conjugated with a 488, 546, or 647 fluorochromes (Life Technologies; AlexaFluor series) at a dilution of 1:400 in BSA. For apoptosis detection in combination with immunofluorescence staining, slides were first incubated with primary and secondary antibody, as described above, followed by apoptosis detection with Fluorometric TUNEL System (Promega).
Immunologic response to decellularized xenografts
Donor tissue preparation: Segments of proximal tracheal rings were isolated from native or perfusion-decellularized laryngotracheal constructs and divided in pieces of similar size. Recipients: nine male Sprague Dawley rats (275–300 g) were randomly assigned to three groups: NAT, n = 3; DEC, n = 3; and SHAM, n = 3. Animals were anesthetized using an induction chamber and maintained with 1–3%. A subcutaneous 1.5 cm2 pocket was created at the midline of the back of all animals. The donor tissue was placed in the pocket and the incision was closed with 4–0 silk. Animals of the SHAM group did not receive a tissue implant. For blood count analysis, whole blood samples were collected from the tail vein with 1 mL lithium heparin tubes (Greiner Bio-One GmbH) on day 5 after implantation.
On day 14, animals were sacrificed, the implants were carefully removed, leaving equal amount of fascia and skin attached, and fixed in PFA overnight. To assess cell infiltration, serial sections of the embedded tissue specimens were cut at 5 μm thickness. The tissue sections were immunohistochemically stained for CD3, CD68, CD80, and CD163. Five representative fields per specimen were imaged at 20 × magnification and used for analysis. A CellProfiler image analysis pipeline was used to quantify CD3+ and CD68+ cells, as well as macrophage subpopulation M1/M2. 8 In total, more than 60,000 cells were analyzed across three groups containing 15 images each.
Human primary cell isolation, characterization, and expansion
Human basal cells (hBCs) were isolated from donated human lungs, not suitable for transplantation and provided by the New England Organ Bank. Tracheal segments were freed from surrounding tissue and transferred into minimum essential medium (MEM) containing 140 mg pronase (Sigma-Aldrich) and 10 mg DNase (Sigma-Aldrich) per 100 mL and incubated at 4°C for 24 to 48 h under constant agitation. The harvested cell suspension was transferred to polypropylene tubes, centrifuged at 300 g for 5 min, resuspended in MEM containing 20% fetal bovine serum (FBS), and repeated. After the third centrifugation step, the resulting cell suspension was resuspended in small airway epithelial growth medium (SAGM; Lonza) supplemented with Rho-associated protein kinase inhibitor (10 μM) and plated on noncoated tissue culture plastic to enrich the epithelial cell population.
Cells were then transferred to previously 10 μg/mL collagen IV-coated flasks and culture medium was changed to SAGM from the second day. Between passage 1 and 2, differential plating was used for further epithelial cell enrichment. At passage 2, cells were characterized using immunofluorescence staining of P63 and E-cadherin and used for further experiments.
Primary human myoblasts (hMBs) were isolated from anonymized skeletal muscle biopsies and fragmented into pieces below 0.5 mm and digested twice in 20 mL of 0.005% trypsin 0.1% Collagenase D. The so obtained cell suspension was preplated on tissue culture dishes for 1 h to enrich for the myoblast fraction over the highly adherent fibroblasts. The supernatant was then seeded for 1 week on Matrigel-coated plates and characterized via fluorescence-activated cell sorting (FACS) to validate cell identity. For FACS analysis, cells were trypsinized, blocked, and resuspended in PBS 1% FBS 0.05 mM EDTA with the following antibodies: CD56 (BioLegend 5.1H11), CD31 (BioLegend WM59), CD146 (BioLegend P1H12). Cells were cultured in the medium referred to as myoblast growth medium (MGM) as follows 9 : megacell 5% FBS, 1% P/S, 1% non-essential amino acid (NEAA), 0.1 mM 2-mercaptoethanol, and 5 ng basic fibroblast growth factor (bFGF).
Human umbilical vein endothelial cells (HUVECs; Lonza) were maintained in 0.1% gelatin-coated (Sigma-Aldrich) flasks with complete EGM-2 (Lonza).
Cell culture tests
HUVECs (100 k/6 well, gelatin coated), hBCs (100 k/6 well, collagen IV coated), and hMBs (100 k/6 well) were plated using conventional EGM-2, SAGM, and MGM, respectively. After complete cell attachment, cells were rinsed with PBS three times, and the culture medium was switched to the following three conditions per cell type: EGM-2, SAGM, and MGM (n = 3 per condition and cell type). Media changes were performed every other day. On day 5, HUVECs were methanol fixed and stained for PECAM-1 and VE-cadherin; hBCs were methanol fixed and stained for P63, E-cadherin; bright field images of myoblasts were taken.
Evaluation of one uniform culture condition: multicell growth medium
For multicellular growth medium (MCGM) testing on HUVECs, the following steps were taken: HUVECs were plated onto gelatin-coated 12-wells in EGM-2 containing 10% FBS (EGM-2-10%). On day 3, confluent HUVEC monolayers were rinsed with PBS three times and the culture medium was replaced by MCGM: SAGM+medium 199 (Gibco) (1:1) supplemented with 5% FBS, 0.5% insulin-transferrin-selenium (Gibco), ascorbic acid (25 μg/mL; Stemcell Technologies), recombinant human vascular endothelial growth factor (20 ng/mL), (bFGF) (20 ng/mL), and 1% antibiotic/antimycotic solution. Cells were maintained in MCGM for 5 days with medium changes performed every other day. HUVECs cultured in EGM-2-10% for 8 days served as controls. On day 8, cells were methanol fixed and stained for PECAM-1, VE-Cadherin.
hBCs were plated onto collagen IV-coated 12-wells in MCGM and maintained for 5 days with alternate day media changes. On day 5, cells were methanol fixed, and stained for E-cadherin, CK5, and P63. hBCs cultured in SAGM for 8 days served as control. For hBC differentiation, cells were plated onto 0.4 μm transwell inserts coated with collagen IV and maintained in submerged culture with MCGM for 5 days. Media was replaced with PneumaCult™-ALI medium (Stemcell Technologies) in the basal chamber only, and maintained for 28 days at Air-Liquid interface (ALI). 10 Media changes were performed every other day. At the end of ALI culture, inserts were PFA fixed, paraffin embedded, and stained for the following markers: E-cadherin, CK5, P63, ACT TUB, FOX J1, MUC 5AC, and CCP.
hMBs were plated onto 12-wells in MCGM and maintained for 5 days with media changes every other day. On day 5, hMBs were rinsed in PBS three times, the culture medium was replaced by myoblast differentiation medium (DMEM, 2% Horse serum, 1% P/A, 1% NEAA), and cells were differentiated for 14 days with medium changes every other day. hMBs were cultured in conventional MGM for 8 days, followed by conventional myoblast differentiation medium as controls. On day 14 of differentiation, cells were methanol fixed and stained for MYC.
Cell proliferation assay
Quantitative assessment of HUVEC proliferation under EGM-2-10% and MCGM was performed as follows: HUVECs were plated onto gelatin-coated six-wells at a density of 10 k/well and maintained for 5 days in EGM-2, EGM-2-10%, and MCGM with medium changes performed on alternate days. At day 5, cells from three wells per condition were counted using an automated system. Cell numbers are normalized to counted cells in EGM-2.
Cell differentiation assay
For quantitative assessment of hMB differentiation, the percentage of MYH-positive cells in groups treated with MCGM or conventional MGM (n = 6 per condition) for 5 days, followed by 14 days of differentiation that was calculated using ImageJ Software (NIH)
Dextran transwell permeability assay
We measured dextran permeability across the HUVEC monolayer using 3.0 μm pore Transwell inserts (12 well format). HUVECs were plated on gelatin-coated inserts at a density of 75,000 cells/well and cultured and submerged in EGM-2-10% for 3 days, followed by 5 days of MCGM. HUVECs cultured in EGM-2-10% for 8 days served as controls (n = 3 per condition). Completing the culture, 50 μL of fluorescein isothiocyanate-conjugated 500-kDa dextran (5 mg/mL; Sigma-Aldrich) was added to the upper chamber and the media fluorescence of the lower chamber was measured after 30 min of incubation at 37°C, 5% CO2 using SpectraMax Microplate Reader (Molecular Devices, Sunnycale, CA) set at 485 nm (excitation)/538 nm (emission). Fluorescence readings were normalized to dextran permeability in transwell inserts without HUVECs.
Whole larynx endothelial cell seeding
Decellularized laryngeal scaffolds (n = 3) were primed by perfusion from both the right and left CCA and right and left EJ at 2 mL/min with 100 mL of EGM-2-10% for 24 h.
Eighty million HUVECs were suspended in 400 mL EGM-2-10% and seeded simultaneously through the four vascular accesses (100 mL each) at 100 mmHg (CCAs) and 60 mmHg (EJs). After 2 h of static culture to allow cell attachment, the culture medium was filtered to remove nonadherent cells and the laryngeal scaffold was perfused from all four vascular accesses at 1 mL/min medium for 2 h, followed by 2 mL/min perfusion rate for 3 days. Subsequently, on day 3, the vascular ports of left and right EJ were removed (open to chamber) and the scaffold was perfused from arterial side only, using 200 mL of MCGM. Medium changes were performed on alternate days. On day 8 of overall culture, larynxes were removed from the seeding chamber and PFA fixed by perfusion.
In vitro reconstruction of epithelialized mucosa
For reepithelialization of acellular laryngeal muscle constructs, we first isolated the luminal mucosa from the decellularized scaffolds using a 6 mm punch biopsy. Mucosa was primed in MCGM for 24 h and then seeded with hBCs at a density of 700,000 cells/cm2. Two hours after seeding, cell suspension was replaced with new MCGM, and constructs were cultured for 5 days with culture medium changes performed daily. On day 5, constructs were PFA fixed for histology (n = 6).
In vitro reconstruction of laryngeal muscle
Laryngeal intrinsic muscle ECM was isolated from perfusion-decellularized larynxes and primed in MCGM for 24 h. For seeding, 2 million hMBs were resuspended in 100 μL MCGM and injected along the fiber direction into the scaffold, using a 29G insulin syringe (Exelint). Repopulated constructs were cultured in MCGM for 24 h under static condition. From day 1, static culture was replaced by dynamic condition using an orbital shaker (Troemner, NJ), and medium was replaced daily. On day 5 of culture, constructs were either PFA fixed for histology or used for in vivo experiments.
3D viability and proliferation assay
To assess the viability of in vitro generated muscle constructs and increase in cellularity, a modified Resazurin (Presto blue; Invitrogen) reduction assay was performed on day 1 and 5. 11 Muscle constructs were transferred to 12-wells and incubated in 1 mL MCGM containing 50 μL (1:20) resazurin for 60 min at 37° under constant agitation. The fluorescence intensity of the metabolized medium was read using a SpectraMax Microplate Reader (Molecular Devices) at 544 nm (ex), 590 nm (em). Unseeded scaffolds served as controls.
In vivo maturation of regenerated laryngeal muscle
In vitro regenerated muscle constructs (n = 6) were transplanted into 6 weeks old, female nude mice. Under general anesthesia, a 1.5 cm skin incision was performed on the anterolateral side of the lower hind limb and the fascia of the tibialis anterior muscle was carefully lifted from the underlying muscle. The regenerated muscle construct was then placed between fascia and muscle, thereby matching the fiber direction of the native muscle. 12 The skin incision was closed with 6-0 silk (Ethicon) and the animals were weaned from anesthesia and single housed. Animals receiving regenerated muscle implants showed no signs of limping or reduced physical activity on day 1 after implantation. Bioengineered muscle constructs were explanted from sacrificed animals on day 14 after implantation, fixed with PFA and embedded in paraffin for histology.
Statistical tests
Student's t-tests and one-way analysis of variance followed by Bonferroni multiple comparison tests were performed using PRISM 6.
Results
Creation of native laryngeal scaffolds
We isolated whole canine larynxes postmortem and proceeded with the cannulation of the vasculature. The larynxes were then mounted in an ad-hoc designed perfusion chamber. We performed pressure controlled perfusion-decellularization through both CCAs using an adapted detergent-based protocol. 13 Over 5 days of 1% SDS perfusion, followed by 6 h deionized water and 6 h 1% Triton X-100, the composite tissue gradually lost opacity as decellularization occurred. Perfusion decellularization followed by clearing of detergents was completed on day 9 (Fig. 1A). Macroscopically, the vocal fold turned transparent and on gross examination appeared to be edematous. A thin, yellow mucosal layer covered the luminal surface of the larynx. Epiglottis, cricoid, and thyroid cartilage retained their shape (Fig. 1B). We confirmed vascular continuity after decellularization with a Computer Tomography Angiography (CTA) (Fig. 1C).

Creation of whole laryngeal scaffolds from native canine larynxes.
4′,6-diamidino-2-phenylindole (DAPI) staining confirmed successful removal of the nuclei from the laryngeal mucosa, exocrine glands, perichondrium, and the outer layer of the cartilage (Fig. 1D, left panel). These findings were supported by a substantial decrease in dsDNA found in mucosa/submucosa, muscle, and hyaline cartilage of decellularized (DEC) compared to native (NAT) specimens (dsDNA ng/mg dWT; NAT vs. DEC: Mucosa: 654.6 ± 227.7 vs. 115.8 ± 38.8, p = 0.0156; Muscle: 966.3 ± 316 vs. 282.7 ± 92.32, p = 0.0228; Cartilage: 141.1 ± 20.01 vs. 53.93 ± 21.32, p = 0.0067) (Fig. 1D, right panel). H&E stain of an axial laryngeal section showed preservation of laryngeal microarchitecture (Fig. 1E). The ultrastructure of the decellularized submucosa, vasculature, muscle, and cartilage was assessed by Russell-Movat pentachrome stain. The observed tissue compartments remained intact and showed loss of cellular components (Fig. 1F).
Biochemical and biomechanical characterization of the laryngeal scaffold
GAG occupies the interfibrillar space of the cartilage ECM provides cartilage with its osmotic properties, which are critical to its ability to resist compressive loads. 14 GAGs were preserved and equally distributed throughout the cartilage, as assessed qualitatively by Safranin-O/Fast green and Toluidine blue stain (Fig. 1G). Analysis of the GAG content did not reveal a significant difference between native and decellularized cartilage (GAG content in μg/mg: NAT 376.4 ± 89.36 vs. DEC 291.5 ± 85.11; p = 0.122, Fig. 1G). To determine the impact of decellularization on the biomechanical properties of cartilage, we performed strain testing. Decellularized cartilage underwent nonsignificant changes in strain over the area of tested tissue (Strain over strain map: NAT 0.228 ± 0.0519 vs. DEC 0.2742 ± 0.0194; p = 0.2488, Fig. 1H).
Immunological response to laryngeal scaffolds
To evaluate the immunogenicity of the decellularized scaffolds, we subcutaneously implanted native and decellularized canine tracheal tissue into immunocompetent rats for 14 days (Fig. 2A). SHAM-operated animals did not receive implants and served as controls. On day 5, white blood cell count showed a significant increase of monocytes in animals receiving native tissue (Monocytes in 103/μL: 0.93 ± 0.25 NAT vs. 0.46 ± 0.11 DEC vs. SHAM 0.5 ± 0.26; NAT vs. DEC, p < 0.005, NAT vs. SHAM, p < 0.005, DEC vs. SHAM, p = ns). Lymphocyte, granulocyte, and total white blood count did not significantly differ between groups (Fig. 2B).
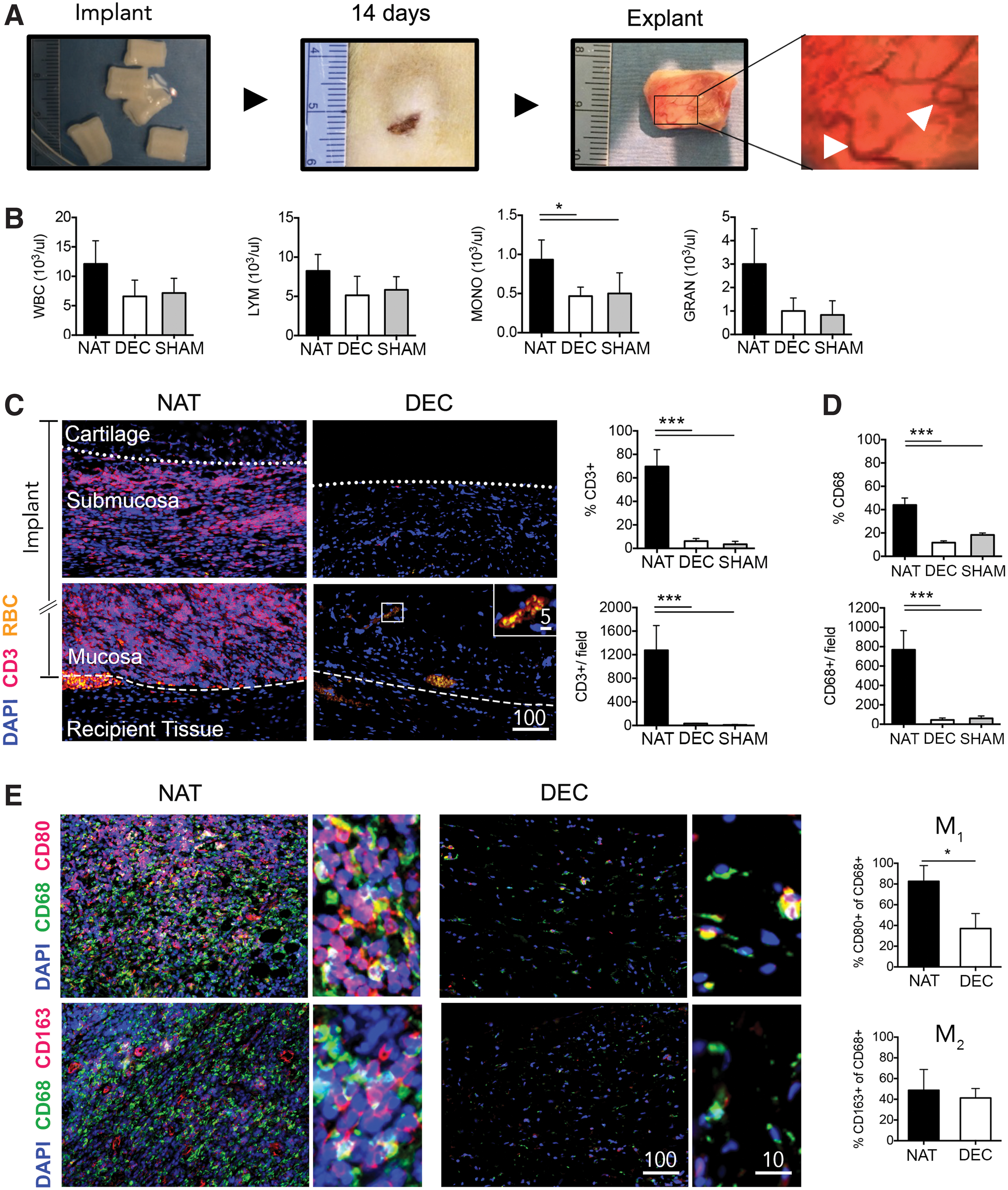
Immunogenic response to laryngeal scaffolds.
On day 14, we removed composite tissue implants and evaluated for cellular infiltration by histology. Both DEC and NAT implants had induced angiogenesis, likely due to a foreign body inflammatory response (Fig. 2A). Consistent with macroscopic findings, histology revealed blood perfused vasculature at the border zone between recipient tissue and implant (Fig. 2C left panel). In addition we saw neovascularization in the mucosa of DEC implants. NAT implants showed diffuse infiltration of CD3+ lymphocytes in the mucosa and submucosa, whereas we observed limited lymphocyte migration to the cartilage (Fig. 2D left panel). In contrast, we detected minimal infiltration of CD3+ cells in the DEC mucosa and submucosa, and absence of CD3+ cells in the cartilage (Fig. 2C left panel).
We analyzed over 60,000 cells, using a CellProfiler image analysis pipeline, for expression of CD3 (Fig. 2C right panel) and found 69.7% ± 14.3% to be positive in NAT, 6.1% ± 2.2% in DEC, and 3.5% ± 2.4% in SHAM (NAT vs. DEC: p < 0.0001; NAT vs. SHAM: p < 0.0001; DEC vs. SHAM, p = 0.533). The total number of CD3+ cells per field were 1276 ± 418 NAT, 30 ± 4 DEC, and 10 ± 6 SHAM (NAT vs. DEC: p < 0.0001; NAT vs. SHAM: p < 0.0001; DEC vs. SHAM, p = 0.9030). We next assessed macrophage (CD68+) infiltration (Fig. 2D). Within implants, 43.9% ± 6% of cells were CD68+ in NAT, 11.7% ± 1.5% in DEC, and 18.3% ± 1.6% in SHAM (NAT vs. DEC: p < 0.0001; NAT vs. SHAM: p < 0.0001; DEC vs. SHAM, p = 0.1950). The total number of CD68+ cells per field were 768 ± 197 in NAT, 44 ± 19 in DEC, and 60 ± 25 in SHAM (NAT vs. DEC: p < 0.0001; NAT vs. SHAM: p < 0.0001; DEC vs. SHAM, p = 0.911).
To assess the macrophage response to implanted decellularized tissue in more detail, we looked for macrophage type 1 (M1: CD68+ CD80+) and type 2 (M2: CD68+ CD163+) polarization (Fig. 2F). While M1 predominance is associated with dense tissue deposition and scarring, M2 results in a more constructive type remodeling. 8 Immunohistological stains revealed a high density of CD68+ cells in NAT specimen with a dominance of M1 macrophages over M2 (Fig. 2E, left panel). M1 macrophages contributed to 82.6% ± 15.1% of CD68+ cells in NAT, compared to 37% ± 15.4% in DEC (p = 0.0196). M2 macrophages contributed to 48.7% ± 20% of CD68+ cells in NAT, compared to 41.2% ± 9.1% in DEC (p = 0.5906) (Fig. 2E, right panel). Overall, native tissue showed a clear M1 predominance, whereas no distinct macrophage phenotype was found in decellularized tissue implants.
Development of a uniform culture condition for bioengineering of laryngeal grafts
To repopulate scaffolds, we chose three primary human cell types. Human umbilical endothelial cells (HUVECs) were used for reendothelialization of whole scaffolds based on a previously described two-step seeding protocol. 15 We obtained human epithelial cells from fresh cadaveric tracheas for reepithelialization of the laryngeal mucosal ECM (Fig. 3A). Epithelial-oriented culture yielded a uniform human epithelial basal cell (hBC) population, characterized by immunofluorescence stain for E-cadherin and P63 (Fig. 3B). In addition, we isolated primary hMBs from discarded muscle biopsies through a mechanical/enzymatic method (Fig. 3A). We subsequently cultured the cells at low density in myoblasts media (Fig. 3C, left panel) and characterized the cell population by three-marker flow cytometry. Isolated cells were positive for CD56 and negative for CD31 and CD45, consistently with the expected cell phenotype (Fig. 3B, right panel).
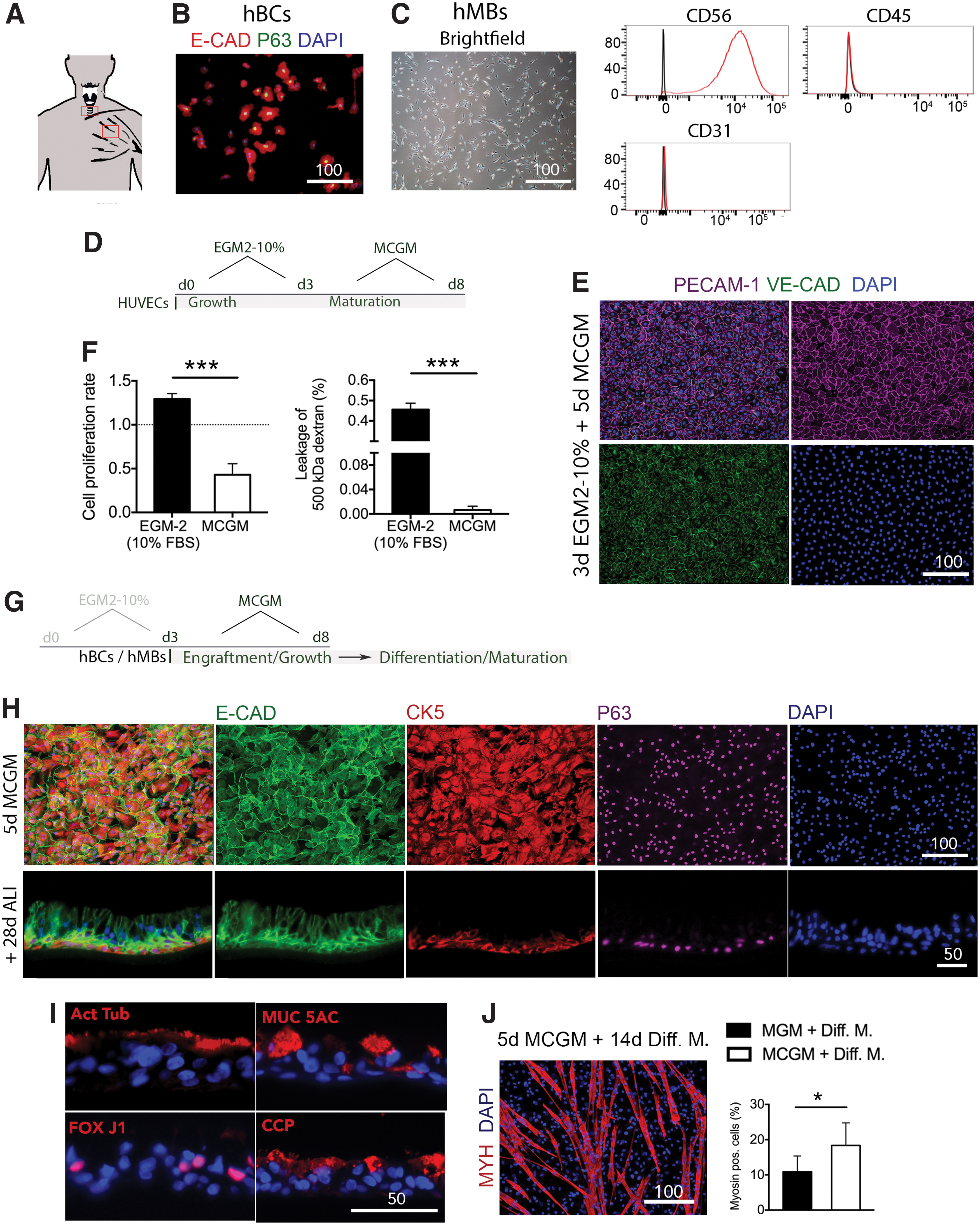
Development of a uniform culture condition for bioengineering of laryngeal grafts.
To allow for repopulation of laryngeal scaffolds with primary human cells, we explored a cell culture medium composition that maintains and matures preformed endothelial structures, while supporting engraftment and growth of hBCs and hMBs for laryngeal tissue formation.
In a pilot study, we tested conventional endothelial, epithelial, and myoblast cell culture media on all three cell types (Supplementary Fig. S1). As expected, neither of the media conditions tested could maintain cell phenotype and/or cell survival of all three different cell types. We chose a composition based on conventional airway epithelial medium, since hBCs were most difficult to maintain in absence of its medium and modified it to support endothelial and myoblast culture. This medium is referred to as MCGM.
For the two-step endothelial cell seeding process, we first cultured HUVECs in conventional endothelial growth medium containing 10% fresh-frozen plasma (EGM-2-10%) until confluency was reached on day 3 (Growth phase), followed by 5 days of MCGM (Maturation phase) (Fig. 3D). After contact inhibition, monolayers could be maintained without cell detachment, loss of cell phenotype and with strong cell–cell adhesion, as revealed by immunofluorescence stain for PECAM-1 and VE-cadherin (Fig. 3D). HUVECs cultured in EGM-2-10% for 8 days served as a control (Supplementary Fig. S2A). In addition, MCGM lead to a significant reduction in cell proliferation, compared to EGM-2-10%. (Cell proliferation rate normalized to EGM-2: EGM-2-10% 1.29 ± 0.04 vs. MCGM 0.43 ± 0.073; p = 0.0004; Fig. 3F, left panel) and confluent monolayers formed a stronger intercellular barrier in MCGM, compared to EGM-2-10% (500 kDa dextran leakage [%]: MCGM 0.0065 ± 0.0062 vs. EGM-2-10% 0.46 ± 0.031; p < 0.001) (Fig. 3D, lower panel). 16
We then evaluated the effect of MCGM on hBCs over 5 days of culture (Fig. 3G). hBCs formed a continuous E-cadherin monolayer and cells homogeneously expressed the basal cell marker P63 and cytokeratin 5 (Fig. 3H, upper panel). hBCs cultured in conventional epithelial medium for 5 days served as control (Supplementary Fig. S2B). To confirm maintenance of differentiation potential, basal cells were cultured in MCGM for 5 days and then differentiated under air/liquid interface culture for 28 days. Cells formed a pseudostratified epithelium composed of a basal cell layer (Fig. 3H, lower panel). In addition, ciliated cells (Acetyl. alpha tubulin+, FoxJ1+), goblet cells (Mucine 5AC+), and club cells (CCP+) were present in the newly formed epithelium (Fig. 3I).
Myoblasts cultured in MCGM versus conventional myoblast growth medium, followed by 14 days of differentiation showed no phenotypical difference (Fig. 3J and Supplementary Fig. S2C). We evaluated myoblast differentiation as the percentage of nuclei contributing to MYH+ fibers, resulting in a significantly higher percentage in MCGM than in conventional myoblast growth medium (MYH+ fibers in %: MCGM 18.36 ± 6.4 vs. conventional medium 10.9 ± 4.49, p = 0.041). Once an optimal uniform culture condition for all three primary cell types was identified, we applied it to bioengineered laryngeal tissue.
Bioengineering of laryngeal grafts (under one uniform culture condition)
The first step in generating vital laryngeal tissue was the development of an endothelialized vascular network within the whole laryngeal scaffold. With this aim, the larynxes were mounted in a custom designed biochamber (Fig. 4A) and seeded with a total of 80 million HUVECs. We have shown that HUVEC seeding from both arterial and venous side significantly benefits endothelial cell coverage. 15 Thus, we chose to deliver cells, simultaneously, through four vascular accesses: the right and left CCAs and the right and left EJs (Fig. 4A, right panel). We perfusion cultured whole larynxes for 3 days in EGM-2-10% to promote HUVEC proliferation. We then applied MCGM culture to endothelialized laryngeal scaffolds to further mature the endothelium and complete the endothelialization process. Immunofluorescence imaging revealed a high density of PECAM-1+ vascular structures within the native ECM (Collagen IV) scaffold (Fig. 4B). We observed endothelial lining in both arterial and venous vascular beds, suggesting successful delivery of cells from arterial and venous vascular accesses. Notably, endothelial delivery reached vascular segments of different calibers (Fig. 4C). This indicates endothelial lining of the hierarchical vascular network.

Bioengineering of laryngeal grafts.
For reepithelialization, we seeded about 700,000 cells/cm2 onto the decellularized mucosa of laryngeal grafts and cultured them for tissue formation in MCGM for 5 days. Immunohistologic characterization showed that hBCs were engrafted to the luminal surface of collagen IV-positive basal membrane of the mucosal ECM, preferentially forming a continuous monolayer (Fig. 4D). The basal cell layer remained positive for E-cadherin and CK5 under MCGM culture (Fig. 4E). Based on the results from the previous cell culture experiments, the newly formed laryngeal mucosa can likely be differentiated into mature pseudostratified epithelium.
To repopulate the laryngeal intrinsic muscular ECM, we seeded a total of 2 million myoblasts per 100 μL injection volume into laryngeal intrinsic muscular ECM and cultured the tissue for 5 days in MCGM. To trace cell viability, in cell culture medium, we measured fluorescence of metabolized resazurin, a surrogate for cell viability and growth. 11 Resazurin metabolism revealed a significant increase in fluorescence over 5 days, indicating cell proliferation (day 1: 1190 ± 246.7 vs. day 5: 2235 ± 580.3; p = 0.0165) (Fig. 4F). Immunohistologically, human lamin A/C (LamAC+) identified human cells densely repopulating the scaffolds on day 5 of culture (Fig. 4G).
In vivo maturation of bioengineered laryngeal grafts
To investigate whether active differentiation to muscle-like tissue can be achieved in vivo, we implanted the in vitro engineered laryngeal intrinsic muscle grafts into nude mice. We chose the subfascial space of the anterior tibialis muscle for implantation because it provides mechanical stimulation and is easily accessible2,12 (Fig. 5A).
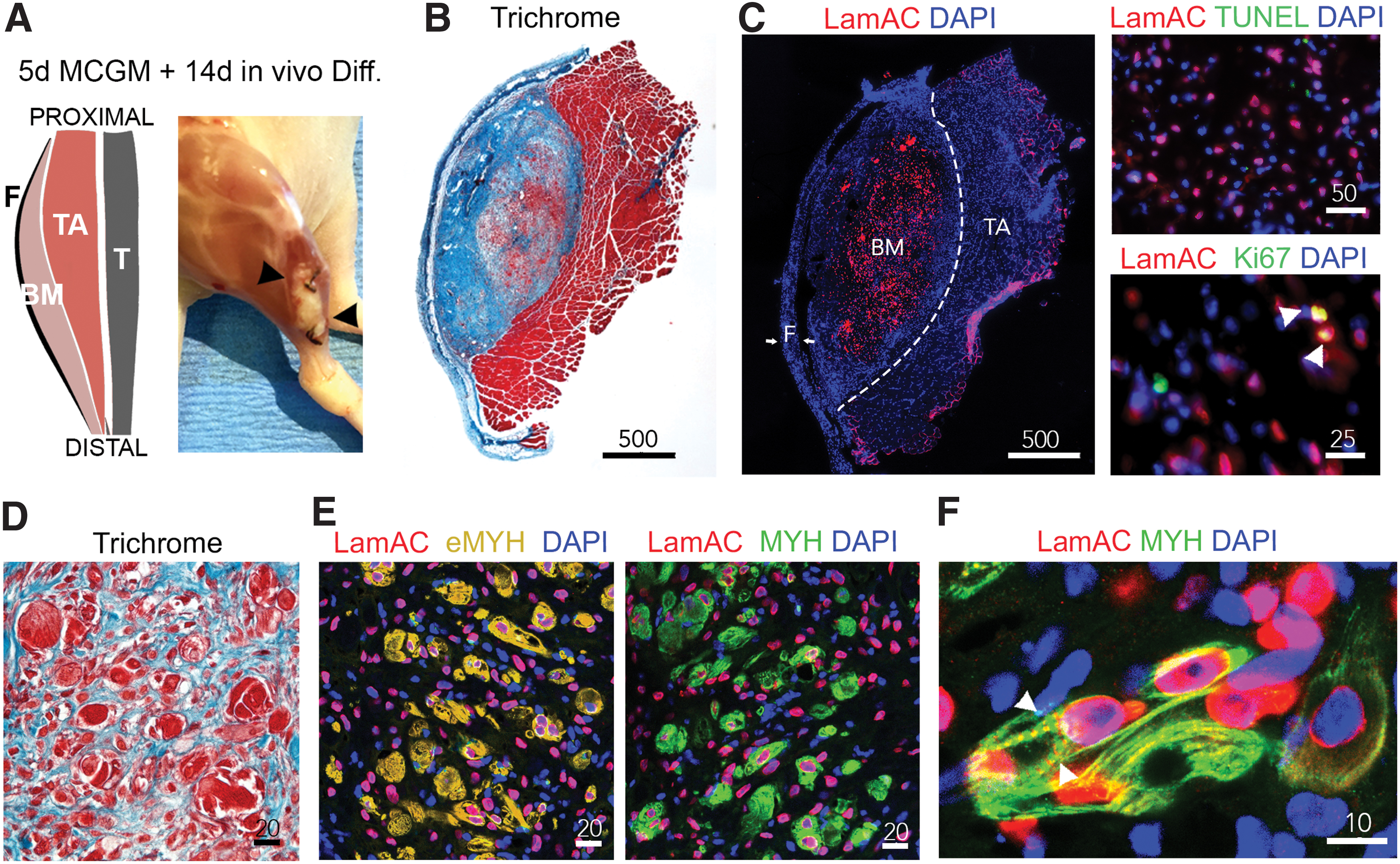
In vivo maturation of bioengineered laryngeal grafts.
On day 14 postimplantation, trichrome stain of cross sections from explanted tissue revealed a defined area of muscle-like tissue within the implant (Fig. 5B). Immunofluorescence images showed the distribution of human identified cells (LamAC+) within the implanted scaffold (Fig. 5C, left panel). Absence of TUNEL indicated viability of LamAC+ cells in the bioengineered muscle constructs. As to be expected, there was little proliferation of LamAC+ cells during the stage of maturation (Fig. 5C, right panel). In contrast to day 5 of ex vivo culture, early developmental fiber formation could be observed (Fig. 5D) and a broad myosin expression of LamAC+ cells, as well as, formation of multinucleated fibers was found (Fig. 5E, lefty panel). The presence of embryonic myosin confirmed an early developmental phenotype (Fig. 5E, right panel). We did not see contribution of recipient myoblast (LamAC−/Myosin+) to muscle formation within implants. In addition, LamAC+ cells were not found beyond the border of the implant (Supplementary Fig. S3). Finally, fibers of organized myosin striation were present in the newly formed muscle-like laryngeal tissue (Fig. 5F).
Discussion
The human larynx is a highly orchestrated organ, with several tissue compartments contributing to the function of swallowing, breathing, and phonation. To achieve laryngotypic organ function various tissue compartments are necessary. In this study, we describe the decellularization of whole larynxes by vascular perfusion, on a clinically relevant scale. We showed ex vivo formation of laryngeal tissue compartments from three primary human cell types under one uniform culture condition and assessed in vivo compatibility and tissue maturation. Immersion decellularization of human larynxes has been achieved by 25 consecutive cycles of a detergent-enzymatic method, which reportedly took several weeks. 17
More recently, acellular laryngeal scaffolds were generated from pigs within 8 days using negative pressure in combination with freeze-thawing and hyper-hypotonic solutions, followed by detergent-enzymatic immersion. 18 While this method reduced the processing time significantly, a loss of mechanical durability of the laryngeal cartilage was reported and attributed to a substantial loss in GAG content during the decellularization process. With our selective perfusion protocol, we minimized exposure of detergents to 5 days, which efficiently eliminates immunogenic properties and preserves the critical GAG content and biomechanical properties of the avascular cartilage.
The immunologic profile of implanted decellularized scaffolds revealed a nonspecific foreign body reaction and negligible antigen-mediated immune response, comparable to SHAM procedures. In contrast, native tissue showed significant lymphocyte and M1 dominant infiltration. Notably, despite remaining chondrocytes within the cartilage, no CD3+ cells infiltrated the cartilage, nor were recruited to the perichondrium. These findings agree with previous reports of possible immune evasion of laryngeal cartilage due to low MHC II expression. 19
Development of true laryngeal tissue is challenging due to the vast number of cell types that resident within the organ, each with special requirements for culture conditions. In this work, we propose a uniform culture condition to support development of laryngeal grafts from three human primary cell types ex vivo. A cell culture medium that supports endothelial maturation as well as cell engraftment and proliferation of epithelial cells and myoblasts is a useful tool for ex vivo laryngeal tissue assembly. By using four vascular accesses for cell delivery, we achieved hierarchical reendothelialization within whole larynxes.
This is, to our knowledge, the first report of a reendothelialized laryngeal scaffold, a crucial step in achieving vital, transplantable laryngeal grafts. 20 The introduction of MCGM allowed for endothelial maturation, as suggested by strengthened endothelial barrier and reduction in mitosis. 21 We next explored development of mucosal epithelial lining from hBCs and achieved a continuous epithelial monolayer on full thickness laryngeal mucosa formed by intercellular adherence junction. Due to the anatomical location of the larynx, orthotopic implants are prone to infection. The epithelium's primary role is a first-line defense by providing a barrier. 22 Thus ex vivo development of a continuous cell layer may minimize risk of infection. 23 Presence of basal cell markers suggests the newly formed tissue could be differentiated into pseudostratified ciliated epithelium in vivo, however, a large animal transplantation model would be needed to assess in vivo epithelial differentiation of laryngeal mucosa.
We further showed that intrinsic laryngeal muscle ECM could be repopulated with primary hMBs ex vivo. A crucial question in the field of organ engineering is the optimal maturation stage at which to transplant grafts regenerated ex vivo. 24 We did not see myofiber formation after ex vivo culture, which was to be expected due to short duration of culture and absence of biochemical or biomechanical stimuli. By providing these stimuli upon heterotopic implantation, we achieved early differentiation to muscle-like tissue. Extended in vivo maturation would likely be needed to develop functional, more mature laryngeal muscle.
The heterotopic implantation site proved the concept of in vivo differentiation of bioengineered muscle constructs. One limitation of this model is that the implanted construct is in close proximity to native muscle. Noteworthy, we did not observe recipient myoblast migration into the implant. 25 An orthotropic transplantation of the bioengineered graft is preferred. However, an animal model matching the size of the donor animal would be required.
The complexity of tissue architecture and cellular composition makes functional replacement of the larynx challenging. Ideally, engineered laryngeal tissue grafts could be constructed ex vivo and further matured orthotopically to replace missing laryngeal function. We achieved early tissue formation of three laryngeal compartments by introducing one uniform culture condition, a key factor in developing multicellular, ready-to-transplant tissue grafts.
Footnotes
Acknowledgment
The authors thank MGH Center for Skeletal Research Core for histological processing and MGH Department of Radiology for CTA image acquisition.
Disclosure Statement
Dr. H.C.O. is founder and stockholder of IVIVA Medical, Inc., This relationship did not affect the present study. The authors P.T.M., M.G., G.R.D., D.E.-L., J.M.C., X.R., S.E.G., B.J.J., J.R.G., G.R.G., and C.J.H. have no competing interest. The funding organization had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data.
Funding Information
The study was funded by the Sara and Charles Fabrikant MGH Research Scholarship and by departmental funds.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.